
Abstract
Cellular stress can lead to the production of reactive oxygen species (ROS) while autophagy, as a catabolic pathway, protects the cells against stress. Autophagy in its turn plays a pivotal role in the pathophysiology of multiple sclerosis (MS). In the current review, we first summarized the contribution of ROS and autophagy to MS pathogenesis. Then probable crosstalk between these two pathways through HIF-1α for the first time has been proposed with the hope of employing a better understanding of MS pathophysiology and probable therapeutic approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple Sclerosis (MS) is an autoimmune inflammatory disease that affects the central nervous system (CNS) and leads to demyelination, neurodegeneration, and axonal loss in young adults (Andhavarapu et al. 2019). The underlying factors that affect the etiology and pathogenesis of MS remain largely unknown, however, the role of some factors like oxidative stress (OS) and autophagy which are involved in cell damage, degradation, and recycling of cellular components have been highlighted extensively (Gilgun-Sherki et al. 2004; Liang and Le 2015; Aleagha et al. 2015; Ahmadi et al. 2016; Karami et al. 2017). OS mainly occurs with the overproduction of reactive oxygen species (ROS) and disturbs the normal function of the immune system and could be a risk factor for the development and progression of autoimmune diseases (Gilgun-Sherki et al. 2001, 2004).
Autophagy, as a lysosome-dependent degradation procedure, plays a critical role in maintaining the homeostasis of neurons (Ravikumar et al. 2010). Defects in the autophagy process can lead to neuroinflammation and neuronal cell death which results in hypoxia and inducing numerous transcription factors (TFs) such as hypoxia-inducible factor-1α (HIF-1α), nuclear factor-κB (NF-κB), cAMP response-element binding protein (CREB), kruppel-like factor 4 (Klf4), interferon regulatory factor-8 (IRF8), nuclear receptor 4a1 (Nr4a1) (Zou and Crews 2006; Kaushik et al. 2010; Yoshida et al. 2014; Shaked et al. 2015; Liang and Le 2015; Movafagh et al. 2015). HIF-1α is a key factor that regulates the transcription of the genes involved in several vital processes including autophagy and mitochondrial production of ROS. HIF-1α can affects autophagy by various pathways including regulating the interaction of Bcl-2 and Beclin1 which leads to the formation of the Bcl-2/Beclin1 complex, the effect on LC3 levels via ATG5 and maintaining the levels of Beclin1 protein (Bohensky et al. 2007; Williams et al. 2010). Based on a review of the literature, HIF-1α has a paradoxical role through gene activation. So this factor can protect or kill the affected cells (Merelli et al. 2018). The present review focuses on the roles of autophagy and ROS and their possible crosstalk in MS pathophysiology through HIF-1α.
ROS and Pathogenesis of MS
Cells produce controlled amounts of ROS to regulate their physiological activity. The regulated levels of ROS act as secondary messengers in various signaling pathways and help in maintaining cell homeostasis (Sauer et al. 2001; He et al. 2017). However, high levels of ROS cause the oxidation of some critical macromolecules such as nucleic acids, proteins, and lipids, which in this way can disturb the normal function of cells and be a risk factor in the development of various diseases. In fact, ROS leads to cellular injury by overwhelming the capacity of antioxidants (Van Horssen et al. 2011). In the nervous system, ROS are produced in different ways, and inflammation, inflammation-induced hypoxia, and mitochondrial dysfunction raise the level of ROS in this system (Beal 1996; Fiorini et al. 2013). Concomitantly, it has been reported that patients with relapsing–remitting MS (RRMS) show elevated levels of oxidized proteins in the CNS compared to healthy individuals (Di Dalmazi et al. 2016). Increased levels of ROS in the brain and spinal cord cause deterioration of oligodendrocytes and axons which ultimately result in active demyelination and neural cell death (Gilgun-Sherki et al. 2001; Ohl et al. 2016). Moreover, the respiratory burst (rapid release of ROS) in activated mononuclear cells (e.g., microglial cells) generates a large amount of ROS in patients with MS which especially makes remarkable damages in mitochondrial DNA and up-regulates the expression of genes that contribute to the OS (Fischer et al. 2012).
ROS are involved in the dysregulation of the blood–brain barrier (BBB) that leads to rapid disease progression because of the enhanced infiltration of monocytes and inflammation (Ortiz et al. 2013). Accordingly, these results show that ROS is also implicated in the early stages of MS disease when the myelin sheath is still undamaged and the microglial cells and lymphocytes infiltration have occurred (Fischer et al. 2012; Ortiz et al. 2013). Moreover, it has been found that high levels of ROS can promote T-cell activity through the arachidonic acid cascade, or injury to myelin or BBB (Haider et al. 2011).
Another example providing evidence for the contribution of OS to MS pathogenesis is the involvement of mesenchymal stem cells in attenuation of MS progression via decreasing the OS and enhancement of antioxidant factors (Shiri et al. 2020). Taken together, high levels of ROS might damage important cells of the nervous and immune systems and ultimately result in ROS‐induced lesions and cell death in the CNS during various stages of MS.
Autophagy
Mechanisms of Autophagy
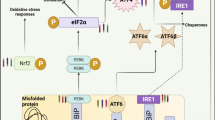
Autophagy is a highly conserved cellular recycling process that is promoted under cellular stress to increase the survival of the cells (Dikic and Elazar 2018). There are three main types of autophagy, which are mechanistically different from each other: macro-autophagy, chaperone-mediated autophagy (CMA), and micro-autophagy (Fig. 1) (Parzych and Klionsky 2014). Macro-autophagy is a breakdown system that associates with the formation of autophagosomes, whilst micro-autophagy is the direct engulfment of a part of the cytoplasm by the lysosomes. These two types of autophagy degrade the organelles, proteins, and lipids non-selectively. In contrast, CMA is a selective (specific) pathway that leads to protein degradation with the help of the KFERQ peptide motif and a cytosolic chaperone called Hsc70. This type of autophagy fails to participate in the degradation of organelles and lipids (Parzych and Klionsky 2014). Macro-autophagy is the most prevalent pathway of autophagy that often referred to as ‘‘autophagy’’ and is distinguished from micro-autophagy and CMA. Autophagy is initiated by various signals that originate from different stress conditions, including hypoxia, starvation, protein aggregation, OS, and endoplasmic reticulum (ER) stress (Dikic and Elazar 2018). When autophagy is started, membrane budding and extension occur and results in the creation of phagophores. Then, double-membrane vesicles, namely autophagosomes, are formed that deliver their contents to the lysosome for degradation and recycling (Parzych and Klionsky 2014). In this stage, the autophagosome outer membrane fuses to the membrane of the lysosome and makes the autolysosome (Yang and Klionsky 2009). In the acidic lumen of the lysosome, the inner membrane of the autophagosome and its contents are degraded by lysosomal hydrolases. Finally, the resulting components of degradation are transferred into the cytoplasm via permeases of the lysosomal membrane and used in different biosynthetic pathways (Yorimitsu and Klionsky 2005).

The main pathways of autophagy: Macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), which are mechanistically different from each other. Macroautophagy acts by the formation of autophagosomes, while microautophagy is the direct confinement of a part of the cytoplasm content by the lysosomes. These two types of autophagy are non-selective pathways. The CMA is a selective pathway that degrades the proteins by the KFERQ peptide motif dependent on the cytosolic chaperone called Hsc70
Autophagy and the Pathogenesis of MS
Autophagy is involved in the homeostasis of the nervous system. Neurons are differentiated cells, which due to their post-mitotic nature and great polarization may be especially sensitive to the accumulation of aggregated or defective compounds, and their survival depends on the process of autophagy. Therefore, the important role of autophagy in the nervous system is mainly related to maintaining the natural balance between the formation and degradation of cellular compounds (Ciechanover 2005; Rubinsztein 2006). Under the physiologic conditions, autophagy is neuroprotective, however, physiopathological increased or decreased activity of autophagy, in turn, can relate to neurodegenerative disorders. Thus, autophagy has a dual role in the nervous system. In literature, autophagy is referred to as a double-edged sword. For example, it has been revealed that autophagy is involved in the homeostasis of amyloid-β (Aβ) under the physiological conditions, and in Alzheimer’s disease (degradation vs. production, respectively) (Tung et al. 2012).
Previous investigations proposed that deficient autophagy is related to neuronal damage in a mouse model of MS (Feng et al. 2017). Another study has shown that the autophagy up-regulation in mice model through inhibitors of mTOR, like Rapamycin, protects the cells from neurodegeneration (Ravikumar et al. 2004). In this regard, it has been shown that Rapamycin, as an autophagy inducer, promotes myelination and increased survival of the neurons (Meikle et al. 2008). In addition, autophagy participates in immune system functions through eliminating cellular pathogens, increasing the antigen-presentation to CD4+ T-cells, and regulating inflammation (Liang and Le 2015). Autophagy also contributes to the proliferation, maturation, and differentiation of lymphocytes via participation in positive and negative selection in the thymus (Yang et al. 2015). It has been reported that the autophagy process is activated in the peripheral blood mononuclear cells (PBMCs) during the acute phase of the disease and alleviated after therapy (Zheng et al. 2014). Moreover, microglial cells, as resident macrophages in the CNS, control intrinsic and extrinsic pathogens and the phagocytosis of the cell debris during demyelination through autophagy-related genes (ATGs). Thus, defective autophagy in microglial cells may inhibit the remyelination of neurons and potentiates continuous neuroinflammation (Sanjuan et al. 2007).
Overexpression of the ATGs including ATG2B, ATG4C, ATG5, ATG11, Bcl2, ULK1, ULK2, ATG-9A, and ATG16L2 in the T cells, demyelinated lesions and blood samples of MS patients have been reported (Igci et al. 2016; Plaza-Zabala et al. 2017; Andhavarapu et al. 2019). The ATG5 plays an important role in the survival of T-cells and involves the proliferation and differentiation of CD4/8+ T-cells and B-cells. Moreover, the ATG5 is significantly increased within the T-cells of MS plaques. The serum levels of ATG5 and Parkin, as biomarkers of autophagic and mitophagic pathways, respectively, are significantly increased in the cerebrospinal fluid (CSF) of patients with MS (Castellazzi et al. 2019). Astrocytes could also be affected by defects in the autophagy pathway (Lee et al. 2009; Wang and Xu 2020). Moreover, in dendritic cells, Atg7 contributes to the activation of T cells, as the Atg7 gene deficient in these cells is accompanied by fewer T-cells presentation in the model of EAE (Plaza-Zabala et al., 2017). Thus, these pieces of evidence indicate that the excess autophagy process contributes to MS pathogenesis. In this regard, demyelination of MS may be potentiated through the extended survival of autoreactive T-cells which these mechanisms can be promoted by autophagy.
On the other hand, mitophagy, as selective autophagy of mitochondria, prevents the excessive generation of ROS and exerts a protective role against MS-induced neurodegeneration (Chen et al. 2008; Shefa et al. 2019). This results in the lower release of toxic molecules including ROS from mitochondria.
Mutual Relationship Between ROS and Autophagy
There are two main sources of ROS: mitochondria that produce ROS as a byproduct of cellular respiration, and the NADPH oxidase complex that actively generates superoxide at the membrane of the neutrophils (Scherz-Shouval and Elazar 2011). ROS and autophagy act in a negative regulatory feedback loop (Azad et al. 2009). It has been shown that mitochondria are involved in autophagy regulation through ROS production (Scherz-Shouval and Elazar 2007; Azad et al. 2009). ROS, as cell signaling molecules, can potentiate the formation of autophagosomes and autophagic degradation. In fact, mitochondria can be involved in the formation of autophagosomes by providing the membrane sources for the biogenesis of autophagosomes. In contrast, autophagy also can decrease oxidative damage and levels of ROS by the elimination of damaged organelles and protein aggregates and the pathways including mitophagy, CMA, and P62 delivery (Chen et al. 2009; Ureshino et al. 2014; Li et al. 2015).
ROS and autophagy act in a negative regulatory feedback loop. In this line, various molecular pathways regulate the interactions between ROS and the process of autophagy. These regulatory mechanisms often contain transcriptional and post-transcriptional regulations, which occur in the nucleus and cytoplasm. In response to excess production of ROS, the HIF-1 transcription factor, p53, FOXO3, and NRF2 are activated, and then induce the transcription of BNIP3/NIX, TIGAR/DRAM, LC3/BNIP3, and p62, respectively (Scherz-Shouval and Elazar 2011). Moreover, PKR-like ER kinase (PERK), an ER stress sensor, is stimulated by ROS which its downstream effectors promote the expression of genes involved in the autophagy pathway. Under hypoxia conditions, the unfolded protein response (UPR) as a HIF-independent pathway is involved in autophagy. This pathway is activated via ER stress sensors of inositol-requiring enzyme 1 (IRE1), activating transcription factor 6 (ATF6) and PERK, and responds to ER stress (Wouters and Koritzinsky 2008). In fact, hypoxia leads to the activation of PERK and then induces the transcription of LC3 and ATG5 by the activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP) transcription factors, which the cell capacity for maintaining autophagy in prolonged hypoxia is enhanced (Rouschop et al. 2010). Moreover, in the cytoplasm, ROS can be involved in the organization of autophagic membranes through the regulation of Atg4 activity (Li et al. 2015).
Hypoxia and HIF-1α in the Pathogenesis of MS
Hypoxia is a state where cells and tissues of the body fail to receive enough oxygen. Hypoxia may exert undesirable effects on the different tissues including the brain. The brain requires a continuous oxygen supply for the maintenance of its normal function. Therefore, prompt detection and response to hypoxic conditions can be very essential. Moreover, the brain is known as an important energy consumer which intense and protracted oxygen deprivation can associate with brain injury through the induction of neurodegeneration and cell death (Merelli et al. 2018). Under deep hypoxia, the cells from CNS, especially oligodendrocytes, are severely and even lethally at risk, and resulting in demyelination. It has been found that hypoxia is present in patients with MS which leads to the creation of neurological deficits including oligodendrocyte loss and demyelination (Lassmann 2003; Desai et al. 2016; Martinez Sosa and Smith 2017). Moreover, the lesions in the brain of patients with MS tend to form in regions that are sensitive to hypoxia (Martinez Sosa and Smith 2017).
HIF is an important transcription factor, which enables adaptive response to hypoxic state in physiological and pathological conditions through activating a large number of genes responsible for oxygen delivery, and other processes. The HIF-1 is a heterodimer composed of two subunits: alpha and beta. The alpha subunit is an inducible and oxygen-sensitive subunit while the beta subunit shows a constitutive expression (Movafagh et al. 2015). Therefore, reactions to the hypoxic states are mainly dependent on HIF-1α. The HIF-1α is a key factor in activating the transcription of genes involved in various aspects of cellular processes such as autophagy, inflammation, metabolism, proliferation, and cell death. In addition, this factor regulates the mitochondrial production of ROS, and it plays an important role in innate and adaptive immunity (Movafagh et al. 2015). In myeloid cells, HIF-1α is required for glycolytic capacity regulation. Lack of the HIF-1α gene may also cause the defect in the aggregation of the myeloid cells, invasiveness, and killing of the bacterial pathogens (Cramer et al. 2003). Moreover, HIF-1α enhances neutrophil survival by triggering the NF-κB-related neutrophilic inflammation and inhibition of apoptosis (Walmsley et al. 2005).
Generally, in the nervous system, the function of HIF-1α can be detrimental via inducing the disruption of BBB or be protective by enhancement of oligodendrocytes and neurons survival (Argaw et al. 2006; Baranova et al. 2007; Vangeison et al. 2008; Weidemann et al. 2009). Regarding the type of the cell in which HIF-1α is expressed, negative or positive impacts on inflammatory demyelination can be predictable. So, the HIF-1α might either have protective impacts on the survival of nerve cells and oligodendrocytes or may intensify the neural damage through enhancement of BBB opening and inflammatory infiltration (Guan et al. 2017).
Interestingly, it has been reported that HIF-1α is upregulated in pre-demyelinating lesions, without observable changes in the white matter of patients with MS (Graumann et al. 2003; Zeis et al. 2008). It has been shown that at the peak of the disease, the expression of HIF-1α is induced in microglia/macrophages, and astrocytes in the spinal cord of experimental autoimmune encephalomyelitis (EAE) mice (Le Moan et al. 2015). In the early lesions of MS, the high level of HIF-1α at mRNA and protein levels makes evidence for the role of this protein in the pathogenesis of the disease (Uurlink 2013). Early hypoxic alterations occurring at pre-demyelinating plaques are linked to lymphocytes’ perivascular accumulation, activation of microglial, disruption of BBB, and mild axonal damage (Marik et al. 2007). HIF-1α overexpression results in exacerbation of vascular permeability and inflammatory responses, whilst the HIF-1α ablation reduces the inflammation (Peyssonnaux et al. 2007; Thiel et al. 2007; Weidemann et al. 2009). Inside the active white matter lesions (WMLs), the expression of HIF-1α is generally minimal or absent (Aboul-Enein et al. 2003; Marik et al. 2007), whereas in oligodendrocytes, especially in the lesions with distal dying-back oligodendrogliopathy, it has a high level of expression (Lassmann 2003).
On the other hand, hypoxia is known to be one of the causes of the inducting process of autophagy (Bellot et al. 2009; Schaaf et al. 2013). Based on the intensity and duration of oxygen deprivation, hypoxia promotes various pathways of autophagy. For example, chronic and moderate hypoxia promotes HIF-1α and PKCδ-JNK1-dependent pathways for autophagy induction (Semenza 2000; Mazure and Pouyssegur 2010). In contrast, the intense and rapid oscillation of oxygen triggers autophagy through HIF-1α independent procedures such as the mTOR-mediated pathway and UPR (Papandreou et al. 2008). Totally, accumulating data indicates that autophagy impairment also occurs in MS and may contribute to neurodegeneration.
Crosstalk Between ROS and Autophagy During MS Pathogenesis
We mentioned several pieces of evidence that support the mutual crosstalk between ROS and autophagy. Here, we proposed a probable link between autophagy and ROS by HIF-1 in MS disease. ROS that is produced under OS conditions can damage the vascular endothelium and decrease the ability of vascular smooth muscles for relaxation and vasodilation. This mechanism might be one of the factors leading to decreased cerebral blood flow and hypoxia in MS (Yang and Dunn 2019). On the other hand, cerebrovascular dysfunction leads to hypo-perfusion and promotes the transient hypoxic condition in the vulnerable regions of the CNS. This phenomenon causes the disruption of BBB and leak of serum proteins such as fibronectin and fibrinogen which eventually leads to the activation of the microglial cells. Afterward, cytotoxic factors such as ROS and NO are released by activated microglial cells that trigger the damage to the oligodendrocytes which in turn, results in cellular death and demyelination (Halder and Milner 2021).
Under hypoxic conditions, HIF-1α also reduces the level of ROS by various mechanisms. These include induction of lactate dehydrogenase A and pyruvate dehydrogenase kinase 1, improvement of efficiency of complex IV via switching of cytochrome c oxidase subunit COX4-1 to COX4-2, the direct regulation of mitochondrial performance through its nuclear-independent activities, shunting the pyruvate away from the mitochondria and the conversion of pyruvate to lactate, induction of microRNA-210 that is able to block the Fe/S clusters assembly needed for oxidative phosphorylation, induction of the BNIP3 and triggering the autophagy (Tormos and Chandel 2010; Li et al. 2019).
In connection with autophagy, HIF-1 induces the BNIP3 gene expression which competes with Beclin-1 for binding to the BCL2, the inhibitory binding partner of Beclin-1. Then autophagy is induced by releasing Beclin-1 from BCL2 (Semenza 2011). Thus, HIF-1α-mediated BNIP3 expression has a key role in hypoxia-induced mitophagy (Zhang et al. 2008). The mitochondrial autophagy, as the metabolic adaptive response, promotes cell survival in protracted hypoxic conditions that is essential to prevent ROS production and cell death (Zhang et al. 2008; Dhuria et al. 2010). In mitochondria, HIF regulates mitochondrial oxidative stress and respiration and vice versa. Of note, mitochondria are known as the major oxygen consumers in the cell. So, mitochondria and HIF are intimately linked in order to regulate each other and respond properly to hypoxic conditions (Tormos and Chandel 2010). In general, HIF-1α may play a dual role as "protective" or "harmful” during the pathogenesis of neurodegenerative diseases which is dependent on the severity of hypoxia and environmental conditions (Merelli et al. 2018).
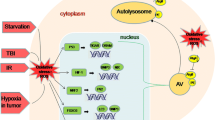
Although determining the exact relationship of ROS, hypoxia, and autophagy in MS requires supportive clinical evidence, based on available data, it seems that HIF-1α can make crosstalk between these processes during the pathogenesis of MS. So that ROS probably exacerbates the situation of hypoxia. Afterward, by activating the HIF-1α under hypoxic conditions, various pathways including autophagy are induced. The enhancement of autophagic degradation can damage nerve cells and leads to demyelination or cell death in the CNS and contributes to MS progression. Generally, hyperactivity or hypoactivity of autophagy is undesirable for the normal function of neurons and these cells need a regulated amount of autophagy to maintain cell survival under different stress conditions. Thereby, it is possible to infer a “ROS—hypoxia—autophagy pathway” in MS pathogenesis (Fig. 2). According to available data, the factors and signaling pathways, which link autophagy and ROS, can be of great importance in MS combinatorial treatment. However, more investigations are required to find whether the crosstalk between autophagy and ROS can be more efficient in MS therapy.

The crosstalk between ROS and autophagy by HIF-1α in the pathogenesis of multiple sclerosis. Cellular stresses lead to increased ROS levels. ROS can intensify the situation of hypoxia. Then, under hypoxic condition, HIF-1α is activated, which induces the various pathways including autophagy. The enhancement of autophagic degradation can result in neuronal cell damage and demyelination. In the figure, green arrows show the protective pathways, while black arrows show the neurodegenerative pathways
Conclusion
ROS production in the brain and autophagy dysregulation can damage cells of the nervous system and are associated with various stages of MS. In this review, for the first time, we suggested crosstalk between ROS and autophagy in MS pathogenesis by HIF-1α. Accordingly, targeting ROS—hypoxia—autophagy cycle may lead to more efficient results in combinatorial treatments of MS.
References
Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Brück W, Lucchinetti C, Schmidbauer M, Jellinger K, Lassmann H (2003) Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol 62:25–33
Ahmadi M, Aleagha MS, Harirchian MH, Yarani R, Tavakoli F, Siroos B (2016) Multiple sclerosis influences on the augmentation of serum Klotho concentration. J Neurol Sci 362:69–72
Aleagha MS, Siroos B, Ahmadi M, Balood M, Palangi A, Haghighi AN, Harirchian MH (2015) Decreased concentration of Klotho in the cerebrospinal fluid of patients with relapsing–remitting multiple sclerosis. J Neuroimmunol 281:5–8
Andhavarapu S, Mubariz F, Arvas M, BeverJr C, Makar TK (2019) Interplay between ER stress and autophagy: a possible mechanism in multiple sclerosis pathology. Exp Mol Pathol 108:183–190
Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, Raine CS, Brosnan CF, John GR (2006) IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol 177:5574–5584
Azad MB, Chen Y, Gibson SB (2009) Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal 11(4):777–790
Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC (2007) Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci 27:6320–6332
Beal M (1996) Aging, energy, and oxidative stress in neurodegenerative diseases. Restor Neurol Neurosci 3:180–181
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM (2009) Hypoxia-induced autophagy is mediated through HIF-induction of BNIP3 and BNIP3L via their BH3-domains. Mol Cell Biol 29:2570–2581
Bohensky J, Shapiro IM, Leshinsky S, Terkhorn SP, Adams CS, Srinivas V (2007) HIF-1 regulation of chondrocyte apoptosis: induction of the autophagic pathway. Autophagy 3:207–214
Castellazzi M, Patergnani S, Donadio M, Giorgi C, Bonora M, Fainardi E, Casetta I, Granieri E, Pugliatti M, Pinton P (2019) Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J Neuroinflamm 16(1):1–8
Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB (2008) Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 15(1):171–182
Chen Y, Azad MB, Gibson SB (2009) Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 16(7):1040–1052
Ciechanover A (2005) Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 6:79–87
Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112:645–657
Desai RA, Davies AL, Tachrount M, Kasti M, Laulund F, Golay X, Smith KJ (2016) Cause and prevention of demyelination in a model multiple sclerosis lesion. Ann Neurol 79(4):591–604
Dhuria SV, Hanson LR, Frey WH (2010) II Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci 99(4):1654–1673
Di Dalmazi G, Hirshberg J, Lyle D, Freij JB, Caturegli P (2016) Reactive oxygen species in organ-specific autoimmunity. Autoimmun Highlights 7(1):1–1
Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19(6):349–364
Feng X, Hou H, Zou Y, Guo L (2017) Defective autophagy is associated with neuronal injury in a mouse model of multiple sclerosis. Bosn J Basic Med Sci 17(2):95
Fiorini A, Koudriavtseva T, Bucaj E, Coccia R, Foppoli C, Giorgi A, Schininà ME, Di Domenico F, De Marco F, Perluigi M (2013) Involvement of oxidative stress in occurrence of relapses in multiple sclerosis: the spectrum of oxidatively modified serum proteins detected by proteomics and redox proteomics analysis. PLoS ONE 8(6):e65184
Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, Mahad D, Bradl M, van Horssen J, Lassmann H (2012) NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135(3):886–899
Gilgun-Sherki Y, Melamed E, Offen D (2001) Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 40(8):959–975
Gilgun-Sherki Y, Melamed E, Offen D (2004) The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol 251(3):261–268
Graumann U, Reynolds R, Steck AJ, Schaeren-Wiemers N (2003) Molecular changes in normal appearing white matter in multiple sclerosis are characteristic of neuroprotective mechanisms against hypoxic insult. Brain Pathol 13:554–573
Guan SY, Leng RX, Tao JH, Li XP, Ye DQ, Olsen N, Zheng SG, Pan HF (2017) Hypoxia-inducible factor-1α: a promising therapeutic target for autoimmune diseases. Expert Opin Ther Targets 21(7):715–723
Haider L, Fischer MT, Frischer JM, Bauer J, Höftberger R, Botond G, Esterbauer H, Binder CJ, Witztum JL, Lassmann H (2011) Oxidative damage in multiple sclerosis lesions. Brain 134(7):1914–1924
Halder SK, Milner R (2021) Hypoxia in multiple sclerosis; is it the chicken or the egg? Brain 144(2):402–410
He L, He T, Farrar S, Ji L, Liu T, Ma X (2017) Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem 44(2):532–553
Igci M, Baysan M, Yigiter R, Ulasli M, Geyik S, Bayraktar R, Bozgeyik İ, Bozgeyik E, Bayram A, Cakmak EA (2016) Gene expression profiles of autophagy-related genes in multiple sclerosis. Gene 588(1):38–46
Karami M, Mehrabi F, Allameh A, Kakhki MP, Amiri M, Aleagha MS (2017) Klotho gene expression decreases in peripheral blood mononuclear cells (PBMCs) of patients with relapsing-remitting multiple sclerosis. J Neurol Sci 381:305–307
Kaushik DK, Gupta M, Das S, Basu A (2010) Krüppel-like factor 4, a novel transcription factor regulates microglial activation and subsequent neuroinflammation. J Neuroinflamm 7(1):1–20
Lassmann H (2003) Hypoxia-like tissue injury as a component of multiple sclerosis lesions. J Neurol Sci 206(2):187–191
Le Moan N, Baeten KM, Rafalski VA, Ryu JK, Coronado PE, Bedard C, Syme C, Davalos D, Akassoglou K (2015) Hypoxia inducible factor-1 in astrocytes and/or myeloid cells is not required for the development of autoimmune demyelinating disease. eNeuro 2
Lee SJ, Cho KS, Koh JY (2009) Oxidative injury triggers autophagy in astrocytes: the role of endogenous zinc. Glia 57(12):1351–1361
Li L, Tan J, Miao Y, Lei P, Zhang Q (2015) ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol 35(5):615–621
Li HS, Zhou YN, Li L, Li SF, Long D, Chen XL, Zhang JB, Feng L, Li YP (2019) HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol 25:101109
Liang P, Le W (2015) Role of autophagy in the pathogenesis of multiple sclerosis. Neurosci Bull 31(4):435–444
Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ (2007) Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain 130:2800–2815
Martinez Sosa S, Smith KJ (2017) Understanding a role for hypoxia in lesion formation and location in the deep and periventricular white matter in small vessel disease and multiple sclerosis. Clin Sci 131(20):2503–2524
Mazure NM, Pouyssegur J (2010) Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol 22(2):177–180
Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ (2008) Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci 28(21):5422–5432
Merelli A, Rodríguez JC, Folch J, Regueiro MR, Camins A, Lazarowski A (2018) Understanding the role of hypoxia inducible factor during neurodegeneration for new therapeutics opportunities. Curr Neuropharmacol 16(10):1484–1498
Movafagh S, Crook S, Vo K (2015) Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J Cell Biochem 116(5):696–703
Ohl K, Tenbrock K, Kipp M (2016) Oxidative stress in multiple sclerosis: central and peripheral mode of action. Exp Neurol 277:58–67
Ortiz GG, Pacheco-Moises FP, Bitzer-Quintero OK, Ramírez-Anguiano AC, Flores-Alvarado LJ, Ramírez-Ramírez V, Macias-Islas MA, Torres-Sanchez ED (2013) Immunology and oxidative stress in multiple sclerosis: clinical and basic approach. Clin Dev Immunol. https://doi.org/10.1155/2013/708659
Papandreou I, Lim AL, Laderoute K, Denko NC (2008) Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ 15:1572–1581
Parzych KR, Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20(3):460–473
Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V (2007) Cutting edge: essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol 178:7516–7519
Plaza-Zabala A, Sierra-Torre V, Sierra A (2017) Autophagy and microglia: novel partners in neurodegeneration and aging. Int J Mol Sci 18(3):598
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36(6):585–595
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90(4):1383–1435
Rouschop KM, Van Den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P (2010) The unfolded protein response protectshuman tumor cells during hypoxia through regulation of theautophagy genes MAP1LC3B and ATG5. J Clin Investig 120:127–141
Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786
Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR (2007) Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450(7173):1253–1257
Sauer H, Wartenberg M, Hescheler J (2001) Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem 11(4):173–186
Schaaf MB, Cojocari D, Keulers TG, Jutten B, Starmans MH, de Jong MC, Begg AC, Savelkouls KG, Bussink J, Vooijs M, Wouters BG (2013) The autophagy associated gene, ULK1, promotes tolerance to chronic and acute hypoxia. Radiother Oncol 108:529–534
Scherz-Shouval R, Elazar Z (2007) ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 17(9):422–427
Scherz-Shouval R, Elazar Z (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36(1):30–38
Semenza GL (2000) HIF-1 and human disease: one highly involved factor. Genes Dev 14:1983–1991
Semenza GL (2011) Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta Mol Cell Res 1813(7):1263–1268
Shaked I, Hanna RN, Shaked H, Chodaczek G, Nowyhed HN, Tweet G, Tacke R, Basat AB, Mikulski Z, Togher S, Miller J (2015) Transcription factor Nr4a1 couples sympathetic and inflammatory cues in CNS-recruited macrophages to limit neuroinflammation. Nat Immunol 16(12):1228–1234
Shefa U, Jeong NY, Song IO, Chung HJ, Kim D, Jung J, Huh Y (2019) Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen Res 14(5):749
Shiri E, Pasbakhsh P, Borhani-Haghighi M, Alizadeh Z, Nekoonam S, Mojaverrostami S, Mahabadi VP, Mehdi A, Zibara K, Kashani IR (2020) Mesenchymal stem cells ameliorate cuprizone-induced demyelination by targeting oxidative stress and mitochondrial dysfunction. Cell Mol Neurobiol 27:1–5
Thiel M, Caldwell CC, Kreth S, Kuboki S, Chen P, Smith P, Ohta A, Lentsch AB, Lukashev D, Sitkovsky MV (2007) Targeted deletion of HIF-1 alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS ONE 2:e853
Tormos KV, Chandel NS (2010) Inter-connection between mitochondria and HIFs. J Cell Mol Med 14(4):795–804
Tung YT, Wang BJ, Hu MK, Hsu WM, Lee H, Huang WP, Liao YF (2012) Autophagy: a double-edged sword in Alzheimer’s disease. J Biosci 37(1):157–165
Ureshino RP, Rocha KK, Lopes GS, Bincoletto C, Smaili SS (2014) Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid Redox Signal 21(1):123–137
Uurlink BHJ (2013) The evidence for Hypoperfusion as a factor in multiplesclerosis lesion development. Mult Scler Int 2013:6
Van Horssen J, Witte ME, Schreibelt G, de Vries HE (2011) Radical changes in multiple sclerosis pathogenesis. Biochem Biophys Acta 1812(2):141–150
Vangeison G, Carr D, Federoff HJ, Rempe DA (2008) The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J Neurosci 28:1988–1993
Walmsley SR, Print C, Farahi N (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med 201:105–115
Wang JL, Xu CJ (2020) Astrocytes autophagy in aging and neurodegenerative disorders. Biomed Pharmacother 122:109691
Weidemann A, Kerdiles YM, Knaup KX, Rafie CA, Boutin AT, Stockmann C, Takeda N, Scadeng M, Shih AY, Haase VH, Simon MC (2009) The glial cell response is an essential component of hypoxia-induced erythropoiesis in mice. J Clin Investig 119:3373–3383
Williams H, Johnson JL, Jackson CL, White SJ, George SJ (2010) MMP-7 mediates cleavage of N-cadherin and promotes smooth muscle cell apoptosis. Cardiovasc Res 87:137–146
Wouters BG, Koritzinsky M (2008) Hypoxia signalling throughmTOR and the unfolded protein response in cancer. Nat Rev Cancer 8:851–864
Yang R, Dunn JF (2019) Multiple sclerosis disease progression: contributions from a hypoxia–inflammation cycle. Mult Scler J 25(13):1715–1718
Yang Z, Klionsky DJ (2009) An overview of the molecular mechanism of autophagy. Curr Top MicrobiolImmunol 335:1–32
Yang Z, Goronzy JJ, Weyand CM (2015) Autophagy in autoimmune disease. J Mol Med 93(7):707–717
Yorimitsu T, Klionsky DJ (2005) Autophagy: molecular machinery for self-eating. Cell Death Differ 12(2):1542–1552
Yoshida Y, Yoshimi R, Yoshii H, Kim D, Dey A, Xiong H, Munasinghe J, Yazawa I, O’Donovan MJ, Maximova OA, Sharma S (2014) The transcription factor IRF8 activates integrin-mediated TGF-β signaling and promotes neuroinflammation. Immunity 40(2):187–198
Zeis T, Graumann U, Reynolds R, Schaeren-Wiemers N (2008) Normal-appearing white matter in multiple sclerosis is in a subtle balance between inflammation and neuroprotection. Brain 131:288–303
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283(16):10892–10903
Zheng L, Xue Q, Ni J, Guo S, Dong W (2014) Levels of Beclin 1 and LC3 in peripheral blood mononuclear cells of patients with multiple sclerosis and neuromyelitisoptica and its significance. Zhonghuayixuezazhi 94(39):3052–3055
Zou J, Crews F (2006) CREB and NF-κB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell Mol Neurobiol 26(4):383–403
Author information
Authors and Affiliations
Contributions
RA and MSEA designed the structure of the manuscript. RA, MSEA, RY, and PM drafted the manuscript. RA, MSEA, RY, and PM finalized the paper. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Informed Consent
No informed consent is needed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Asgari, R., Yarani, R., Mohammadi, P. et al. HIF-1α in the Crosstalk Between Reactive Oxygen Species and Autophagy Process: A Review in Multiple Sclerosis. Cell Mol Neurobiol 42, 2121–2129 (2022). https://doi.org/10.1007/s10571-021-01111-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-021-01111-5