
Abstract
Over 90% of all cancers are carcinomas, malignancies derived from cells of epithelial origin. As carcinomas progress, these tumors may lose epithelial morphology and acquire mesenchymal characteristics which contribute to metastatic potential. An epithelial-to-mesenchymal transition (EMT) similar to the process critical for embryonic development is thought to be an important mechanism for promoting cancer invasion and metastasis. Epithelial-to-mesenchymal transitions have been induced in vitro by transient or unregulated activation of receptor tyrosine kinase signaling pathways, oncogene signaling and disruption of homotypic cell adhesion. These cellular models attempt to mimic the complexity of human carcinomas which respond to autocrine and paracrine signals from both the tumor and its microenvironment. Activation of the epidermal growth factor receptor (EGFR) has been implicated in the neoplastic transformation of solid tumors and overexpression of EGFR has been shown to correlate with poor survival. Notably, epithelial tumor cells have been shown to be significantly more sensitive to EGFR inhibitors than tumor cells which have undergone an EMT-like transition and acquired mesenchymal characteristics, including non-small cell lung (NSCLC), head and neck (HN), bladder, colorectal, pancreas and breast carcinomas. EGFR blockade has also been shown to inhibit cellular migration, suggesting a role for EGFR inhibitors in the control of metastasis. The interaction between EGFR and the multiple signaling nodes which regulate EMT suggest that the combination of an EGFR inhibitor and other molecular targeted agents may offer a novel approach to controlling metastasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
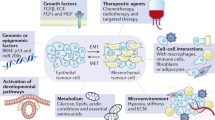
Human cancers rely on multiple overlapping signal transduction pathways to activate and regulate cellular proliferation, survival and migration programs. The epithelial-to-mesenchymal transition (EMT) is a critical process in embryonic development for metazoan organisms and a similar process has also been shown to play a role in oncogenic progression and metastasis. Tumor metastasis involves a sequential series of processes which promote and regulate the escape of migratory cancer cells to generate metastatic lesions at distant sites. The process begins in the primary tumor, where tumor cells dysregulate homotypic cell adhesion, downregulate cell adhesion proteins such as E-cadherin, and upregulate proteins characteristic of a more motile, mesenchymal-like phenotype such as vimentin. This process requires transcriptional reprogramming to suppress E-cadherin expression via transcription factors associated with EMT (for review see [1]). Tumor cells undergoing EMT have been shown to undergo “cadherin switching”, downregulating E-cadherin and compensating with alternate cadherin proteins such as N cadherin [2]. There is evidence that the downregulation of E-cadherin and upregulation of proteins characteristic of a mesenchymal phenotype may occur preferentially at the invasive edge of a tumor [3]. Initiation of metastasis requires this initial disruption of cell–cell junctions and gain of cellular motility, permitting individual cells to migrate away from the primary tumor. In order to migrate through the surrounding extracellular matrix (ECM) cells may upregulate secreted proteases such as the matrix metalloproteinases (MMPs). These motile, invasive cells may then cross the endothelial cell barrier and intravasate into the bloodstream. Once in the bloodstream, these mesenchymal-like cancer cells can travel to distant sites where they extravasate the endothelial cell wall to colonize in a new, supportive niche. Primary tumor cells of specific cellular origins have been shown to preferentially colonize specific tissues, although the reasons for this are not entirely clear. However it is commonly accepted that once a metastatic tumor cell has implanted in a niche supportive of proliferation, that cell may undergo a mesenchymal-to-epithelial transition (MET). Consistent with this, the emerging metastatic tumor often resembles the primary tumor from which it derived both in cellular phenotype and multi-cellular architecture. It is not clear whether EMT-like changes are required for all steps in metastasis, and the possibility remains that EMT is a necessary but insufficient step in cancer metastasis.
Cellular biomarkers of EMT
A hallmark of EMT is loss of E-cadherin, a key mediator of cell–cell junctions. Numerous studies have shown a high correlation between loss of E-cadherin, the gain of vimentin and tumor invasiveness in cancer cells and patient tumors (e.g. [4–6]). A down regulation of E-cadherin most frequently results from transcriptional repression, mediated by zinc finger, forkhead domain and bHLH factors including Zeb1/TCF8/δEF1, Zeb2 (Sip1), Snail, Slug, FOXC2 and Twist. The expression of Snail, Slug and specific bHLH transcription factors have been implicated in cell survival and acquired resistance to chemotherapy [7–13]. However loss of E-cadherin alone does not constitute EMT, as cells which harbor a mutation in E-cadherin and have lost functional cell–cell junctions do not acquire the additional morphological and transcriptional changes associated with EMT [14, 15]. These changes include acquisition of cellular markers characteristic of a mesenchymal cell such as vimentin and fibronectin, expression of E-cadherin-repressing transcription factors, and frequently the acquisition of a migratory or “scattering” morphology. Loss of E-cadherin appears to be a prerequisite for tumor progression and not just a consequence of tumor dedifferentiation. In transgenic mice which spontaneously develop pancreatic tumors, E-cadherin expression was shown to decrease with tumor progression, but maintenance of E-cadherin expression during tumorigenesis arrested tumor development at the benign adenoma stage while expression of a dominant negative E-cadherin induced early metastasis [16]. Ectopic expression of E-cadherin is sufficient to suppress tumor cell invasion in vitro and tumor progression in vivo while knock-down of E-cadherin converts cells from non-invasive to invasive [17]. However, ectopic expression of E-cadherin does not restore an epithelial phenotype in cells which overexpress the transcriptional repressor Twist [18]. This implies that restoration of E-cadherin to mesenchymal cells may be insufficient to reverse EMT and confer an epithelial phenotype. Taken together, these observations are consistent with the hypothesis that E-cadherin has a tumor suppressive function and is not simply a marker of tumor differentiation.
Accumulating evidence suggests that EMT occurs at the level of transcriptional reprogramming and chromatin remodeling [18–23]. Several transcription factors have been implicated in the transcriptional repression of E-cadherin, through interaction with specific E-boxes (reviewed in [24]). The zinc finger proteins Snail, Slug, Zeb-1 and Zeb-2 (SIP1) as well as the bHLH factors Twist and E12/E47 have been shown to repress E-cadherin and markers of cell polarity. Ectopic expression of Twist or Zeb-1 is sufficient to downregulate endogenous E-cadherin and induce EMT [18, 20, 25–27]. Snail has been shown to activate the transcription of the mesenchymal biomarkers vimentin and fibronectin. Moreover, expression of Snail has been shown to induce transcriptional downregulation of E-cadherin, to upregulate vimentin, fibronectin and Zeb1 and to promote a fibroblastic, invasive cellular phenotype [28]. The fact that Zeb1 is activated by Snail implies cooperativity between these factors although Zeb1 does not appear to be directly downstream of Snail. Elegant in vivo and in vitro studies using a series of breast carcinoma cell lines with distinct metastatic properties identified Twist as a key regulator of metastasis [18]. Overexpression of Twist in non-cancerous epithelial cells induces expression of mesenchymal cell markers, repression of E-cadherin and an EMT-like phenotype. Consistent with this, suppression of Twist inhibits metastasis in a mouse mammary carcinoma model [18].
Cellular signals promoting EMT-like transitions
Activation of these transcription factors, leading to initiation of EMT and consequently metastasis, may be triggered by a variety of extra- and intracellular signals. One of the first factors observed to induce EMT was “scatter factor” or HGF [29, 30]. Since this early observation, a large number signaling pathways have been shown to induce EMT in vitro. These include growth factors (EGF, VEGF, TGF-β, Wnt, SDF, PGE2), cytokines (ILEI, interleukins), integrin signaling, extracellular matrix proteins (MMPs), inflammatory signals (COX-2), and potentially stress stimuli such as hypoxia, signaling through non-receptor tyrosine kinases such as Src and oncogenic activation of receptor tyrosine kinases RTKs) [31–35]. These EMT-activating signals can be paracrine, emanating from infiltrating stroma, or autocrine, produced by the tumor cells themselves. One intriguing report suggests that EMT may not only promote the migration of primary breast cancer tumor cells, but may also lead to the formation of nonmalignant stromal cells, and this reciprocal interaction may help explain the poor prognosis of some cancers which show evidence of EMT [36].
In addition to effects on proliferation and migration, EMT activators have been shown to promote cell survival through inhibition of apoptosis. Interestingly, TGF-β has been shown to be a potent activator of apoptosis in many cell types including epithelial cells [37, 38]. When treated with TGF-β, most fetal hepatocytes undergo apoptosis, however a fraction survive. Those hepatocytes which have survived TGF-β treatment, at least in part through resistance to apoptosis, exhibit phenotypic and genomic changes characteristic of an EMT, including increased vimentin expression and higher levels of proto-oncogene transcripts as well as elevated pAkt and Bcl-XL [39, 40]. EMT-like transitions have been also been shown to shown to confer resistance to TGF-β induced apoptosis mammary epithelial cells [41, 42]. Evidence from several independent research groups suggests that the EMT-inducing transcription factors Snail and Slug can induce expression of anti-apoptotic genes while down-regulating pro-apoptotic pathways in both epithelial cells and hematopoetic progenitor cells [43–45]. This EMT-related inhibition of apoptosis may provide selective advantage for tumor cells which are transitioning to a mesenchymal-like state. The activation of the PI3K/Akt cell survival pathway through alternate RTKs may protect against TGF-β induced apoptosis while driving pathways critical to carcinogenesis.
RTKs, such as EGFR, c-Met, IGF-1R, FGF receptors and the non-RTK c-Src have been reported to induce phosphorylation of E-cadherin and associated catenins, resulting in their degradation [46], providing a link between oncogenic activation of these kinases and induction of EMT. Thus a rationale exists for the prevention of EMT-like transitions and tumor metastasis through inhibition of these oncogenic kinases in early-stage carcinomas.
IGF-1R signaling and EMT
Several lines of evidence implicate IGF-1R signaling as an important driver of EMT. In mammary epithelial cells, constitutively active IGF-IR caused cells to undergo EMT which was associated with dramatically increased migration and invasion, and this transition was mediated by the induction of Snail and downregulation of E-cadherin [47]. Multiple groups have demonstrated that IGF-1R activation or overexpression correlates with increased invasion and metastasis [48–51]. These effects are mediated, at least in part, by its ligand, IGF-1. IGF-1 is known to influence cell adhesion to the substratum and integrin-mediated cell motility [52]. Furthermore, IGF-1 stimulation can induce the phosphorylation and transcriptional activation of β-catenin and dissociation of E-cadherin from the cell membrane [53]. In addition to disruption of homotypic cell adhesion, IGF-1 has also been shown to promote tumor invasiveness via secretion of matrix metalloproteinases [54] or crosstalk with integrin signaling pathways [55].
IGF-1R is ubiquitously expressed but is frequently overexpressed in tumors, including melanomas, pancreas, prostate and kidney (reviewed in [56]). Perhaps most relevant is not the expression level of IGF-1R but its function in cancer cells. IGF-1R signaling promotes Akt phosphorylation and protection from apoptosis, which is predicted to limit the efficacy of standard of care chemotherapies. Thus there is a strong rationale for development of IGF-1R targeted therapies, and IGF-1R inhibition might be expected to enhance the effect of cytotoxic chemotherapies or other molecular targeted therapies. Several IGF-1R targeted therapeutics are currently in early clinical trials. An array of monoclonal antibodies are currently in Phase I or Phase II trials, including CP-753,871 (Pfizer), AMG0479 (Amgen), R1507 (Genmab/Roche), IMC-A12 (Imclone), AVE-1642 (ImmunoGen/Sanofi-Aventis), MK0646 (Merck) and SCH717454 (Schering-Plough). Two low molecular weight inhibitors of the IGF-1R tyrosine kinase have entered Phase I trials: OSI-906 (OSI Pharmaceuticals) and INSM-18 (Insmed). Given the role of IGF-1R signaling in cell survival and EMT, one might hope that these therapies might target both the primary tumor as well as emerging metastatic cells.
EGFR promotion of EMT-like transitions
EGFR function is frequently dysregulated in epithelial tumors, and EGFR signaling has been shown to play an important role both in cancer progression and in EMT-like transitions. EGF has been shown to promote tumor cell migration and invasion, at least in part through dephosphorylation and inactivation of FAK [57–60]. EGF treatment of tumor cells overexpressing EGFR also leads to downregulation of caveolin-1 which leads to loss of E-cadherin, transcriptional activation of β-catenin and enhanced invasiveness [61]. Thus inhibition of EGFR might be expected to restrain EMT in certain cellular contexts. In support of this hypothesis, ligand-independent, constitutively active forms of EGFR can increase motility and invasiveness of tumor cells and EGFR inhibitors have been shown to inhibit cancer cell migration in vitro [62–65]. In oral squamous cell carcinoma cells EGFR inhibition resulted in a transition from a fibroblastic morphology to a more epithelial phenotype as well as accumulation of desmosomal cadherins at cell–cell junctions [66]. Taken together, these summarized observations suggest that inhibition of EGFR affects tumor growth through inhibiting EGFR-dependent mitogenic stimulation but may also restrain invasion and metastasis by re-establishing intercellular contacts between tumor cells. Such inhibition of EMT, and potentially metastasis, may translate to improved overall patient survival in the clinical setting.
EGFR inhibitors in cancer therapy
EGFR is widely expressed by cells of both epithelial and mesenchymal lineages, and the degree of EGFR expression is variable [67]. EGFR overexpression has been reported in multiple human cancers including non-small cell lung (NSCLC), head and neck (HNSCC), pancreas, breast and central nervous system (CNS), and has been shown to correlate with poor survival [68]. Several selective EGFR and Her family antagonists have been shown to offer clinical benefit, including erlotinib (OSI Pharmaceuticals/Genetech/Roche), gefitinib (Astra Zeneca) and lapatinib (GlaxoSmithKline). Erlotinib is approved for the treatment of NSCLC patients who have failed two or three previous rounds of chemotherapy. Erlotinib is also approved in the USA and Europe for the treatment of pancreatic cancer in combination with gemcitibine. Lapatinib, a dual inhibitor of EGFR and Her2, has been shown to delay progression of trastuzumab-refractory breast cancer and is used in combination with capecitibine for patients who have received prior therapy with an anthracycline, a taxane, and trastuzumab. Anti-EGFR antibodies have also shown clinical utility, including cetuximab (Imclone/Bristol Myers) and panitumamab (Abgenix/Amgen) which are approved for the treatment of EGFR-expressing, metastatic colorectal carcinoma. Additional small molecule dual EGFR-Her2 inhibitors which bind irreversibly are in earlier stages of clinical development. The current EGFR inhibitors have provided significant clinical benefit when compared to the current standard of care, however not all patients derive a benefit in terms of overall survival or RECIST criteria. For example, in the BR.21 trial which compared the efficacy of erlotinib as a single agent in comparison to placebo in NSCLC patients who did not respond to chemotherapy, the overall response rate was only 8.9% while the hazard ratio for treatment benefit associated with overall survival was 0.7 [69]. The median survival among patients who were treated with erlotinib was 6.7 months compared to 4.7 months for those treated with placebo. These data suggest both that RECIST did not directly correlate with potential survival benefit and that some, but not all, patients clearly benefited from erlotinib. This observation spurred research to identify biomarkers to predict patient response to erlotinib and potentially other EGFR antagonists, and to identify the mechanistic basis for the differential response.
EMT and sensitivity to EGFR inhibitors
Parallel efforts from independent research groups employed gene expression and proteomic profiling to identify biomarkers which correlated with sensitivity to erlotinib [70–72]. Each of these groups used panels of established NSCLC cell lines and xenografts to determine commonalities within those cell lines for which erlotinib inhibited growth in vitro and in vivo, as compared to those cell lines which were insensitive to erlotinib. Those cell lines which were classified as sensitive, having greater than 50% maximal inhibition of proliferation, expressed the canonical epithelial markers E-cadherin and γ-catenin and displayed the classic cobblestone epithelial morphology and tight cell–cell junctions of epithelial cells. Conversely, those cell lines which were relatively insensitive to erlotinib lacked those epithelial markers and expressed proteins characteristic of mesenchymal cells, including vimentin, fibronectin and Zeb-1 and exhibited a more fibroblastic, scattered morphology. These changes are consistent with cells which have undergone EMT. These observations were later extended to other tumor types and EGFR antagonists, including pancreatic, colorectal [5], head and neck [73], bladder [74] and breast [75] suggesting that EMT status may be a broadly applicable indicator of sensitivity to EGFR inhibitors.
An important clinical substantiation of this hypothesis resulted from a retrospective analysis of TRIBUTE, a NSCLC phase III randomized trial which compared the combination of erlotinib with chemotherapy to chemotherapy alone [76]. This trial failed to show significant clinical benefit for the concurrent administration of erlotinib and chemotherapy, however subset analysis of E-cadherin levels in patient samples using immunohistochemistry was revealing. Patients with tumor samples showing strong E-cadherin staining had a significantly longer time to progression (hazard ratio 0.37) and a nonsignificant increase in overall survival when treated with the combination of erlotinib and chemotherapy as compared to chemotherapy alone [71]. Notably, expression of EGFR itself, as measured by IHC was a poor predictor of response to EGFR antagonists, both in the clinic and in cultured cell lines [70, 77]. Recent studies suggest that it is not the abundance of receptor, but rather the activation of the EGFR signaling axis that mediates sensitivity to EGFR inhibition, at least in vitro [78–80]. Collectively, these compelling in vitro data and clinical findings indicate that expression of E-cadherin or vimentin, and EMT as a process, may be viable biomarkers to predict efficacy of EGFR inhibitors in cancer patients. Clearly further clinical evaluation is warranted and is currently underway at multiple sites.
Bypassing EGFR-dependent activation of PI3-kinase and Ras pathways
The mechanism by which EMT results in insensitivity to EGFR antagonists appears to derive from the acquisition of alternative routes to activation of the PI3 kinase-Akt-mTOR and Ras-Raf-Mek-Erk pathways. It has been reported that cells which have transitioned to a mesenchymal-like state may have upregulated the PI3K-Akt cell survival pathway to circumvent apoptosis [39], and consequently have decreased sensitivity to an inhibitor of the MAPK proliferative pathway. Human cancer cells which exhibit mesenchymal characteristics express lower levels of EGF ligands, suggesting that these cells have become dependent upon alternate signaling pathways [71]. However fetal rat hepatocytes which have undergone TGF-β -mediated EMT have upregulated EGFR ligand expression [81], and in this system inhibition of EGFR does not block TGF-β -mediated EMT [82]. Thus in fetal hepatocytes, EGFR activation appears to be dispensable for the EMT process, however in human cancer cells EGFR signaling is an important driver of EMT. The expression EGFR and reported downregulation of ligand in mesenchymal NSCLC cells suggests that it is not the expression of the growth factor receptor, but rather the usage of that receptor, which governs sensitivity to targeted inhibitors. For example it has been shown that Her3, a heterodimerization partner of EGFR, allows EGFR to effectively activate the PI3 kinase-Akt pathway [83]. It has also been shown that Her3 RNA transcripts and protein are attenuated or lost during EMT-like transitions, depriving EGFR of PI3 kinase coupling [70, 80]. There is substantial evidence that cancer cells can readily shift the cellular equilibrium to rely on alternate growth factor and adhesion signaling pathways in response to EMT-like transitions. For example, inhibition of the Ras-MAPK proliferative pathway can lead to increased activation of the PI3K-Akt cellular survival pathway [75, 84, 85]. Therefore it is not surprising that increased IGF-1R signaling has been associated with insensitivity to EGFR inhibitors [86, 87]. High concentrations of the ligand IGF-1 were shown to inhibit apoptosis caused by the EGFR antagonist erlotinib, possibly due to an increased reliance on the IGF-1R/PI3K signaling axis.
Interestingly, while IGF-1R can promote EMT-like transitions it appears to be less frequently used by mesenchymal-like carcinoma cells as a sole driver of the PI3 kinase pathway. IGF-1R activation drives upregulation of the PI3K-Akt survival pathway and promotes epithelial-to-mesenchymal transition, and these changes may account for the reduced cellular sensitivity to EGFR antagonists. However, committed mesenchymal-like NSCLC, colon and pancreas adenocarcinomas are not dependent on IGF-1R for proliferation or survival [75], suggesting that, like EGFR, IGF-1R signaling is an important driver in epithelial cells and can promote EMT, but once these cells have transitioned to a mesenchymal state they are no longer reliant on IGF-1R. This hypothesis would suggest that the combination of an EGFR antagonist such as erlotinib and an inhibitor of IGF-1R could synergistically inhibit proliferation and potentially drive apoptosis in early stage tumors with an epithelial phenotype. Several in vitro studies have provided data which supports this hypothesis [86, 88–91]. Furthermore, since blockade of either IGF-1R [92, 93] or EGFR [94–96] signaling results in decreased metastasis in vivo, partnering EGFR and IGF-1R antagonists might also improve overall patient survival in early disease in epithelial tumors dependent on EGFR and IGF1R signaling.
Once a cancer cell has transitioned to a mesenchymal-like phenotype, cellular dependence on IGF-1R and EGFR signaling is reduced and alternate growth factor pathways are activated. Recent data suggest that EMT-like transitions can promote the novel acquisition of alternate receptor tyrosine kinase (RTK) autocrine and paracrine loops [97], such as PDGFR, which can exert proliferative and anti-apoptotic actions. PDGFRα and β are restricted to cells of mesodermal origin [98] and autocrine PDGFR signaling has been described in non-epithelial tumors such as gliomas [99, 100]. However high expression has been observed in multiple tumor types including ovarian [101] prostate [102] and breast [103] carcinomas, suggesting that PDGFR may be detectable in stroma and in tumor cells which have undergone EMT. Autocrine PDGFR expression has been shown to promote progression of breast and ovarian cancer and contribute to maintenance of a mesenchymal-like cell phenotype [101, 103, 104]. Thus, mesenchymal cells which have acquired alternate signaling pathways, such as PDGFR, to activate PI3K signaling and prevent apoptosis may be sensitized to specific molecular targeted therapies. The acquisition of these receptor signaling pathways which are predominantly used in mesenchymal cells may overlap on a common set of required signaling nodes, although this remains to be determined. Nevertheless, this observation provides support for rationally designed combinations of targeted therapies to impede the complex, heterogeneous signaling pathways that exist within a tumor.
In summary, we propose a model in which carcinomas undergo an epithelial-to-mesenchymal-like transition, potentially triggered by dysregulated growth factor signaling or inflammation-mediated activation of the PI3 kinase and Ras pathways [105]. Tumor cells with a mesenchymal-like phenotype become less reliant on EGFR, IGF-1R, Met and Ron signaling pathways. Over time, mesenchymal-like tumor cells appear to become more committed to a mesenchymal phenotype through epigenetic changes and can upregulate alternate receptor tyrosine kinase pathways as mechanisms for survival signaling and escape from anoikis. The interactions of EGF receptor signaling with other cellular pathways regulating mitogenic, survival and migration cues has clinical implications as we try to identify and develop treatments which not only target the primary tumor cells but also the mesenchymal-like cells deriving from EMT-like transitions that can promote cancer metastasis and recurrence.
References
Peinado H, Olmeda D, Cano A (2007) Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7(6):415–428
Maeda M, Johnson KR, Wheelock MJ (2005) Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J Cell Sci 118(Pt 5):873–887
Brabletz T, Jung A, Reu S et al (2001) Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA 98(18):10356–10361
Willipinski-Stapelfeldt B, Riethdorf S, Assmann V et al (2005) Changes in cytoskeletal protein composition indicative of an epithelial-mesenchymal transition in human micrometastatic and primary breast carcinoma cells. Clin Cancer Res 11(22):8006–8014
Buck E, Eyzaguirre A, Barr S et al (2007) Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther 6(2):532–541
Umbas R, Schalken JA, Aalders TW et al (1992) Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Res 52(18):5104–5109
Yin T, Wang C, Liu T et al (2007) Expression of snail in pancreatic cancer promotes metastasis and chemoresistance. J Surg Res 141(2):196–203
Kajiyama H, Shibata K, Terauchi M et al (2007) Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 31(2):277–283
Cheng GZ, Chan J, Wang Q et al (2007) Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res 67(5):1979–1987
Zhang Z, Xie D, Li X et al (2007) Significance of TWIST expression and its association with E-cadherin in bladder cancer. Hum Pathol 38(4):598–606
Hosono S, Kajiyama H, Terauchi M et al (2007) Expression of Twist increases the risk for recurrence and for poor survival in epithelial ovarian carcinoma patients. Br J Cancer 96(2):314–320
Leroy P, Mostov KE (2007) Slug is required for cell survival during partial epithelial-mesenchymal transition of HGF-induced tubulogenesis. Mol Biol Cell 18(5):1943–1952
Cho HJ, Baek KE, Saika S et al (2007) Snail is required for transforming growth factor-beta-induced epithelial-mesenchymal transition by activating PI3 kinase/Akt signal pathway. Biochem Biophys Res Commun 353(2):337–343
Laux H, Tomer R, Mader MT et al (2004) Tumor-associated E-cadherin mutations do not induce Wnt target gene expression, but affect E-cadherin repressors. Lab Invest 84(10):1372–1386
Lombaerts M, van Wezel T, Philippo K et al (2006) E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J Cancer 94(5):661–671
Perl AK, Wilgenbus P, Dahl U et al (1998) A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392(6672):190–193
Vleminckx K, Vakaet L Jr, Mareel M et al (1991) Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 66(1):107–119
Yang J, Mani SA, Donaher JL et al (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117(7):927–939
Cano A, Perez-Moreno MA, Rodrigo I et al (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2(2):76–83
Batlle E, Sancho E, Franci C et al (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2(2):84–89
Hennig G, Lowrick O, Birchmeier W et al (1996) Mechanisms identified in the transcriptional control of epithelial gene expression. J Biol Chem 271(1):595–602
Hennig G, Behrens J, Truss M et al (1995) Progression of carcinoma cells is associated with alterations in chromatin structure and factor binding at the E-cadherin promoter in vivo. Oncogene 11(3):475–484
Birchmeier W, Behrens J (1994) Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta 1198(1):11–26
Nieto MA (2002) The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol 3(3):155–166
Aigner K, Dampier B, Descovich L et al (2007) The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26(49):6979–6988
Eger A, Aigner K, Sonderegger S et al (2005) DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 24(14):2375–2385
Perez-Moreno MA, Locascio A, Rodrigo I et al (2001) A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem 276(29):27424–27431
Guaita S, Puig I, Franci C et al (2002) Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J Biol Chem 277(42):39209–39216
Tamagnone L, Comoglio PM (1997) Control of invasive growth by hepatocyte growth factor (HGF) and related scatter factors. Cytokine Growth Factor Rev 8(2):129–142
Montesano R, Matsumoto K, Nakamura T et al (1991) Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell 67(5):901–908
Thiery JP (2002) Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2(6):442–454
Thiery JP (2003) Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol 15(6):740–746
Vincent-Salomon A, Thiery JP (2003) Host microenvironment in breast cancer development: epithelial-mesenchymal transition in breast cancer development. Breast Cancer Res 5(2):101–106
Onoue T, Uchida D, Begum NM et al (2006) Epithelial-mesenchymal transition induced by the stromal cell-derived factor-1/CXCR4 system in oral squamous cell carcinoma cells. Int J Oncol 29(5):1133–1138
Waerner T, Alacakaptan M, Tamir I et al (2006) ILEI: a cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell 10(3):227–239
Petersen OW, Nielsen HL, Gudjonsson T et al (2003) Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol 162(2):391–402
Derynck R, Akhurst RJ, Balmain A (2001) TGF-beta signaling in tumor suppression and cancer progression. Nat Genet 29(2):117–129
Moses HL, Yang EY, Pietenpol JA (1990) TGF-beta stimulation and inhibition of cell proliferation: new mechanistic insights. Cell 63(2):245–247
Valdes F, Alvarez AM, Locascio A et al (2002) The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol Cancer Res 1(1):68–78
Sanchez A, Alvarez AM, Lopez Pedrosa JM et al (1999) Apoptotic response to TGF-beta in fetal hepatocytes depends upon their state of differentiation. Exp Cell Res 252(2):281–291
Robson EJ, Khaled WT, Abell K et al (2006) Epithelial-to-mesenchymal transition confers resistance to apoptosis in three murine mammary epithelial cell lines. Differentiation; Res Biol Divers 74(5):254–264
Gal A, Sjoblom T, Fedorova L et al (2007) Sustained TGFbeta exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene. doi:10.1038/sj.onc.1210741
Vega S, Morales AV, Ocana OH et al (2004) Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 18(10):1131–1143
Inoue A, Seidel MG, Wu W et al (2002) Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2(4):279–288
Wu WS, Heinrichs S, Xu D et al (2005) Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell 123(4):641–653
Fujita Y, Krause G, Scheffner M et al (2002) Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 4(3):222–231
Kim HJ, Litzenburger BC, Cui X et al (2007) Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol 27(8):3165–3175
Lopez T, Hanahan D (2002) Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 1(4):339–353
Doerr ME, Jones JI (1996) The roles of integrins and extracellular matrix proteins in the insulin-like growth factor I-stimulated chemotaxis of human breast cancer cells. J Biol Chem 271(5):2443–2447
Stracke ML, Engel JD, Wilson LW et al (1989) The type I insulin-like growth factor receptor is a motility receptor in human melanoma cells. J Biol Chem 264(36):21544–21549
Bornfeldt KE, Raines EW, Nakano T et al (1994) Insulin-like growth factor-I and platelet-derived growth factor-BB induce directed migration of human arterial smooth muscle cells via signaling pathways that are distinct from those of proliferation. J Clin Invest 93(3):1266–1274
Leventhal PS, Shelden EA, Kim B et al (1997) Tyrosine phosphorylation of paxillin and focal adhesion kinase during insulin-like growth factor-I-stimulated lamellipodial advance. J Biol Chem 272(8):5214–5218
Playford MP, Bicknell D, Bodmer WF et al (2000) Insulin-like growth factor 1 regulates the location, stability, and transcriptional activity of beta-catenin. Proc Natl Acad Sci USA 97(22):12103–12108
Zhang D, Brodt P (2003) Type 1 insulin-like growth factor regulates MT1-MMP synthesis and tumor invasion via PI 3-kinase/Akt signaling. Oncogene 22(7):974–982
Shen MR, Hsu YM, Hsu KF et al (2006) Insulin-like growth factor 1 is a potent stimulator of cervical cancer cell invasiveness and proliferation that is modulated by alphavbeta3 integrin signaling. Carcinogenesis 27(5):962–971
Bohula EA, Playford MP, Macaulay VM (2003) Targeting the type 1 insulin-like growth factor receptor as anti-cancer treatment. Anticancer Drugs 14(9):669–682
Price JT, Wilson HM, Haites NE (1996) Epidermal growth factor (EGF) increases the in vitro invasion, motility and adhesion interactions of the primary renal carcinoma cell line, A704. Eur J Cancer 32A(11):1977–1982
Shibata T, Kawano T, Nagayasu H et al (1996) Enhancing effects of epidermal growth factor on human squamous cell carcinoma motility and matrix degradation but not growth. Tumour Biol 17(3):168–175
Lu Z, Jiang G, Blume-Jensen P et al (2001) Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol Cell Biol 21(12):4016–4031
Salamanca CM, Maines-Bandiera SL, Leung PC et al (2004) Effects of epidermal growth factor/hydrocortisone on the growth and differentiation of human ovarian surface epithelium. J Soc Gynecol Invest 11(4):241–251
Lu Z, Ghosh S, Wang Z et al (2003) Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell 4(6):499–515
Cole GW Jr, Alleva AM, Reddy RM et al (2005) The selective epidermal growth factor receptor tyrosine kinase inhibitor PD153035 suppresses expression of prometastasis phenotypes in malignant pleural mesothelioma cells in vitro. J Thorac Cardiovasc Surg 129(5):1010–1017
Lal A, Glazer CA, Martinson HM et al (2002) Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res 62(12):3335–3339
Seton-Rogers SE, Lu Y, Hines LM et al (2004) Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc Natl Acad Sci USA 101(5):1257–1262
Mateus AR, Seruca R, Machado JC et al (2007) EGFR regulates RhoA-GTP dependent cell motility in E-cadherin mutant cells. Hum Mol Genet 16(13):1639–1647
Lorch JH, Klessner J, Park JK et al (2004) Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J Biol Chem 279(35):37191–37200
Wells A (1999) EGF receptor. Int J Biochem Cell Biol 31(6):637–643
Grandis JR, Sok JC (2004) Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol Ther 102(1):37–46
Shepherd FA, Rodrigues Pereira J, Ciuleanu T et al (2005) Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 353(2):123–132
Thomson S, Buck E, Petti F et al (2005) Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res 65(20):9455–9462
Yauch RL, Januario T, Eberhard DA et al (2005) Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res 11(24 Pt 1):8686–8698
Witta SE, Gemmill RM, Hirsch FR et al (2006) Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 66(2):944–950
Frederick BA, Helfrich BA, Coldren CD et al (2007) Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther 6(6):1683–1691
Shrader M, Pino MS, Brown G et al (2007) Molecular correlates of gefitinib responsiveness in human bladder cancer cells. Mol Cancer Ther 6(1):277–285
Buck E, Eyzaguirre A, Franklin M et al (2007) The EGFR inhibitor erlotinib sensitizes tumor cells to IGF-1 receptor inhibition by promoting Akt signaling through the IGF-1R-IRS1-Akt axis. Proceedings of American Association for Cancer Research Annual Meeting, Los Angeles, CA
Herbst RS, Prager D, Hermann R et al (2005) TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol 23(25):5892–5899
Ono M, Hirata A, Kometani T et al (2004) Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther 3(4):465–472
Arteaga CL (2002) Epidermal growth factor receptor dependence in human tumors: more than just expression? Oncologist 7(Suppl 4):31–39
Moulder SL, Yakes FM, Muthuswamy SK et al (2001) Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Res 61(24):8887–8895
Buck E, Eyzaguirre A, Haley JD et al (2006) Inactivation of Akt by the epidermal growth factor receptor inhibitor erlotinib is mediated by HER-3 in pancreatic and colorectal tumor cell lines and contributes to erlotinib sensitivity. Mol Cancer Ther 5(8):2051–2059
Del Castillo G, Murillo MM, Alvarez-Barrientos A et al (2006) Autocrine production of TGF-beta confers resistance to apoptosis after an epithelial-mesenchymal transition process in hepatocytes: role of EGF receptor ligands. Exp Cell Res 312(15):2860–2871
Murillo MM, del Castillo G, Sanchez A et al (2005) Involvement of EGF receptor and c-Src in the survival signals induced by TGF-beta1 in hepatocytes. Oncogene 24(28):4580–4587
Engelman JA, Janne PA, Mermel C et al (2005) ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci USA 102(10):3788–3793
Desbois-Mouthon C, Cacheux W, Blivet-Van Eggelpoel MJ et al (2006) Impact of IGF-1R/EGFR cross-talks on hepatoma cell sensitivity to gefitinib. Int J Cancer 119(11):2557–2566
Barr SM, Buck EA, Thomson S et al (2007) The combination of small molecule inhibitors of EGFR and IGF-1R is synergistic in HNSCC and ovarian cancer cell lines. Proceedings of American Association of Cancer Research Annual Meeting. Los Angeles, CA
Chakravarti A, Loeffler JS, Dyson NJ (2002) Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res 62(1):200–207
Jones HE, Goddard L, Gee JM et al (2004) Insulin-like growth factor-I receptor signalling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr Relat Cancer 11(4):793–814
Sutter AP, Hopfner M, Huether A et al (2006) Targeting the epidermal growth factor receptor by erlotinib (Tarceva) for the treatment of esophageal cancer. Int J Cancer 118(7):1814–1822
Slomiany MG, Black LA, Kibbey MM et al (2007) Insulin-like growth factor-1 receptor and ligand targeting in head and neck squamous cell carcinoma. Cancer Lett 248(2):269–279
Desbois-Mouthon C, Danan C, Amselem S et al (1996) Severe resistance to insulin and insulin-like growth factor-I in cells from a patient with leprechaunism as a result of two mutations in the tyrosine kinase domain of the insulin receptor. Metabolism 45(12):1493–1500
Morgillo F, Woo JK, Kim ES et al (2006) Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res 66(20):10100–10111
Dunn SE, Ehrlich M, Sharp NJ et al (1998) A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res 58(15):3353–3361
Samani AA, Fallavollita L, Jaalouk DE et al (2001) Inhibition of carcinoma cell growth and metastasis by a vesicular stomatitis virus G-pseudotyped retrovector expressing type I insulin-like growth factor receptor antisense. Hum Gene Ther 12(16):1969–1977
Choi YJ, Nam SJ, Son MJ et al (2006) Erlotinib prevents pulmonary metastasis in curatively resected breast carcinoma using a mouse model. Oncol Rep 16(1):119–122
Angelucci A, Gravina GL, Rucci N et al (2006) Suppression of EGF-R signaling reduces the incidence of prostate cancer metastasis in nude mice. Endocr Relat Cancer 13(1):197–210
Shintani S, Li C, Mihara M et al (2003) Gefitinib (‘Iressa’), an epidermal growth factor receptor tyrosine kinase inhibitor, mediates the inhibition of lymph node metastasis in oral cancer cells. Cancer Lett 201(2):149–155
Thomson S, Petti F, Haley J (2007) Multiple mechanisms of insensitivity to Erlotinib in mesenchymal-like NSCLC cell lines. Proceedings of the American Association for Cancer Research Anual Meeting, Los Angeles, CA; 48
Heldin CH, Westermark B, Wasteson A (1981) Specific receptors for platelet-derived growth factor on cells derived from connective tissue and glia. Proc Natl Acad Sci USA 78(6):3664–3668
Westermark B, Heldin CH, Nister M (1995) Platelet-derived growth factor in human glioma. Glia 15(3):257–263
Dai C, Celestino JC, Okada Y et al (2001) PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev 15(15):1913–1925
Matei D, Emerson RE, Lai YC et al (2006) Autocrine activation of PDGFRalpha promotes the progression of ovarian cancer. Oncogene 25(14):2060–2069
Ko YJ, Small EJ, Kabbinavar F et al (2001) A multi-institutional phase ii study of SU101, a platelet-derived growth factor receptor inhibitor, for patients with hormone-refractory prostate cancer. Clin Cancer Res 7(4):800–805
Carvalho I, Milanezi F, Martins A et al (2005) Overexpression of platelet-derived growth factor receptor alpha in breast cancer is associated with tumour progression. Breast Cancer Res 7(5):R788–R795
Jechlinger M, Sommer A, Moriggl R et al (2006) Autocrine PDGFR signaling promotes mammary cancer metastasis. J Clin Invest 116(6):1561–1570
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6(5):392–401
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Barr, S., Thomson, S., Buck, E. et al. Bypassing cellular EGF receptor dependence through epithelial-to-mesenchymal-like transitions. Clin Exp Metastasis 25, 685–693 (2008). https://doi.org/10.1007/s10585-007-9121-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-007-9121-7