
Abstract
Nucleosome positioning plays an essential role in various fundamental cellular processes by modulating the accessibility of DNA to binding proteins. Understanding the mechanisms and precise recognition of nucleosome positioning along genomic sequences are substantially important for elucidating regulations of cellular processes such as DNA replication, recombination, and gene transcription. In this report, we present a knowledge-based model for calculation of deformational energy of nucleosomal DNA and apply the model to the prediction of nucleosome positioning along the genome of Saccharomyces cerevisiae accurately. The model successfully predicted genome-wide in vitro nucleosome positions. When combined with quadratic discriminant classifier, the model achieved an accuracy of 92.9 % in discriminating in vitro nucleosome forming sequences from nucleosome inhibiting sequences. Our result supports the debated notion that the nucleosome positioning in the genomic sequences is guided primarily by deformational properties of DNA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The nucleosome is the basic repeating unit of chromatins in eukaryotes. Each nucleosome consists of a histone octamer and a 147-bp core DNA that is sharply bent and tightly wrapped ∼1.65 times around the histone octamer in a left-handed superhelix. Nucleosomes are separated by DNA fragments, referred to as linker DNA, and constitute the high-order structure of chromatin (Richmond and Davey 2003; Richmond et al. 1984). The nucleosome positioning affects accessibility of a concrete DNA sequence either by burying it in the chromatin structure, when the sequence is in the inward position thus is inaccessible to other proteins, or by exposing it when the sequence is in the outward position or in a linker region thus is accessible to protein binding (Jenuwein and Allis 2001; Kornberg and Lorch 1999; Wyrick et al. 1999; Xu and Olson 2010). Therefore, a clear understanding of the mechanisms and precise recognition of nucleosome positioning along genomic sequences are essential for decoding the chromatin structure and function.
Nucleosome positioning is known to be affected by internal and external factors. The internal factors are encoded in the genomic sequences, such as the ∼10-bp periodicities of dinucleotides and the intrinsic sequence preference to form nucleosome (Bao et al. 2006; Peckham et al. 2007; Rapoport et al. 2011; Satchwell et al. 1986; Segal et al. 2006; Segal and Widom 2009). The external factors include chromatin remodeler, DNA methylation, histone variation and post-translational modification, polymerase II binding, etc. (Mavrich et al. 2008a; Schones et al. 2008; Zhang et al. 2009). Contrast to a consensus in the effect of external factors, the intrinsic preference of DNA sequence to form nucleosomes has been an issue of debate for the past few years. Segal et al. proposed a genomic code for nucleosome positioning in which the intrinsic DNA sequence preference can explain ∼50 % of the in vivo nucleosome positions (Segal et al. 2006). However, Zhang et al. (2010), based on their experimental data, alleged that DNA sequence property is not the major determinant of nucleosome positioning. Regardless of the debate about the genomic code, the significant similarity between the in vitro and in vivo maps of nucleosome organization demonstrated the sequence dependency of nucleosome positioning (Kaplan et al. 2009).
Intrinsic property of DNA sequences influences nucleosome formation in many ways. Satchwell and Segal et al. demonstrated the ∼10-bp periodicity of AA/TT/TA dinucleotides that oscillate in phase with each other and out of phase with ∼10-bp periodic GC dinucleotides (Satchwell et al. 1986; Segal et al. 2006), where phase refers to rotational position relative to the position inward to histone octamers. These ∼10-bp periodicities can be attributed to the bending preferentially towards minor groove for AA/TT/TA and towards major groove for GC dinucleotides. In the nucleosomal DNA, the minor groove facing inward relative to histone octamer occurs at every DNA helical repeat (∼10 bp) when the major groove of DNA ∼5 bp away faces outwards. Thus, the same periodicity of occurrence for AA/TT/TA and GC dinucleotides but with different phases lowers the bending energy for DNA wrapping around the histone octamers. The sequence-dependent DNA bendability (or flexibility) is also reflected in nucleosome positioning (Bao et al. 2006; Peckham et al. 2007; Segal and Widom 2009); for example, poly (dA:dT) tracts, which are known to be much rigid, disfavor nucleosome formation and are enriched in nucleosome-depleted regions (NDRs), especially in the upstream of TATA-less promoter (Bao et al. 2006; Field et al. 2008; Segal and Widom 2009). In spite of the sequence dependency, there appears to be no specific motif responsible for nucleosome formation. Thus, it is likely that a composite signal produced by the combinations of dinucleotide periodicity (Segal et al. 2006), phases within the period (Satchwell et al. 1986), and DNA bendability (Olson et al. 1998) determines the sequence preference of nucleosome positioning.
In recent years, experimental mapping of nucleosome organization genome-wide has been obtained for Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, and human (Barski et al. 2007; Lee et al. 2007; Mavrich et al. 2008b; Yuan et al. 2005). In the mapping, the nucleosome organization is analyzed by using micrococcal nuclease (MNase) that preferentially digests the linker regions, followed by microarray hybridization. The experimental mapping of nucleosome positions has some drawbacks; for example, the biases of MNase cleavage specificity, composition-dependent labeling, and hybridization biases inherent to the microarray procedure were generally not assessed, and thus cannot provide highly reliable maps of one base-pair resolution. Such studies nevertheless provided datasets that facilitate further investigation on the mechanisms of nucleosome positioning.
A variety of models have been proposed for prediction of nucleosome positioning that are classified into categories of bioinformatics (Peckham et al. 2007; Segal et al. 2006; Xing et al. 2011; Zhao et al. 2010), physical statistics (Schwab et al. 2008; Scipioni et al. 2009), and energetics of nucleosomal core particles (Miele et al. 2008; Morozov et al. 2009; Tolkunov and Morozov 2010). Bioinformatics models based exclusively on the sequences use a large number of nucleosomal datasets to train their algorithms or to extract common traits of nucleosomal DNA, which are used to predict nucleosome positioning (Peckham et al. 2007; Xing et al. 2011; Zhao et al. 2010). Although they can predict nucleosome formation tendency in a large scale, most bioinformatics models are unable to assign exactly the center dinucleotide step (or the dyad axis) and the boundaries for an individual nucleosome; thus, their practical application is limited. A probabilistic model based on ∼10-bp periodicity of some dinucleotide elements along the nucleosome DNA has been employed to predict nucleosome positions (Segal et al. 2006), and the validity of the model was questioned (Zhao et al. 2010). DNA bendability matrix, which reflects the phase relationships between various dinucleotides within the period, was used for sequence-directed mapping of the nucleosomes on the sequences (Gabdank et al. 2010).
Recently, energetic models to predict nucleosome positioning have attracted much attention (Miele et al. 2008; Morozov et al. 2009; Tolkunov and Morozov 2010). In principle of physics, DNA sequences forming nucleosome core particles with histone octamers should have lower free energy than the linker DNAs do. Thus, efforts were made to calculate the deformation energy of nucleosomal DNA for nucleosome positioning prediction. So far, only one model in this category by Morozov et al. (2009) has gone far enough for predicting genome-wide nucleosome positioning.
In this report, we present a knowledge-based model for calculation of nucleosomal DNA deformational energy from the geometric parameters, i.e., roll, tilt, slide, and twist, and predetermined force constants. The model predicts nucleosomal position with high accuracy, and the prediction ability is tested by using well-positioned nucleosomal DNA sequences. When applied to specific genomic regions of interest, our model rediscovered distinct nucleosome-depleted regions (NDRs) around transcription start sites (TSS), transcription end sites (TES), and ARS consensus sequences (ACS) for DNA replication. The model also successfully predicted genome-wide in vitro nucleosome positions. Combined with quadratic discriminant classifier using deformation energy as input, the model achieved a high accuracy of 92.9 % in predicting nucleosome-forming tendency of DNA sequences, which is comparable to that obtained by a bioinformatics classifier based on k-mer frequency (Zhao et al. 2010). Thus, our model has important implications for the nature of nucleosome positioning.
Methods
The geometry of DNA double helix
We adopt the system (Dickerson 1989) recommended by the Cambridge Convention to describe the geometry of DNA double helix, in which each base pair is viewed as a rigid board, and its position relevant to its neighbor is specified by roll, tilt, twist, slide, shift, and rise (Fig. 1).

Definition of moving coordinate system and nucleic acid structure parameters according to the Cambridge Convention (Dickerson 1989)
Deformation energy calculation
Nucleosomal DNA bending appeared to be due to periodic variations in both roll and tilt in the crystal structure 1kx5 analyzed by Richmond et al. using the program Curves (Richmond and Davey 2003). The periodic changes reflected the helix twisting that altered the rotational position of each dinucleotide step relative to the dyad. In addition to the general trend of periodic changes, variations in the roll and tilt for each step were also dependent on the property of individual dinucleotide. Crystal structure data also showed that the majority contributions to the pitch of superhelix come from slide. Moreover, it was revealed that DNA bending in the nucleosome is achieved using the roll–slide–twist degree of freedom that was previously identified as the major degree of freedom in naked DNA oligomers (Battistini et al. 2010).
In this report, nucleosomal DNA deformation is viewed as forced bending and shearing by DNA–histone interaction. We adopted approximations that the torque F b and shearing force F s are uniformly distributed along the DNA.
We consider DNA bending to be analogous to the bending a rod of multiple segments with variable stiffness. For a bending force exerted by the histone octamer on a segment of the DNA, the deformation energy at each step along the sequence depends on both the corresponding dinucleotide flexibility and the phasing of the dinucleotide with respect to the dyad. In this section, we detail the method for the calculation of the energy required to bend DNA around histone octamer when DNA bending in the nucleosome is assumed to be due to deformations in roll, tilt, and slide.
At a dinucleotide step i (integer number),
Thus, the bending energy can be evaluated by
where \( \rho (i) \) and \( \tau (i) \) are, respectively, the actual roll and tilt angle at dinucleotide step i, and \( {\rho_0}(i) \) and \( {\tau_0}(i) \), which are dependent on the dinucleotide at step i, are, respectively, the roll and tilt without torque; k ρ (i) and k τ (i) are the dinucleotide-dependent force constants; Ω i is the accumulated twist (ω) at the center of step i, counted from the dyad point. In the context of 147-bp core DNA in the X-ray crystal such as 1kx5, the structure is symmetrical about the dyad that is located at the center nucleotide, and the dinucleotide steps from the dyad are labeled as \( i=\pm 1,\pm 2,\pm 3,\cdots, \pm 73 \) towards upstream and downstream directions. The step \( \pm 1 \) are half-step away from the dyad, thus the accumulated twist is calculated as follows:
where L, an odd number, is the length of nucleosomal DNA.
In this report, we determined F b by its relationship with the total bending angle of the core DNA. In nucleosomes, the 147-bp core DNA bends around histone octamer about 597° (α) under the stress of F b, and the α is the total contribution of ρ and τ for each step, i.e.,
Combining with Eq. (1) leads to
or
For the initial calculation, ω(i) = 360°/10.4 was used for all the dinucleotide steps. The final ω(i) was corrected using its correlation (Olson et al. 1998) with ρ by
The coefficients a i and b i in the equation for the 10 dinucleotides are listed in Table 1.
The calculation is charted in Fig. 2.

Flow chart for calculating of bending energy E b, rolls, and twists
Similarly, we use the following relationship to obtain an expression for the shearing force F s.
The superhelical DNA in the nucleosomes has a radius of 41.9 Å and a pitch of 25.9 Å (Richmond and Davey 2003). The 25.9 Å pitch is largely from the contribution of slide. So we have
Combining Eqs. (7) and (8) leads to
or
where F s is the shearing force, k s (i) is the force constant, s(i) and s 0(i) are the slides of the step i with and without stress of F s, respectively, and S is the total displacement, i.e., the pitch. We use Ω i to calculate F s in the Eq. (9) as well as E s by
The total deformation energy is estimated by
The nucleosome occupancy is then defined based on Boltzmann distribution as
In the calculation, gas constant R = 8.31 J/(K mol) and room temperature T = 300 K are used.
The empirical parameters of our model for deformation energy calculation consist of 10 dinucleotide-dependent force constants; three dinucleotide-dependent averages for roll, tilt, and slide; and 20 coefficients (Table 1) obtained by the correlation between twist and roll. The dinucleotide-dependent parameters ρ 0, τ 0, and s 0 (the average roll, tilt, and slide) and force constants k ρ (i), k τ (i), and k s (i) are taken from the work of Morozov et al. (2009) with minor modification, and are listed in the Table 2.
IDQD predictor
To compare our deformation energy-based prediction of nucleosome occupancy with a model in which the frequencies of k-mers in DNA sequence are used as input (Zhao et al. 2010), a machine learning method, IDQD, is introduced. Among the many statistics used, IDQD (ID, increment of diversity; QD, quadratic determinant) method showed superior performance. In the IDQD analysis, each DNA segment is viewed as a point vector in a multi-dimensional state space whose coordinates are the occurrence frequency of each of the k-mers. Two sets of vectors constituted of nucleosome-forming sequences and nucleosome-forbidding sequences or positive and negative sets serve as standards; then the distance of the point vector representing a segment to be classified to the average point vectors in the positive and negative sets are evaluated as criterion for classification. The detail is outlined below.
Increment of diversity (ID)
In the state space, the diversity measure for a DNA segment X: {n 1, n 2,…, n d } is defined as
where \( N=\sum\limits_{i=1}^d {{n_i}} \), and \( {n_i} \) indicates the frequency of the ith oligomer in the DNA segment; d is the dimension of the state space, e.g., for 4-mers, d is 44. Diversity measure is a description of sequence randomness for subregions of k nucleotides in a DNA segment; D(X) is zero when all the subregions are the same oligomer, and it reaches the maximum when all the possible oligomers occur with the same frequency.
For two segments, X: {n 1, n 2,…, n d } and Y: {m 1, m 2,…, m d }, in the same state space, the increment of diversity is defined as
where \( D\left( {X+Y} \right) \) is the diversity measure of the mixed source X + Y:{n 1 + m 1, n 2 + m 2, …, n d + m d }. It can be proved that \( ID\left( {X,Y} \right)\geqslant 0 \). The increment of diversity quantitatively describes the similarity of two segments (Laxton 1978; Li and Lu 2001). The smaller the increment of diversity is, the more similar the two corresponding segments are in term of occurrence frequencies of oligomers.
Quadratic discriminant (QD) analysis
In this study, a quadratic discriminant function for classifying a DNA segment is based on Mahalanobis distances of the segment in the test set to positive set and negative set (Zhang 1997).
To evaluate Mahalanobis distance, each vector involved must be converted to a vector in a feature space whose coordinates are the increments of diversity with respect to the positive set and the negative set. Thus, for a defined k-mer, each segment is represented by two numbers as its coordinates. In general, let \( {X_j}=\left[ {{x_{j1 }},\,{x_{j2 }}, \cdot s, {x_{jn }}} \right] \) be the jth vector in the dataset (j = 1, 2, …p; p is the size of the dataset) in the n–D feature space, herein x ji is the ID with respect to the positive (or negative ) training set. The mean vector averaged over the dataset is denoted as \( \overline{X}=\left[ {{{\overline{x}}_1},{{\overline{x}}_2}, \cdot s, {{\overline{x}}_n}} \right] \), where \( {{\overline{x}}_i}=\sum {_{j=1}^p{x_{ji }}/N,i=1,2,\cdots, n} \). A covariance matrix of the dataset is denoted as
where \( {c_{ji }}=\sum\limits_{s=1}^p {{{{\left( {{x_{sj }}-{{\overline{x}}_j}} \right)\left( {{x_{si }}-\overline{{{x_i}}}} \right)}} \left/ {{\left( {p-1} \right),{c_{ji }}={c_{ij }}}} \right.}} \).
The squared Mahalanobis distance of any vector \( Y=\left[ {{y_1},{y_2},\cdots, {y_n}} \right] \) to the positive set is defined as
Similarly, the Mahalanobis distance of the vector Y to the negative set δ 2 can be defined.
The quadratic discriminant function (Zhang 1997) that determines the class of a DNA segment in the test set is given by
where p and q represent the sample sizes of positive and negative sets, respectively, δ i (i = 1, 2) is the squared Mahalanobis distance of Y to the positive set and negative training set respectively, and \( |{C^i}| \) is the determinant of covariance matrix \( {C^i} \).
Generally, the positive and negative test sets can be discriminated at the threshold \( \xi =0 \), that is, the discriminating rule assigns Y to positive class if \( \xi >0 \), otherwise to negative class. The threshold of \( \xi \), however, is not necessarily zero due to the difference in sample size between positive and negative datasets. Optimal threshold of \( \xi \) for discriminating the two datasets is determined empirically (Lu et al. 2010).
When deformation energy of a DNA segment is used as an input for QD analysis, the class (nucleosome positioning or nucleosome forbidding) of the segment is determined by comparing the actual \( \xi \) and the threshold of \( \xi \).
Correlation analysis
We measured the degree of association between experimental nucleosome occupancy and our prediction using Pearson’s correlation.
Results
Prediction of DNA structure parameters at each step in the nucleosome
One way to test the energetic model for nucleosome positioning is to compare the prediction of the base-step parameters with the measured data in the high-resolution nucleosome crystal structures (NCP147). Figure 3 illustrates the comparison for calculated roll with measured roll in crystal 1kx5, which contributes most to the bending angle of nucleosome DNA; the correlation was 0.67. In contrast, the correlation between calculated and measured tilt and slide were poor (0.18 and −0.11, respectively). We did not include slide in the deformation energy calculation because the poor prediction for slide is likely to contribute negatively to our deformation energy calculation. We used Eq. (4) (see “Methods” section) to calculate the nucleosomal DNA deformation energy throughout the paper.

Comparison of calculated roll of bended DNA sequence NCP147 and experimentally measured roll in crystal 1kx5
Test of the model
A nucleosomal DNA sequence, compared to the immediate vicinity sequences, should have deformation energy minimum so that it can form a stable nucleosomal core particle. Requirement for minima of nucleosomal DNA deformation energy is a critical test for theoretical models.
We tested our model first with one nucleosome crystal. NCP147 DNA is one of the few DNA sequences that can co-crystallize with histone octamers. In the crystals of core particles, about 10 bp at each end are not involved in the contact with the histone octamers, and therefore the sequence dependency of nucleosome positioning is merely reflected in the central 129-bp part of the nucleosomal DNA. Thus, the 147-bp DNA of NCP147 can accommodate core particles whose dyad steps are from 65 to 83. We calculated the deformation energy for all 19 potential core particles and plotted the energy against the nucleotide coordinates. As shown in Fig. 4, the core particle with dyad at the center coincided with the crystal dyad has the minimum deformation energy.

Calculated DNA deformation energy of 129-bp windows within NCP147, whose centers were located from base-pair position 65 to 83
To assess the prediction ability of our model for nucleosome positioning, we applied it to a genomic region in the chromosome 12 of S. cerevisiae containing an experimentally determined nucleosome (Mavrich et al. 2008a). The calculated deformation energy based on a 129-bp sliding window is shown in the Fig. 5. In general, the calculated energy was oscillating with the nucleotide coordinates. There were regions where the outer profile of oscillating energy looked like bulges with oscillation period of about 10 nucleotides most frequently. We interpret the bulges as the translational settings of nucleosomes and energy oscillation within each bulge reflects the rotational effect of DNA relative to the histone octamers (phase). The multiple local energy minima should specify the center position of settings of nucleosome dislocation. Flanking the bulges are the sequences with higher local energy minima and less defined oscillating period that we interpret as linkers. In Fig. 5, the center of the energy minimum profile (yellow dashed line) is close to the position of an experimentally determined nucleosome (red arrow head). The bioinformatics method FineStr (Gabdank et al. 2010), which was developed for nucleosomal positioning prediction at single-nucleotide accuracy, identified two nucleosomal centers (black arrowheads) in the region distance away from the measured position (red arrowhead).

Calculated deformation energy for a DNA fragment of S. cerevisiae chromosome 12 from nucleotide 459050 to 459350 containing a nucleosomal DNA with the highest occupancy score in the mapping experiment (Mavrich et al. 2008a). Yellow dashed line is the profile of local energy minima fitted by hand; the red arrowhead indicates the center of experimentally determined nucleosome; the black arrowheads indicate the nucleosomal centers (nucleotide positions 459101 and 459303) predicted by the program FineStr (Gabdank et al. 2010) in this region
We further tested the model by comparing deformation energy between nucleosomal DNA and linker DNA in the genome. For the stability of nucleosomes, the deformation energy of nucleosomal sequences would be lower than linker DNA. We selected 2,000 genomic sequences of well-positioned nucleosomes, from the experimental data of Lee et al. (2007), and extend them at each end by 200 bp. After being aligned around the center, the deformation energy for the sequences was calculated by using a 129-bp sliding window. The average energy profile obtained by our model shows that the centered windows of the sequences has the lowest deformation energy compared with the windows centered away from the middle (Fig. 6a).

a The average deformation energy around the centers of 2,000 well-defined nucleosomal DNAs from published data, whose centers are aligned at the coordinate 0. b Deformation energy profile for the experimentally validated nucleosomal DNA sequences (409 in vitro sequences, 199 in vivo sequences) that were used to construct a model for nucleosome–DNA interactions by Segal et al. (2006)
Moreover, we examined the deformation energy profile for the experimentally validated nucleosomal DNA sequences that were used to construct a model for nucleosome–DNA interactions by Segal et al. (2006). When the sequences were aligned around their center, calculated deformation energy of the central window coincided with the location of the trough (Fig.6b). In both Fig. 6a and b, the calculated energy oscillated with a periodicity of 10–11 bases.
The bulge profile and periodical oscillation of calculated deformation energy in Figs. 5 and 6b are a reminiscence of course grains and fine grains observed in experiments (Albert et al. 2007), where multiple nucleosomal positions (fine grains) were found to located in a small region (course grain) with the dyad centers separated about 10-bp apart.
Rediscovery of nucleosome-depleted region (NDR) in the vicinity of regulatory sites in S. cerevisiae by our model
One way for cells to regulate gene expression is the presence of NDR around transcription start site (TSS) and transcription end site (TES) in various organisms (Field et al. 2008; Lee et al. 2007; Yuan et al. 2005). The NDRs near TSS facilitate the binding of transcription factors to DNA, and the NDR near TES contributes to the DNA looping, which may increase RNA polymerase transcriptional activity by promoting more efficient template clearance. We applied our model to genomic regions around TSS and TES of 5,015 validated transcripts that were taken from the work by Lee et al. (2007). Consistent with previous reports, a pronounced NDR was found at ∼80 bp upstream of the TSS and at ∼80 bp downstream of TES. As shown in Fig. 7, nucleosomes are much more depleted in upstream promoter regions of high transcription genes than low transcription genes, and positioned more frequent in coding regions of high transcription genes than low transcription genes. These are in agreement with the experimental finding that nucleosome occupancy in promoter is correlated inversely with transcriptional activity, whereas nucleosome occupancy in coding region is correlated positively with transcriptional activity (Lee et al. 2007). A more distinct NDR in the intergenic downstream of TES was observed for high transcription genes than low transcription genes.

Model predicted nucleosome occupancies around a transcription start and b end sites, and c DNA replication ACS in ARS. In (a) and (b), 5,015 validated transcripts were taken from the work by Lee et al. (2007). In (c), the sequences of 1-kb regions around 47 replication origins were downloaded from the SGD database
The DNA replication activity appears remarkably affected by chromatin organization and remodeling (Berbenetz et al. 2010; Eaton et al. 2010). In the budding yeast S. cerevisiae, DNA replication starts from the replication origins and proceeding along both directions. Replication origins were defined as autonomously replicating sequence (ARS). Each ARS in yeast contains a short essential consensus sequence (ACS) of ∼11 bp, which provides the binding site for the origin recognition complex (Kornberg and Lorch 1999). Early studies reported that ACS was located in NDR and the placement of the nucleosome adjacent to ACS was crucial for replication initiation (Berbenetz et al. 2010; Eaton et al. 2010). We calculated the nucleosome position profile in 1-kb regions around 47 replication origins distal to chromosome ends, whose sequences were downloaded from the SGD database (Cherry et al. 1998). Our analysis shows that nucleosomes are depleted around the center of ACS (Fig. 7c), indicating that the physical properties of DNA determine the nucleosome depletion at ACS, which favors the replication machinery recruitment and activation.
Genome-wide prediction of nucleosome occupancy
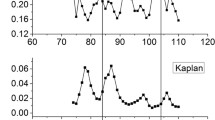
The genome-wide nucleosome occupancy (see “Methods” for definition) in yeast was predicted by using a sliding window of 147 bp with step of 1 bp along the genome. As shown in Table 3, the predicted nucleosome occupancy has higher correlations with in vitro occupancy than with in vivo occupancy, a result as expected since in vitro nucleosome occupancy is affected only by sequence property. The prediction power of our model was demonstrated by a comparison of the predicted with the experimentally measured nucleosome occupancy profile for a 3,500-bp genomic region indicated in the work of Kaplan et al. (2009). As shown in Fig. 8, the nucleosome occupancy profile obtained by our model is in close agreement with in vitro and in vivo nucleosome occupancy. Pearson’s correlation between our prediction and in vitro map is 0.72, which is higher than the correlation (R = 0.70) between the probabilistic model prediction (Segal et al. 2006) and in vitro map.

Comparison of calculated nucleosome occupancy (lowest panel) with experimental data (up and middle panels) that were taken from Kaplan et al. (2009)
Prediction of nucleosome occupancy by deformation energy based on the quadratic discriminant classifier
The energetic model when combined with a powerful bioinformatics statistic tool, QD, can predict nucleosome occupancy with high accuracy. A classifier is developed using quadratic discriminant (QD) algorithm (Zhang 1997) based on the deformation energy difference between nucleosomal sequences and nucleosome inhibiting sequences. The QD classifier based on deformation energy was used to distinguish 20,000 in vitro nucleosome positioning sequences and 20,000 nucleosome inhibiting sequences that were taken from Kaplan’s data (Kaplan et al. 2009). The nucleosome positioning sequences and nucleosome inhibiting sequences were defined in the following way. First, we assume that in vitro nucleosome occupancy scores given for each genomic position in yeast represent the nucleosome formation probabilities of the corresponding 147-bp sequences centered at the genomic position. Then 20,000 non-overlapping sequences of 147 bp having the highest nucleosome occupancy scores were defined as nucleosome positioning sequences, and 20,000 non-overlapping sequences of 147 bp having the lowest nucleosome occupancy scores as nucleosome inhibiting sequences; they were also called positive dataset and negative dataset, respectively. The deformation energies for the sequences were used as input to the QD classifier, and the 5-fold cross-validation was done to examine the performance of our approach. As shown in Table 4, QD classifier based on deformation energy achieved a high accuracy of 92.9 % and correlation coefficient of 0.857. IDQD is known to be a powerful bioinformatics classifier that usually outperforms other machine-learning methods (Zhang and Luo 2003). So, we performed IDQD prediction to compare with our energy-based classifier. 4-mer frequencies in sequences were used as sequence attribute, as previous works indicated that the prediction for nucleosome positioning based on 4-mer frequencies achieved the highest accuracy (Xing et al. 2011; Zhao et al. 2010). In the IDQD model, each sequence is represented by a vector of two dimensions, of which coordinates are the two 4-mer-dependent IDs (increments of diversity) with respect to the positive and negative training set. Then the two IDs are used as inputs of QD classifier. In the deformation energy-based QD model, each sequence is represented by a vector of one dimension, of which coordinate is the deformation energy for the sequence. Then the deformation energy is used as input of the QD classifier. As shown in Table 4, the accuracy of energy-based prediction is slightly lower than that of IDQD prediction, but the high prediction accuracy based on deformation energy implicates an important direction in the nucleosome positioning prediction.
Discussion
In this report, we formulated from the geometric parameters of the DNA double helix the energy of DNA deformation that is required to bend a DNA fragment of given sequence to a superhelical structure around the histone core. The geometric parameters include the roll, tilt, slide, and twist of each dinucleotide step at the equilibrium state, and the elastic force constants of the deformations of dinucleotides deviated from the equilibrium state. The nucleosome occupancy landscape then was evaluated from calculated deformation energy in windows centered at each nucleotide position along the genome sequence.
The unique feature of our model is that DNA elastic force constants, preferential direction of bending, and positions relative to the step inwards, or the phase, of the dinucleotide steps were combined into the formula for deformation energy calculation (Eqs. 4–5, 9, and 10). Thus, our modeling is integration of influence factors for nucleosome formation including dinucleotide flexibility, periodicity, and phasing.
Contrast to a strong correlation for roll between calculated and experimental data, the correlations for tilt and cumulative twist are poor, which is caused by the poor correlations of the crystal structure parameters of NCP147 with cumulative twist. For example, given that the correlation between slide and the sinusoidal function of the cumulative twist for NCP147 is weak, Eq. (7) will generate poor predictions for slide. The poor prediction for twist is due to Eq. (6), which is not an ideal reflection of the correlation between actual values of roll and twist for NCP147.
In general, the more flexible the sequence is, the easier it is to bend and form nucleosome. Periodicity of dinucleotides is closely correlated with their phases relative to the dyad. If dinucleotides occur at ideal phases in the sequence, they will facilitate the bending of DNA helix around histone octamer. Because the curvature is largely due to the base-step roll, the dinucleotides with extreme roll values show strong periodicity in the nucleosomal DNA. These factors are incorporated in our model and the potential of nucleosome formation is predicted by using deformation energy. In the crystals of core particles, about 10 bp at each end are not involved in the contact with the histone octamers, and thus the sequence dependency of nucleosome positioning is merely reflected in the central 129-bp part of the nucleosomal DNA. According to our model, the deformation energy for the central 129-bp part should be the minimum among all 129-bp parts of the NCP147 DNA.
Our model can easily explain two intriguing observations that impose challenges on theoretical models. One challenge is related to the rotational positioning. When the nucleosomal DNA is displaced by a few times of its period, 10.4 bases, the direction of DNA bending should be kept (rotational positioning) (Drew and Travers 1985), thus the displaced DNA is also able to form nucleosomes. This is indeed the case in nucleosome reconstitution experiments (Bao et al. 2006; Drew and Travers 1985; Flaus et al. 1996), where the alternative positions from the dyad with increments of 10 bp are physically eligible because they differ mildly in the stability of the complexes. Our model readily predicts the energy oscillation with 10.4-bp periodicity around the optimal position.
Another challenge is the determination of the dyad axis of the nucleosome. The central dinucleotide step defines which of the remaining dinucleotide steps in the molecule are outward therefore exposed, a crucial information for the interaction of the nucleosomes with externally approaching molecules. Our deformation energy model predicted several alternative positions for a given nucleosome, separated by multiples of nucleosome DNA period, and also implicated the central position of nucleosomal DNA, as illustrated by the minimum deformation energy in Figs. 4, 5, and 6. Our model has important implication for delocalization of nucleosomes or chromatin remodeling. The energy profiles shown in Figs. 4, 5, and 6 imply that the possible repositioning would occur in the loci that are multiples of ∼10 bp away from the original position.
The model can be improved in the future works in several aspects: (1) taking into account the coupling of the parameters, such as roll, tilt, and slide, to construct a more accurate high-resolution mapping of the nucleosomes at a fine-grain level. (2) Correcting the structural parameters used in model for each base step in the relevant tetranucleotide context to account for the effects of conformational coupling that vary with sequence context. (3) Taking into account the inter-influence among the base-step parameters. For example, the contributions of the local base-step parameters roll and tilt to the global curvature are modulated by the cumulative helical twist relative to the dyad. These contributions will be further modulated by variations in slide and shift.
In conclusion, in this report, we presented a knowledge-based model for calculation of the deformational energy required for the bending of a DNA fragment around histone octamer using only intrinsic structural parameters of DNA and predicted the nucleosome occupancy along the genome of S. cerevisiae with high accuracy. We demonstrated the predictive power of our model by correlating it with experimentally determined nucleosome occupancy data. This further demonstrated the ability of our model to capture the sequence dependency of chromatin architecture. We shall make efforts in our future work to provide an improved model for predicting the nucleosome positioning with one base-pair accuracy.
Abbreviations
- MNase:
-
Micrococcal nuclease
- NDRs:
-
Nucleosome-depleted regions
- TSS:
-
Transcription start sites
- TES:
-
Transcription end sites
- ARS:
-
Autonomously replicating sequence
- ACS:
-
ARS consensus sequences
- ID:
-
Increment of diversity
- QD:
-
Quadratic determinant
- ORC:
-
Origin recognition complex
References
Albert I, Mavrich TN, Tomsho LP et al (2007) Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446:572–576
Bao Y, White CL, Luger K (2006) Nucleosome core particles containing a poly(dA.dT) sequence element exhibit a locally distorted DNA structure. J Mol Biol 361:617–624
Barski A, Cuddapah S, Cui K et al (2007) High-resolution profiling of histone methylations in the human genome. Cell 129:823–837
Battistini F, Hunter CA, Gardiner EJ, Packer MJ (2010) Structural mechanics of DNA wrapping in the nucleosome. J Mol Biol 396:264–279
Berbenetz NM, Nislow C, Brown GW (2010) Diversity of eukaryotic DNA replication origins revealed by genome-wide analysis of chromatin structure. PLoS Genet 6
Cherry JM, Adler C, Ball C et al (1998) SGD: Saccharomyces Genome Database. Nucleic Acids Res 26:73–79
Dickerson RE (1989) Definitions and nomenclature of nucleic acid structure parameters. J Biomol Struct Dyn 6:627–634
Drew HR, Travers AA (1985) DNA bending and its relation to nucleosome positioning. J Mol Biol 186:773–790
Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM (2010) Conserved nucleosome positioning defines replication origins. Genes Dev 24:748–753
Field Y, Kaplan N, Fondufe-Mittendorf Y et al (2008) Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput Biol 4:e1000216
Flaus A, Luger K, Tan S, Richmond TJ (1996) Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc Natl Acad Sci U S A 93:1370–1375
Gabdank I, Barash D, Trifonov EN (2010) Single-base resolution nucleosome mapping on DNA sequences. J Biomol Struct Dyn 28:107–122
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293:1074–1080
Kaplan N, Moore IK, Fondufe-Mittendorf Y et al (2009) The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458:362–366
Kornberg RD, Lorch Y (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98:285–294
Laxton RR (1978) The measure of diversity. J Theor Biol 70:51–67
Lee W, Tillo D, Bray N et al (2007) A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 39:1235–1244
Li QZ, Lu ZQ (2001) The prediction of the structural class of protein: application of the measure of diversity. J Theor Biol 213:493–502
Lu J, Luo L, Zhang L, Chen W, Zhang Y (2010) Increment of diversity with quadratic discriminant analysis—an efficient tool for sequence pattern recognition in bioinformatics. Open Access Bioinf 2:89–96
Mavrich TN, Ioshikhes IP, Venters BJ et al (2008a) A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18:1073–1083
Mavrich TN, Jiang C, Ioshikhes IP et al (2008b) Nucleosome organization in the Drosophila genome. Nature 453:358–362
Miele V, Vaillant C, Daubenton-Carafa Y, Thermes C, Grange T (2008) DNA physical properties determine nucleosome occupancy from yeast to fly. Nucleic Acids Res 36:3746–3756
Morozov AV, Fortney K, Gaykalova DA, Studitsky VM, Widom J, Siggia ED (2009) Using DNA mechanics to predict in vitro nucleosome positions and formation energies. Nucleic Acids Res 37:4707–4722
Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB (1998) DNA sequence-dependent deformability deduced from protein–DNA crystal complexes. Proc Natl Acad Sci U S A 95:11163–11168
Peckham HE, Thurman RE, Fu Y et al (2007) Nucleosome positioning signals in genomic DNA. Genome Res 17:1170–1177
Rapoport AE, Frenkel ZM, Trifonov EN (2011) Nucleosome positioning pattern derived from oligonucleotide compositions of genomic sequences. J Biomol Struct Dyn 28:567–574
Richmond TJ, Davey CA (2003) The structure of DNA in the nucleosome core. Nature 423:145–150
Richmond TJ, Finch JT, Rushton B, Rhodes D, Klug A (1984) Structure of the nucleosome core particle at 7 A resolution. Nature 311:532–537
Satchwell SC, Drew HR, Travers AA (1986) Sequence periodicities in chicken nucleosome core DNA. J Mol Biol 191:659–675
Schones DE, Cui K, Cuddapah S et al (2008) Dynamic regulation of nucleosome positioning in the human genome. Cell 132:887–898
Schwab DJ, Bruinsma RF, Rudnick J, Widom J (2008) Nucleosome switches. Phys Rev Lett 100:228105
Scipioni A, Morosetti S, De Santis P (2009) A statistical thermodynamic approach for predicting the sequence-dependent nucleosome positioning along genomes. Biopolymers 91:1143–1153
Segal E, Fondufe-Mittendorf Y, Chen L et al (2006) A genomic code for nucleosome positioning. Nature 442:772–778
Segal E, Widom J (2009) Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr Opin Struct Biol 19:65–71
Tolkunov D, Morozov AV (2010) Genomic studies and computational predictions of nucleosome positions and formation energies. Adv Protein Chem Struct Biol 79:1–57
Wyrick JJ, Holstege FC, Jennings EG et al (1999) Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature 402:418–421
Xing Y, Zhao X, Cai L (2011) Prediction of nucleosome occupancy in Saccharomyces cerevisiae using position-correlation scoring function. Genomics 98:359–366
Xu F, Olson WK (2010) DNA architecture, deformability, and nucleosome positioning. J Biomol Struct Dyn 27:725–739
Yuan GC, Liu YJ, Dion MF et al (2005) Genome-scale identification of nucleosome positions in S. cerevisiae. Science 309:626–630
Zhang L, Luo L (2003) Splice site prediction with quadratic discriminant analysis using diversity measure. Nucleic Acids Res 31:6214–6220
Zhang MQ (1997) Identification of protein coding regions in the human genome by quadratic discriminant analysis. Proc Natl Acad Sci U S A 94:565–568
Zhang Y, Moqtaderi Z, Rattner BP et al (2009) Intrinsic histone–DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol 16:847–852
Zhang Y, Moqtaderi Z, Rattner BP et al (2010) Evidence against a genomic code for nucleosome positioning. Reply to "Nucleosome sequence preferences influence in vivo nucleosome organization". Nat Struct Mol Biol 17:920–923
Zhao X, Pei Z, Liu J, Qin S, Cai L (2010) Prediction of nucleosome DNA formation potential and nucleosome positioning using increment of diversity combined with quadratic discriminant analysis. Chromosome Res 18:777–785
Acknowledgments
We thank Yong-Qiang Xing for his helpful discussions. This work was supported by grants from the National Natural Science Foundation (61102162), Inner Mongolia Natural Science Foundation (2010MS0510), the Research Program of Higher Education of Inner Mongolia Autonomous Region (NJ10098), and the Innovation Fund of Inner Mongolia University of Science and Technology (2009NC005).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Dean A. Jackson.
This work was supported by grants from the National Natural Science Foundation (61102162) Inner Mongolia Natural Science Foundation (2010MS0510), the Research Program of Higher Education of Inner Mongolia Autonomous Region (NJ10098), and the Innovation Fund of Inner Mongolia University of Science and Technology (2009NC005).
Rights and permissions
About this article
Cite this article
Wang, JY., Wang, J. & Liu, G. Calculation of nucleosomal DNA deformation energy: its implication for nucleosome positioning. Chromosome Res 20, 889–902 (2012). https://doi.org/10.1007/s10577-012-9328-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-012-9328-6