
Abstract
Emerging evidence suggests that gap junctional intercellular communication (GJIC) and expression of connexins (Cx) contribute to the metastatic potential of breast cancer cells. To more directly address this, an aggressive bone metastasis breast cancer cell line, MDA-MET (MET), was stably transfected with human Cx43 cDNA (MET/Cx43+). Focusing on clone 28 of MET/Cx43+, we demonstrated that GJIC, Cx43 protein and Cx43 mRNA were significantly increased in MET/Cx43+ cells relative to MET, the plasmid control for the Cx43 transfectants (MET/HY) and a metastatic breast cancer cell that is less metastatic to bone than MET, MDA-MB-231. Cx26 mRNA was also increased in MET/Cx43+ clone 28 cells while mRNA for Cx32, Cx37, Cx40 and Cx45 were not detected in any of the breast cancer cell lines examined. MET/Cx43+ clone 28 invasiveness was decreased by 33% relative to MET/HY, while their ability to migrate was unchanged. The ability of MET/Cx43+ clone 28 cells to adhere to hFOB and HUV-EC-C cells was decreased approximately 30% and 70%, respectively, relative to MET and MET/HY. E-cadherin and N-cadherin proteins were not detected in MET, MDA-MB-231, MET/Cx43+ clone 28 and MET/HY cells. However, OB-cadherin protein levels were decreased approximately 43% in MET/Cx43+ clone 28 relative to MET/HY cells. These findings suggest that GJIC and Cx43 expression contribute to breast cancer cell adhesion and migration, possibly through a mechanism involving OB-cadherin, and these changes in turn regulate the metastatic potential of breast cancer cells, especially to bone.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gap junctions are membrane-spanning channels that facilitate intercellular communication by allowing small signaling molecules to pass from cell to cell. Gap junctions are formed by plasma membrane connexons, each of which is comprised of six connexins (Cx). Thus far, at least 20 Cxs have been identified in mammalian species. Within the connexin family, Cx43, Cx26 and Cx32 have been most intensively investigated in human breast tissue [1]. As early as the 1960s, the loss of gap junctional intercellular communication (GJIC) was described in cancer cells [2]. Emerging evidence suggests that GJIC and connexin expression contribute to cancer cell metastasis [3–6]. For instance, previous studies from our laboratory suggest that increased heterotypic GJIC between breast cancer cells and osteoblastic cells, relative to homotypic communication between breast cancer cells and themselves, contributes to metastatic potential [4]. Furthermore, Pollman et al. [6] recently demonstrated that increasing Cx43 expression in HBL100, a normal breast epithelial cell line, increased adherent migration through endothelial cells. On the other hand, highly metastatic MDA-MB-435 cells express no Cx43 whereas non-metastatic breast epithelial cells express abundant Cx43 and expressing the metastasis suppressing gene BRMS-1 restores Cx43 expression in MDA-MB-435 cells [3]. Thus, while accumulating data suggest a role for connexins and GJIC in metastasis, what this role is, and indeed whether connexins and GJIC potentiate or ameliorate metastatic potential, is unclear. To address this we expressed Cx43 in a bone homing variant of the breast cancer cell line MDA-MB-231.
We focused on Cx43 and selected a highly bone-metastatic breast cancer cell line (MET) as a cell model. MET is a breast cancer cell line with an aggressive bone metastatic phenotype developed by repeated passage, in immune compromised mice, of the less metastatic breast cancer cell line MDA-MB-231. After inoculation, the metastatic tumor cells from bone were isolated, grown in tissue culture, and then re-inoculated into the left ventricle of female nude mice. The cells again metastasized to bone, were expanded and re-inoculated until no micrometastases were detected in other organs, only in bone. MET cells exhibited enhanced early adhesion to type IB collagen, were more invasive through Matrigel™ and had a high level of interleukin-8 (IL-8) expression [7]. To examine the role of Cx43 in breast cancer cell metastasis, we transfected MET cells with Cx43 cDNA and examined GJIC, connexin expression, invasion, migration and adhesion. Furthermore, we examined cadherin, focal adhesion kinase (FAK), paxillin and CT10 regulator of kinase protein (Crk) proteins to explore the adhesion signaling pathway which may contribute to the bone-metastatic potential of breast cancer cells.
Material and methods
Reagents
All reagents were of analytical grade and commercially available. Anti-Cx43 monoclonal and polyclonal antibodies were purchased from Zymed (San Francisco, CA). MET cell lines were provided by Dr. Larry J. Suva (University of Arkansas, Little Rock, AR.); cells from the human osteoblastic cell line, hFOB 1.19, were provided by Steven Harris (Mayo Clinic, Rochester, MN.) [8]; 435/BRMS-1 cells (MDA-MB-435 cells transfected with the breast cancer suppressor gene BRMS-1) were provided by Danny R. Welch (University of Alabama, Birmingham, AB). MDA-MB-231, MDA-MB-435, hTERT (non-tumorigenic, non-metastatic human breast epithelial cell line, hTERT-HME1) and HUV-EC-C (human umbilical vein endothelial cell line) cells were purchased from ATCC (Rockville, MD). pcDNA3.1/hygromycin B (-), hygromycin B, ampicillin, Top10 One Shot kits and Lipofectamine reagents were purchased from Invitrogen (Carlsbad, CA.), Bam H1, Xba-I, KpnI and T4 DNA ligase from Boehringer Mannheim (Indianapolis, IN), and Qiaquick Gel Extraction, EndoFree Plasmid and RNeasy kits from Qiagen (Valencia, CA).
Construction of MET cells expressing Cx43
A 1,817 bp fragment of human Cx43 cDNA [9] was ligated into the Xba1 and Kpn1 sites of the pcDNA3.1/hygro (-) vector in the sense orientation. The sense orientation structure of pcDNA3.1/hygro (-)/human Cx43+ was analyzed and confirmed by sequence analysis (sequencer ABI model 377; Forster, CA) and was named Cx43+. The plasmid vector control pcDNA3.1/hygro (-) was named HY. For transfection, MET cells were plated at 1 × 104 cells/mm2 in 60 mm dishes containing 5 ml of Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS; 9/1, v/v, DMEM/FBS) and incubated at 37°C and 5% CO2 for 36 h. Cx43+ or HY (2 μg) were pre-complexed with 6 μl Plus reagent (Lipofectamine kit) and incubated at room temperature for 15 min. Lipofectamine (12 μl) was added to DMEM (240 μl), mixed with the pre-complex containing either Cx43+ or HY, brought to a final volume of 260 μl with transfection medium and incubated at room temperature for 15 min. Prior to the addition of the transfection medium, the MET cells were washed once and maintained in 2 ml DMEM. The transfection medium (260 μl) was added to each MET cell dish, mixed gently and incubated at 37°C and 5% CO2. After a 3 h incubation, 2.26 ml of DMEM supplemented with 20% FBS (8/2, v/v) was added to the cells and incubated at 37°C and 5% CO2. Following this 24-h incubation, the medium was replaced with selection medium containing 300 μg/ml hygromycin B and cultured for 10–14 days and media replaced at three day intervals until resistant colonies were clearly visible. Resistant colonies (approximately 100–200 cells) were trypsinized in plastic colonizing cylinders. The cells were sub-cultured and cultured in maintenance medium containing 150 μg/ml hygromycin B until a sufficient number of cells (approximately 1 × 107) was obtained. Routine growth conditions included incubation at 37°C and 5% CO2 with media replacement at 3 day intervals. Twenty clones of MET cells transfected with Cx43+ (MET/Cx43+) and eight clones of MET cells transfected with HY (MET/HY) were obtained. All clones were characterized and screened by real time reverse transcriptase polymerase chain reaction (RT-PCR) with Cx43 primers and probes, Western blot with Cx43 antibody and cell coupling by fluorescence activated cell sorting (FACS). The MET/Cx43+ clone expressing the greatest amount of Cx43 mRNA and protein (clone 28) and a plasmid control (HY2) were selected for focused study.
Invasion and migration assay
A BioCoat FluoroBlok invasion system and FluoroBlok Multiwell insert system (Becton Dickinson Labware, Bedlard, MA) were used to assess invasion and migration of the cells according to the manufacturer’s protocol. Briefly, in using the BioCoat FluoroBlok invasion system, 0.5 ml of PBS was added to the interior of the inserts and incubated in a 37°C incubator (without CO2) for 2 h. MET/Cx43+ clone 28, MET/HY, MET and MDA-MB-231 cells were cultured in DMEM with 10% FBS (9/1, v/v) for assays. On the day of experiment, 2.5 × 104 cells in 0.5 ml of DMEM was added to the top chamber of BioCoat FluoroBlok invasion system or FluoroBlok Multiwell insert system and 0.75 ml of culture medium containing 10% FBS (9/1; v/v) was added to the bottom chamber as a chemoattractant. Chambers with cells were incubated for 22 h. The invading or migrating cells to be placed within the wells were stained with 1 mM calcein and incubated at 37°C and 5% CO2 for one hour. Cell invasion and migration were assessed using a fluorescence plate reader at excitation and emission wave lengths of 485 and 530 nm, respectively.
Adhesion assay
To assess the adhesion capacity of breast cancer cells, cells from the human osteoblastic cell line, hFOB 1.19, and the human umbilical vein endothelial cell line, HUV-EC-C, were selected as cancer cell adhesion targets. Cells were cultured in 100 mm dishes until confluent and labeled with 5 μM carboxyfluorescein diacetate, succinimidyl ester (CFDA SE; V#-12883, Molecular Probes, Eugene, OR). The dishes were covered with aluminum foil to prevent exposure to light and incubated at 37°C and 5% CO2 for 15 min. Media with dye was then replaced with regular culture media (DMEM) and incubated for another 30 min. Dye-labeled cells were washed with PBS, trypsinized and suspended in DMEM. 5 × 105 of dye-labeled cells were added to a 24-well plate covered with a monolayer of adhesion target cells (hFOB cells or HUV-EC-C). Following a 2 h incubation, wells were washed three times with PBS to remove non-adherent cells. Adhesion was assessed by using a fluorescence plate reader at excitation and emission wave lengths of 485 and 530 nm, respectively.
Cell coupling
GJIC was assessed with a double label FACS assay described previously [3, 4]. Briefly, donor cells were double-labeled for 30 min with the gap junction permeable dye calcein AM (10 μg/ml) and the lipophilic dye DiI (8.75 μg/ml) (Molecular Probes) in Hank´s Balanced Salt Solution (HBSS; Invitrogen) containing 1% bovine serum albumin (BSA; 1/100, w/v)) and Pluronic F-127 (Molecular Probes). After one washing with PBS, 1 × 104 of the double-labeled donor cells were dropped onto acceptor cells and incubated for 24 h at 37°C and 5% CO2. The co-cultured cells were then harvested by trypsinization. After fixation with 2% paraformaldehyde for 15 min, cells were resuspended in FACS buffer and analyzed by FACS. Using the same procedure, double-labeled donor cells were dropped onto unlabeled acceptor cells of the same cell type for assessment of homotypic GJIC.
Quantitative analysis of protein levels by Western blot
Western blots were utilized to examine protein levels. Total protein, including membrane and cytosolic fractions, isolated from each cultured cell clone and cell line were analyzed by Western blot with antibodies against Cx43, E-cadherin, N-cadherin, OB-cadherin, focal adhesion kinase (FAK), paxillin and CT10 regulator of kinase protein (Crk) and re-probed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as previously described [9]. Equivalent amounts of each protein (40 μg) were applied to 10% pre-cast SDS-polyacrylamide gels (Bio-Rad, Hercules, CA.) at 100 volts for 1.5 h and transferred to nitrocellulose membranes at 40 volts overnight at 4°C. The membrane was blocked with 5% non-fat milk TTBS solution containing 0.05% Tween-20, Tris-Buffered Saline (TBS) (Bio-Rad) for one hour before an incubation of 3 h at room temperature with each antibody diluted 1:1,000–1:500 in 5% non-fat milk TTBS solution. The membranes were washed three times with TTBS solution and incubated for one hour with goat anti-mouse (for monoclonal antibodies) or goat anti-rabbit (for polyclonal antibodies) IgG linked horseradish peroxidase (Bio-Rad) diluted 1:3,000 in 5% non-fat milk TTBS solution. After three additional washes with TTBS solution, the membranes were soaked in enhanced chemiluminescence (ECL) detection reagents (Amersham, UK). The membranes were then air-dried and exposed to X-ray film. Densitometry measurements were performed using a Bio-Rad Q-one program (Bio-Rad). The densities of the proteins of interest were normalized to GAPDH density.
Quantitative real-time RT-PCR
Total RNA was isolated from each cell line using a Qiagen RNeasy mini kit. Gene expression was quantified by real-time RT-PCR using an Applied Biosystems 7300 RealTime PCR System as previously described [9]. Primers and probes were designed using sequence data from Genbank and the real-time PCR probe/primer design software Primer Express (version 1.0, Applied Biosystems). For real time qPCR analysis, DNAse-treated RNA was reverse-transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems) and the protocol provided with the kit. Quantification by real-time RT-PCR was done by adding 10 or 20 ng of cDNA in a reaction with 2X TaqMan Universal PCR Master Mix (Applied Biosystems) in a volume of 20 μl. Primers were added at a concentration of 400 nM and a probe which was labeled with a 5′ FAM and a 3′ Black Hole Quencher (Biosearch Tech, Novato, CA) was added at 200 nM. The amplification protocol consisted of 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and one minute at 60°C. Ct values of genes of interest and the reference gene 18 s were calculated using the delta Ct method to determine relative levels of gene expression for Cx26, Cx32, Cx37, Cx43, Cx40, Cx45 mRNA.
Statistical analysis
All experiments were repeated at least four times in duplicate and in some cases triplicate. Four clones of 435/Cx43+, two clones of 435/hy and other cell lines were tested in all experiments. Results were reported as the mean ± SEM. One-way analysis of variance (ANOVA) followed by a student-Newan-Keuls test, was used to analyze all data. P < 0.05 was considered significant.
Results
Connexin expression and cell-to-cell communication
Cx43 protein and Cx43 mRNA were expressed by several clones of MET/Cx43+ including C1, C10, C13, C28 and by hFOB and hTERT cells, however, very little, if any, Cx43 protein was detected in MET, MDA-MB-231 or MET/HY plasmid control clones (Fig. 1a, b). We report results for MET/Cx43+ clone 28 as it consistently expressed Cx43 protein to the greatest degree. However, similar results were obtained for the other three clones for all assays examined. Abundant Cx43 mRNA was detected in MET/Cx43+ clone 28 and hTERT while significantly less was detected in MET, MDA-MB-231 and MET/HY plasmid control clone (Fig. 1c). Steady state Cx26 mRNA levels were also increased in MET/Cx43+ clone 28 and hTERT relative to MET and MET/HY (Fig. 1d). Cx32, Cx37, Cx40 and Cx45 were not detected in any of the breast cancer cell lines examined (data not shown). GJIC was significantly greater in hTERT cells relative to all other cells examined. Among the breast cancer cell lines there was a trend for an increase in GJIC in MET/Cx43+ clone 28 but this did not reach statistical significance (Fig. 1e).

Connexin expression and GJIC. (a) Western blot analysis of Cx43 and GAPDH in 4 of 20 clones of MET-231 cells transfected to express Cx43 (MET/Cx43+; C1, C10, C13, and C 28), two clones of the plasmid control for this transfection (MET/HY; HY2 and HY8); the parental cell line for this transfection (MET); human fetal osteoblastic cells, hFOB 1.19 (hFOB); MDA-MB-231 cells (231) and non-metastatic breast epithelial cells (hTERT). A 43-kd band of Cx43 protein was detected in C1, C10, C13, C28, hFOB and hTERT cells, but not in HY2, HY8, MET and 231 cells. Results are typical of five gels. Clone 28 had consistently high levels of Cx43 expression and therefore was examined in subsequent experiments. (b) Densitometric analysis of Western blots. Values represent the mean ± SEM of Cx43/GAPDH protein band density ratio. *Significantly different than C28, n = 5. (c and d) Quantitative real time RT-PCR analysis of steady state levels of Cx43 mRNA (c) and Cx26 mRNA (d). Cells were cultured until confluent before RNA isolation. For c, Cx43 mRNA levels, *significantly different than C28 and hTERT and # significantly different than C28, n = 3–6. For d, Cx26 mRNA, *significantly different from hTERT and # significantly different from 231, n = 3–12 (e) Homotypic GJIC. Cells were cultured and subjected for dual-label dye transfer assay and assessed by FACS as described in Materials and Methods. *Significantly different than hTERT. Values are mean ± SEM coupled cells per donor cell, n = 5
Expression of cadherin, FAK, paxcillin and Crk proteins
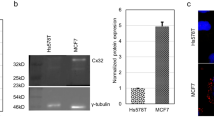
We did not detect E- or N-cadherin by Western blot analysis in any of the breast cancer cell lines examined. However, OB-cadherin was detected in all cell lines examined (Fig. 2). OB-cadherin protein levels were greater in all breast cancer cell lines examined compared to hTERT. Among breast cancer cell lines, OB-cadherin levels were reduced in MET/Cx43+ clone 28 relative to MET/HY, MET and MDA-MB-231. Western blot analysis also revealed that FAK, paxillin and Crk were expressed by all cell lines examined but there were no consistent changes in the level of these proteins amongst cell lines (data not shown).

Western blot analysis of OB-cadherin protein expression. Proteins isolated from cells and analyzed by Western blot with monoclonal antibodies against OB-cadherin and GAPDH. Cell lines are as in Fig. 1. (a) Representative gel. (b) Quantification of Western blots. Values represent the mean ± SEM ratio of Cx43/GAPDH protein band density ratio, n = 4. *Significantly different than HY2, MET and 231
Adhesion, invasion and migration
MET/Cx43+ clone 28 adhered less well to hFOB 1.19 (Fig 3a) although this did not quite reach statistical significance. However, the decreased adhesion of MET/Cx43+ clone 28 to HUV-EC-C (Fig. 3b), relative to MET/HY or MET cells, was statistically significant. We also examined the adhesion to HUV-EC-C of three additional Cx43 clones. While there was a general decrease in adhesion to HUV-EC-C of all Cx43 expressing clones the degree to which adhesion decreased was variable and did not correlate significantly with Cx43 expression levels. Invasion of MET/Cx43+ clone 28 through Matrigel™ was decreased relative to invasion of MET/HY or MET cells (Fig. 3c). Migration was similar in all breast cancer cell lines examined (Fig. 3d).

Cellular adhesion, invasion and migration. Adhesion of breast cancer cells to (a): hFOB or (b) HUV-EC-C cells was assessed as described in methods and materials. Values represent arbitrary fluorescent units from adhered fluoroscein-labeled breast cancer cells, n = 8–20. *Significantly different than either HY2 or MET cells. To assess cellular invasion (c) and migration (d), invading or migrating cells were stained with calcein and invasion or migration assessed by quantifying fluorescence. (c) Values represent mean ± SEM arbitrary florescent units of calcein labeled cells, n = 8–28. *Significantly different than MET cells. There were no significant differences in migration or adhesion on hFOB cells
Discussion
We found that Cx43 levels were dramatically reduced, relative to non metastasizing breast epithelial cells, in all non-transfected breast cancer cell lines examined, confirming our earlier reports [3]. Additionally, Cx43 expression was further decreased in the more bone homing MET than in the more generally metastatic MDA-MB-231. These results suggest that a decrease in Cx43 expression contributes to metastasis to bone. GJIC was also reduced in the breast cancer cell lines but was not different between MET/HY and MDA-MB-231 suggesting that GJIC is related to overall, but not bone, metastasis. Over expression of Cx43 in MET/Cx43+ clone 28 cells increased GJIC such that it was greater than that in MET/HY and MDA-MB-231, although this did not reach statistical significance, but it did not return GJIC levels to those seen in non-metastasizing breast epithelial cells. This suggests that decreases in the expression of connexins other than Cx43 may contribute to the decreased GJIC observed in breast cancer cell lines. Indeed, we found lower expression of Cx26 in MDA-MB-231 cells relative to non-tumorigenic, non-metastasizing breast epithelial cells, hTERT, and still lower Cx26 in MET/HY relative to MDA-MB-231. Restoring Cx43 did not restore Cx26, nor did it lower Cx32, expression. Taken together these results suggest that a decrease in Cx43 and Cx26 are related to breast cancer metastatic potential.
Breast cancer cells metastasize to bone mainly through nutrient arteries, which communicate with a sinusoidal network in the bone marrow [10]. They express cellular adhesion molecules to establish contact with sinus endothelial walls and secrete proteolytic enzymes to degrade the endothelial wall and pass into the bone. Additionally, to reach the bone extracellular matrix invading breast cancer cells must also cross a layer of bone lining osteoblastic cells which, it is generally believed, cover all bone surfaces [11]. Therefore, in this study we examined adhesion of breast cancer cells to an endothelial cell line, HUV-EC-C, as well as an osteoblastic cell line, hFOB 1.19. We found that MET/HY and MET cells adhered to both HUV-EC-C and hFOB 1.19 cells to a greater degree than did MDA-MB-231 cells. Furthermore, expressing Cx43 in MET cells decreased adhesion to both target cell types. While adhesion to hFOB was decreased in Cx43 expressing clone 28 and 231 cells relative to HY2 and MET cells this did not reach statistical significance. However, this trend did reach statistical significance as regards adhesion to HUV-EC-C. We interpret this to mean that Cx43 contributes more to adhesion to HUV-EC-C cells than to hFOB cells. Invasion followed a similar trend as adhesion but migration was similar in all cell lines examined. It is important to note that HUV-EC-C may be a less than ideal model to study adhesion and invasion in the bone microenvironment. For instance, previous studies have demonstrated distinct differences in expression of cell adhesion molecules among endothelial cell types including bone marrow endothelial cells and HUV-EC-C [12, 13]. Therefore, future studies will examine breast cancer cell adhesion to bone marrow endothelial cells.
Restoring Cx43 in MET cells only partially restored GJIC and this was not statistically significant. While this does not rule out the possibility of increased GJIC in clone 28 cells contributing to their decreased, relative to MET, adhesion and invasion, it does suggest non-GJIC functions of Cx43 may contribute to their decreased adhesion and invasion. One possibility could be Cx43 contribution to hemichannel function.
Some caution should be taken in interpreting these results. Comparing the behavior of 231 to that of either MET or the Cx43 expressing clone 28, can be misleading. While MET and clone 28 have nearly identical genetic backgrounds, their backgrounds are likely quite different than that of 231. A more appropriate comparison is MET to clone 28 which reveals changes in cell behavior more likely a result of changes in only Cx43 expression. When one compares MET or MET/HY to clone 28 an inverse relationship between Cx43 protein, mRNA, GJIC and adhesion and invasion is apparent.
These results suggest that expressing Cx43 decreases breast cancer cell adhesion to those cells comprising the tissue through which they must migrate during extravasation and colonization.
Our results are somewhat inconsistent with those of Pollmann et al. [6]. These investigators found that expressing Cx43 in non-metastatic human breast epithelial cells (HBL100) did not affect adherence to human microvascular endothelial cells and actually increased migration of HBL100 cells through an endothelial monolayer. The reason for this discrepancy is unclear. One possibility is that we examined the effect of Cx43 expression on breast cancer cells whereas Pollmann et al. [6] examined HBL100 cells, which are non-metastatic, do not display GJIC and express no known connexins. It is possible that the dramatically different phenotype of MDA-MB-231 cells and HBL100 explains the different effect of Cx43 expression.
To examine the mechanism underlying the decreased adhesion of MET/Cx43+ clone 28 cells we examined the expression of the cell–cell adhesion molecules E-, N- and OB-cadherins. Since connexin expression and gap junction function are related to cell–cell adhesion and cadherins are adhesion molecules that are involved in Cx43 formation of functional gap junctions [14], we examined the cadherin profile of the breast cancer cell variants examined in this study. Our goal was to determine whether the differences observed in heterotypic cell–cell adhesion might be related to changes in cadherin expression as has been previously suggested [4]. While several cadherins are expressed by epithelial cells, we focused on E-cadherin, N-cadherin and OB-cadherin for several reasons. We focused on E-cadherin because it is present in cells of epithelial origin. E-cadherin is absent or downregulated in most invasive breast and other epithelial carcinomas [15] and replaced by aberrant expression of other cadherins [16]. N-cadherin is involved in increased motility, invasion and metastasis of breast cancer cells [17, 18] and is normally expressed by cells of mesenchymal origin, including osteoblasts. Osteoblasts also express OB-cadherin [19], and since previous results from our laboratory suggest that MDA-MB-231 display osteomimicry [4] we also examined OB-cadherin in breast cancer cell lines. We did not detect E- or N-cadherin in any of the breast cancer cell lines examined. However, MET, MET/HY and MDA-MB-231 cells expressed abundant OB-cadherin, the expression of which was reduced by forced expression of Cx43 in MET/Cx43+ clone 28 cells. These findings suggest that Cx43 may regulate invasion and adhesion by interfering with OB-cadherin protein expression. Because it is associated with breast cancer cell metastasis and an aggressive phenotype [20, 21] we also examined the expression of FAK. However, we found no consistent changes in FAK expression in any of the cell lines examined suggesting changes in FAK do not contribute to the effect of Cx43 on heterotypic adhesion of breast cancer cells to bone or endothelial cells.
In summary, our studies demonstrate that Cx43 is downregulated in metastatic breast cancer cells and especially so in the bone homing variant MET. More importantly, bone homing MET cells adhere to a greater degree to either osteoblastic hFOB cells or endothelial cells, and show a greater degree of Matrigel™ invasion, than do MET-231 cells. Restoring Cx43 expression decreases this heterotypic cell–cell adhesion and invasion. Finally, OB-cadherin is decreased in the less adhesive and less invasive MET/Cx43+ clone 28 cells relative to the other MDA-MB-231 variants suggesting that it may contribute to the anti-adhesive anti-invasive properties of Cx43.
References
Pozzi A, Risek B, Kiang DT et al (1995) Analysis of multiple gap junction gene products in the rodent and human mammary gland. Exp Cell Res 220(1):212–219
Loewenstein WR, Kanno Y (1966) Intercellular communication and the control of tissue growth: lack of communication between cancer cells. Nature 209(29):1248–1249
Saunders MM, Seraj MJ, Li Z et al (2001) Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Research 61(5):1765–1767
Kapoor P, Saunders MM, Li Z et al (2004) Breast cancer metastatic potential: correlation with increased heterotypic gap junctional intercellular communication between breast cancer cells and osteoblastic cells. Int J Cancer 111(5):693–697
Ito A, Katoh F, Kataoka TR et al (2000) A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. J Clin Invest 105(9):1189–1197
Pollmann MA, Shao Q, Laird DW et al (2005) Connexin 43 mediated gap junctional communication enhances breast tumor cell diapedesis in culture. Breast Cancer Res 7(4):R522–R534
Bendre MS, Gaddy-Kurten D, Mon-Foote T et al (2002) Expression of interleukin 8 and not parathyroid hormone-related protein by human breast cancer cells correlates with bone metastasis in vivo. Cancer Res 62(19):5571–5579
Harris SA, Enger RJ, Riggs BL et al (1995) Development and characterization of a conditionally immortalized human fetal osteoblastic cell line. J Bone Miner Res 10(2):178–186
Li Z, Zhou Z, Saunders MM et al (2006) Modulation of connexin43 alters expression of osteoblastic differentiation markers. Am J Physiol Cell Physiol 290(4):C1248–C1255
De Bruyn PP (1981) Structural substrates of bone marrow function. Semin Hematol 18(3):179–193
Chole RA, Tinling SP (1994) Bone lining cells of the mammalian cochlea. Hear Res 75(1–2):233–243
Schweitzer KM, Vicart P, Delouis C et al (1997) Characterization of a newly established human bone marrow endothelial cell line: distinct adhesive properties for hematopoietic progenitors compared with human umbilical vein endothelial cells. Lab Invest 76(1):25–36
Hillyer P, Mordelet E, Flynn G et al (2003) Chemokines, chemokine receptors and adhesion molecules on different human endothelia: discriminating the tissue-specific functions that affect leucocyte migration. Clin Exp Immunol 134(3):431–441
Trosko J, Ruch R (1998) Cell–cell communication in carcinogenesis. Front Biosci 3:d208–d236
Perl AK, Wilgenbus P, Dahl U et al (1998) A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392(6672):190–193
Moll R, Mitze M, Frixen UH et al (1993) Differential loss of E-cadherin expression in infiltrating ductal and lobular breast carcinomas. Am J Pathol 143(6):1731–1742
Nieman MT, Prudoff RS, Johnson KR et al (1999) N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol 147(3):631–644
Hazan RB, Phillips GR, Qiao RF et al (2000) Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol 148(4):779–790
Kawaguchi J, Kii I, Sugiyama Y et al (2001) The transition of cadherin expression in osteoblast differentiation from mesenchymal cells: consistent expression of cadherin-11 in osteoblast lineage. J Bone Miner Res 16(2):260–269
van Nimwegen MJ, van de Water B (2007) Focal adhesion kinase: a potential target in cancer therapy. Biochem Pharmacol 73(5):597–609
Lark AL, Livasy CA, Dressler L et al (2005) High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod Pathol 18(10):1289–1294
Acknowledgements
We thank Dr. Deborah S Grove for Real time RT-PCR technical support. This work was supported by the National Institutes of Health, National Cancer Institute grant R01-CA 90991.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, Z., Zhou, Z. & Donahue, H.J. Alterations in Cx43 and OB-cadherin affect breast cancer cell metastatic potential. Clin Exp Metastasis 25, 265–272 (2008). https://doi.org/10.1007/s10585-007-9140-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-007-9140-4