
Abstract
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is a long non-coding RNA contributing to protect the blood–brain barrier (BBB) after stroke. We searched for small molecules that may up-regulate MALAT1 and focused on polydatin (PD), a natural product, as a possible candidate. PD enhanced MALAT1 gene expression in rat brain microvascular endothelial cells, reducing cell toxicity and apoptosis after oxygen and glucose deprivation (OGD). These effects correlated with reduction of inflammatory factors and enhancement of expression of BBB markers. We found opposite changes after MALAT1 silencing. We determined that C/EBPβ is a key transcription factor for PD-mediated MALAT1 expression. PPARγ activity is involved in MALAT1 protective effects through its coactivator PGC-1α and the transcription factor CREB. This suggests that PD activates the MALAT1/CREB/PGC-1α/PPARγ signaling pathway to protect endothelial cells against ischemia. PD administration to rats subjected to brain ischemia by transient middle cerebral artery occlusion (tMCAO) reduced cerebral infarct volume and brain inflammation, protected cerebrovascular endothelial cells and BBB integrity. These effects correlated with increased expression of MALAT1, C/EBPβ, and PGC-1α. Our results strongly suggest that the beneficial effects of PD involve the C/EBPβ/MALAT1/CREB/PGC-1α/PPARγ pathway, which may provide a novel therapeutic strategy for brain ischemic stroke.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cerebral ischemic stroke is one of the leading causes of death and long-term disability worldwide, and in most cases, it is without effective treatment (Wang et al. 2017; Schellinger et al. 2004). Therefore, it is urgent to search for novel and effective treatments for this disease.
Under normal physiological conditions, cerebrovascular endothelial cells play a leading role in maintaining the integrity of the blood–brain barrier (BBB) and brain homeostasis (Hawkins and Davis 2005; Sandoval and Witt 2008). Cerebral ischemia injures cerebrovascular endothelial cells and damages the BBB, Injured vascular endothelial cells express immunoglobulin superfamily adhesion molecules (VCAM-1, ICAM-1, etc.) and selectin family members (E-selectin, P-selectin, etc.), and secrete TNF-α, IL-1β, IL-6, IL-8, MCP-1 and other inflammatory mediators, which can cause neuroinflammation and thus neuronal damage (Ruetzler et al. 2001; Huang et al. 2006). The combination of brain endothelial cell injury, endothelial cell inflammation, increased cerebral vascular permeability and blood–brain barrier leakage increases neuronal damage (DEL and Zoppo 2006; Zhang et al. 2012; Yin et al. 2014) For these reasons, the protection of cerebrovascular endothelial cells has become an important therapeutic strategy to prevent cerebrovascular dysfunction against ischemic stroke (Fisher 2008).
Long non-coding RNAs (lncRNA) play an important role in the occurrence and development of many diseases including stroke (Qureshi and Mehler 2012). Elucidating of the regulatory mechanisms of lncRNA may enhance the understanding of the molecular mechanisms of ischemic stroke and help to establish a more complete treatment strategy. Recent studies reported changes in lncRNA expression profile in cultured rat brain microvascular endothelial cells (rBMVEC) submitted to oxygen and glucose deprivation (OGD) (Zhang et al. 2016). In vivo and in vitro experiments revealed significant differences in the expression of some lncRNAs in cerebral microvascular endothelial cells after cerebral ischemia and that the up-regulation of the lncRNA MALAT1 (Metastasis-associated lung adenocarcinoma transcript 1) was the most significant. Subsequent in vitro and in vivo experiments further confirmed that knockout of MALAT1 exacerbates cerebrovascular endothelial cell damage after cerebral ischemia (Zhang et al. 2017). These studies suggest that MALAT1 may play an important role in the pathological process of ischemic stroke. Other studies have found that lncRNA MALAT1 is also involved in the regulation of endothelial angiogenesis. Michalik et al. found that silencing MALAT1 in vivo reduced vascular endothelial cell proliferation and inhibited neovascularization (Michalik et al. 2014), which indicates that MALAT1 could promote the proliferation of endothelial cells in vivo. In addition, Liu et al. found that MALAT1 silencing can also regulate the proliferation, migration and angiogenesis of retinal vascular endothelial cells (Liu et al. 2014), further indicating the important role of MALAT1 in regulating endothelial cell function. However, the specific molecular mechanisms of MALAT1 in cerebral vascular endothelial cells damaged by ischemia are not fully understood. We propose that finding small molecular probes that increase MALAT1 expression in cerebral microvascular endothelial cells may lead to further developing these probes as therapeutic drugs to ameliorate endothelial injury and brain ischemia.
We constructed human umbilical vein endothelial cells (HUVEC) stably expressing a luciferase reporter gene driven by MALAT1 promoter (MALAT1-pro-luc-HUVEC). These cells can be used as a screening assay to search for small molecular compounds that directly regulate the expression of MALAT1 in endothelial cells. Using these cells, we screened our in-house natural product library. We found a natural product, polydatin (PD), which could directly up-regulate the expression of MALAT1 in our system. PD is a monomeric active ingredient isolated from Chinese traditional medicine Polygonum cuspidatum which has beneficial effects on cardiovascular and cerebrovascular system (Jinmo et al. 2010). PD reduces cerebral ischemia, but the mechanisms of this effect have not been clarified. In this study, we found that PD can not only directly regulate the expression of MALAT1 in cerebral vascular endothelial cells, but also protect cerebral vascular endothelial cells from OGD injury in vitro and from cerebral ischemia in vivo. This suggests that PD may ameliorate endothelial and brain injury after cerebral ischemia by up-regulating MALAT1 expression in cerebral vascular endothelial cells. The discovery that PD up-regulates MALAT1 expression will also provide experimental evidence, indicating that MALAT1 up-regulation could be a new target for the treatment of ischemic stroke.
Methods
Reagents
The natural compound PD (polydatin, powder purity over 98%) was obtained from Yunnan Xili Company (China). Pioglitazone (agonist of PPARγ), GW9662 and T0070907 (antagonists of PPARγ) were obtained from Selleck (Houston, TX, USA). Compound 666-15 (inhibitor of CREB) and SR-18292 (inhibitor of PGC-1α) were purchased from MedChemExpress (MCE). For all the experiments, these compounds were prepared by diluting the stock with culture medium (the final concentration of DMSO was less than 0.1%). Cell culture medium and supplements were purchased from Invitrogen (Carlsbad, CA, USA). All other regular reagents were obtained from Sigma-Aldrich.
Isolation and Culture of Rat Brain Microvascular Endothelial Cells
Primary rat brain microvascular endothelial cells (rBMVEC) were isolated from 3 to 4 weeks old Sprague–Dawley (SD) rats. The rats were killed by cervical dislocation and sterilized in 75% ethanol for 5 min, then the whole brain was quickly removed and placed in cold Hank’s Balanced Salt Solution (HBSS) solution. The cerebral cortex was carefully separated and then shredded followed by digestion by type II collagenase and DNAase (Sigma-Aldrich, St. Louis, USA) for 1.5 h at 37 °C. The digested brain tissue was mixed with 20% BSA solution and centrifuged for 8 min (1000×g). The precipitation after centrifugation was digested again by type II collagenase and dispersase (Sigma-Aldrich, St. Louis, USA) for 1 h at 37 °C. After digestion, the precipitation obtained by centrifugation was resuspended in endothelial cell medium (ECM, ScienCell, San Diego, CA, USA) containing puromycin (final concentration 4 µg/mL) then transferred to a culture dish pre-coated with type I collagen and cultured in a humidified atmosphere of 5% CO2/95% air for 48 h, then replaced with an ECM medium without puromycin. When the cultures reached 80% confluency, the purified endothelial cells were passaged and subjected to the following subsequent experiments.
Cell Culture
Human embryonic kidney cells (HEK-293T) and human umbilical vein endothelial cells (HUVEC) were obtained from ATCC (Manassas, Virginia, USA) and cultured in Dulbecco’s minimum essential medium (DMEM) with 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA), 100 U/mL penicillin and 100 µg/mL streptomycin. All cells were maintained in incubator with an atmosphere of 95% air and 5% CO2.
Drug Treatment
In the in vitro studies, HEK-293T cells were pretreated with the CREB inhibitor 666-15 (1 µM) (Xie et al. 2015) or the PGC-1α inhibitor SR18292 (50 µM) (Sharabi et al. 2017) for 2 h, and then incubated with PD (20 µM) for another 24 h, followed by RT-PCR or luciferase reporter gene assay experiments. In the OGD cellular model, rBMVEC cells pretreated with PPARγ antagonist GW9662 (20 µM) or T0070907 (10 µM) for 3 h (Xu et al. 2015; Zilleßen et al. 2016), were incubated with PD (20 µM) for 24 h, and then placed in an OGD condition for another 12 h, followed by RT-PCR, Western blotting, TUNEL assay or immunostaining experiments.
Western Blotting Analysis
To determine the levels of protein expression, HUVEC and rBMVEC cells obtained from different treatment groups were washed with cold PBS and collected with Tris-Glycine SDS lysis buffer (Chang et al. 2017), and animal tissues were lysed with RIPA buffer (Vazyme, Jiangsu, China) (Xu et al. 2015). The mixtures were collected as whole cell extracts. Cell extracts were separated by 10% SDS-PAGE, and then transferred onto PVDF membranes. The membranes were blocked for 1.5 h with 3% bovine serum albumin (BSA) in Tris-buffered saline plus Tween 20 (TBST, pH 7.4) and then probed with the specific primary antibodies of rabbit anti-C/EBPβ (1:1000), rabbit anti-phospho-CREB (1:500), rabbit anti-CREB (1:1000), rabbit anti-PPARγ (1:1000), rabbit anti-PGC-1α (1:1000), and mouse anti-β-actin (1:2000) overnight at 4 °C. C/EBPβ and β-actin antibodies were purchased from Santa Cruz and other antibodies were obtained from Abclonal. After several washes with TBST, the membranes were incubated with the secondary antibodies of anti-rabbit IgG (1:10,000; SunShineBio, China) or anti-mouse IgG (1:10,000; SunShineBio, China) for 1 h at room temperature, and then blots were visualized using chemiluminescence with a Bio-Rad ChemiDoc XRS (Bio-Rad, Hercules, CA, USA). The resulting bands were quantified using densitometric analysis and were normalized to the levels of β-actin protein.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
To determine the levels of gene expression, HEK-293T, HUVEC, and rBMVEC cells obtained from different treatment groups were washed with cold PBS and total RNA was extracted using Trizol reagent (Vazyme, Nanjing, China). Reverse transcription was performed using a cDNA synthesis kit (Vazyme, Nanjing, China) following standard techniques. For PCR analysis, 5 µL of cDNA was used as a template and amplified using specific primers (Table 1). Each PCR mixture contained 5 µL of cDNA, 1 µL of each primer (10 µmol/L), 3.5 µL of ddH2O, 10.5 µL of SYBR Green Premix (Vazyme, Nanjing, China). The relative quantitative expression was measured in triplicate on a Real-time PCR Applied Biosystems Step One Plus Detection System (USA) where the 18S rRNA levels served as a control. The amplification conditions were 95 °C for 5 min, 40 cycles of 95 °C for 10 s, 60 °C for 30 s. The specificity of amplification was confirmed by a melting curve and the relative gene expression was analyzed by the 2−ΔΔCT method (Kenneth and Thomas 2001). For the specific experimental operation of RT-PCR, we refer to the previous literature (Guo et al. 2017).
Detection of Toxicity of PD by MTT and LDH Assay
HUVEC and rBMVEC cells were pretreated with different concentrations of PD at a density of 60–70% for 24 h. Then the culture medium was collected and lactate dehydrogenase (LDH) was measured by the LDH assay kit (Beyotime Biotechnology) according to the manufacturer’s instructions, and the remaining cells were incubated with 0.5 mg/mL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) for 4 h in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. After 4 h, the medium was replaced with DMSO, and absorbance was measured at 570 nm in a BD plate-reader.
Oxygen and Glucose Deprivation (OGD)
HUVEC and rBMVEC cells were pretreated with PD at a density of 60–70% for 24 h, and then the complete medium was replaced with glucose-free Earle’s balanced salt solution (EBSS, pH 7.4) and transferred to a hypoxia chamber containing a mixture of 95% N2 and 5% CO2 for 12 h. Control cells without OGD were maintained under normal conditions. After OGD, the culture medium (EBSS) was collected for LDH detection by the LDH assay kit, and the remaining cells were collected for Western blotting and RT-PCR to analyze the expressions of related proteins and genes, respectively.
Luciferase Reporter Gene Assay
All luciferase reporter gene experiments were performed on HEK-293T cells and MALAT1-pro-luc-HUVEC. Cells cultured in 96-well plates pre-coated by Poly-l-Lysine (Sigma-Aldrich, St. Louis, USA) at 70% confluence were transfected with 100 ng reporter gene plasmid and 25 ng of β-galactosidase (β-Gal) plasmid as internal control using Lipofectamine™ 3000 (Invitrogen) according to the manufacturer’s instructions. After 24 h of transfection, cells were treated with different concentrations of compounds for another 24 h. Cells in each well were lysed with Reporter Lysis Buffer (150 µL) from the luciferase reporter assay kit (Promega) for 20 min at 37 °C, and then the cell lysis solution (80 µL) was transferred into a 96-well white plate, followed by addition of luciferase assay buffer (40 µL) to measure luciferase activity in accordance with the Luciferase Reporter Assay System instructions. The remaining cell lysis solution was tested for β-Gal activity using a β-Gal reporter assay kit purchased from Beyotime Institute of Biotechnology.
Knockdown and Overexpression of Target Genes
Small interfering RNA (siRNA) was used to knockdown the expression of C/EBPβ, and the overexpression of C/EBPβ was mediated by the plasmid pcDNA3.1-C/EBPβ. The knockdown efficiency of MALAT1 by siRNA in primary rBMVEC cells is very low, so antisense LNA GapmeR of MALAT1 (300635-101, Exiqon), a method to specifically knockdown lncRNA expression in cells was employed to silence the expression of MALAT1 in primary rBMVEC cells, and the method of siRNA was used for knockdown of MALAT1 in HEK-293T cell and HUVEC cell lines. The overexpression of MALAT1 was mediated by pcEGFP-C1-MALAT1. The silencing of PGC-1α was also achieved by the siRNA method. The transfection of siRNA, GapmeR and plasmids was achieved using Lipofectamine™ 3000 obtained from Invitrogen. GapmeR is a potent antisense oligonucleotide used for highly efficient inhibition of mRNA and lncRNA function. The specific experimental procedures for cell transfection have been described in previous published literature (Ruan et al. 2018). The GapmeR and siRNA sense strands were listed in Table 2.
Measurement of Apoptotic Cells Level by TUNEL Staining
The culture medium from rBMVEC cells was collected after OGD treatment for the detection of LDH release, and apoptotic cells were measured by Apoptosis Fluorescein Detection kit (TUNEL), which was purchased from Beyotime Institute of Biotechnology. Briefly, the cells fixed on the crawler were infiltrated in 4% of paraformaldehyde for 10 min. The cells were then washed 3 times for 2 min each with PBS (pH 7.4), then cells were infiltrated in the TUNEL reagents for 60 min followed by washing with PBS. After washing, an anti-fluorescent quencher was added dropwise to the cells and the total number of TUNEL-positive cells was counted under a fluorescence microscope set at excitation and emission wavelengths of 550 nm and 570 nm, respectively. The apoptosis rate of cells was calculated by morphometric analysis Image-pro plus (Media cybernetics, MD, USA).
Immunofluorescence Staining
Primary rBMVEC cells exposed to drug treatment were plated on coverslips pre-coated with type I collagen. For immunofluorescence staining, cells were fixed with 4% paraformaldehyde in PBS for 10 min, permeabilized in PBS containing 0.1% Triton X-100 for 10 min and blocked with 5% bovine serum albumin. Then coverslips were incubated with anti-ZO-1 antibody (1:100; Abclonal) overnight at 4 °C. Nuclei were labeled with Hoechst 33,342 (Beyotime, Haimen, China) at room temperature. After washing three times with PBS, the cells were incubated with Alexa Fluor 488 goat anti-rabbit IgG (Life, USA) for 60 min. The fluorescence was then examined using a confocal microscope (Olympus FV1000, Japan). The specific experimental procedures for immunofluorescence staining refer to the previous literature (Chang et al. 2017). The intensity of fluorescence was calculated by morphometric analysis Image-pro plus (Media cybernetics, MD, USA).
Transient Middle Cerebral Artery Occlusion (tMCAO) in SD Rats
Transient middle cerebral artery occlusion (tMCAO) was performed as previously described (Wang et al. 2018a, 2018b). The rats were randomly divided into three groups, sham, vehicle, and PD treatment. Total of 112 male SD rats were used in this study. Each group consisted of 8–12 successful treated rats. The right common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA) of individual rats were visualized. A monofilament nylon suture (diameter of approximately 0.26 mm) with a round tip was inserted into the ICA through the ECA stump and gently advanced to the MCA. After 2 h of MCAO, the filament was withdrawn to restore blood flow (reperfusion). Cerebral blood flow (CBF) was measured using a Laser doppler flowmetry machine, and following the manufacturer’s instruction (MoorFLPI-2, Moor Instruments Inc. Delaware, USA). Briefly, anesthetized rats were placed in the prone position with the skull exposed but unopened. The CBF was measured in both cerebral hemispheres and recorded immediately after the tMCAO surgery. CBF was analyzed by the MoorFLPI software and shown with arbitrary units in a 16-color palette. Body temperature was remained at 37 °C with a temperature control system. In addition, all rats had free access to food and water. Rats in the administration group were injected with a 0.1 mL volume via a single intravenous dose of PD (30 mg/kg, dissolved in saline) at 10 min before surgery, and vehicle group was injected with solvent under the same conditions. The rats were tested for behavioral changes and scored 24 h after the operation, subsequently sacrificed and the entire brain was taken for subsequent experiments. All procedures were performed according to the US National Institutes of Health (NIH) Guide for the Care and Use of Laboratory. Animals published by the US National Academy of Sciences (http://oacu.od.nih.gov/regs/index.htm) and were approved by the Administration Committee of Experimental Animals in Jiangsu Province and the Ethics Committee of China Pharmaceutical University.
Measurement of Neurological Performance
At 24 h after tMCAO, the surviving rats were tested for behavioral changes and scored as described previously, with minor modifications (Longa et al. 1989). Neurological performance scores were assessed using a 5-point scale: 0, no observable neurologic deficits; (1) failure to extend the left forepaw (a mild focal neurologic deficit); (2) circling to the contralateral side (a moderate focal neurologic deficit); (3) falling to the left (a severe focal deficit); and (4) unable to walk spontaneously.
Measurement of Infarct Size
After assessing the neurological deficit, the rat brain was collected and placed on ice, then cut into coronal sections of 2 mm. Sections were soaked in 2% TTC (2,3,5-triphenyltetrazolium chloride) (Sigma-Aldrich) phosphate buffer for 20 min at 37 °C in the dark. The infarcted tissue was stained white, while the normal brain tissue was red. The percentage of cerebral infarction volume per rat according to the following formula: (total infarction area/total area of brain slice) × 100%. The infarct areas and the total brain areas were calculated by morphometric analysis with Image-pro Plus (Media cybernetics, MD, USA) (Wang et al. 2016).
Evaluation of Evans Blue Dye Extravasation
Evans Blue (2% in saline, 4 mL/kg; Sigma, St. Louis, MO, USA) was intravenously administered 24 h after tMCAO, and then 1 h later, rats were perfused with saline to remove intravascular dyes. The sample was homogenized in 50% trichloroacetic acid solution. The supernatant was obtained by centrifugation and diluted fourfold with ethanol. The fluorescence intensity was measured with a fluorescence detection system (Magellan V6.6; Safire 2, Tecan excitation 620 nm and emission 680 nm).
Rat Cerebral Microvessel Isolation
Rats in the sham, vehicle, and the PD administration groups were euthanized. The brains were rapidly removed and placed in a cold PBS solution. The right brain was shredded and placed in a tube containing PBS followed by homogenization. The homogenized brain tissue was centrifuged several times at 720×g to remove blood cells, and then the centrifuged homogenate tissue was spread on top of 16% dextran, followed by centrifugation at 4500×g for 20 min to collect the lowest layer (cerebral microvascular tissue). The upper homogenate tissue repeats this procedure to obtain more vascular precipitation. The obtained vascular deposits were stored in a − 80 °C refrigerator for subsequent Western blotting and RT-PCR experiments. In this section, we refer to the previously published literature (Wang et al. 2016) for specific experimental operations.
Statistical Analysis
All results are expressed as the mean ± SEM. Statistical analysis was performed using GraphPad Prism 5 software (San Diego, CA) with either unpaired two-tailed t-test or one-way ANOVA as appropriate. The statistical test used and n-numbers are indicated in each figure legend. The differences were considered significant if the p < 0.05.
Results
PD Up-Regulates the Expression of MALAT1 in rBMVEC and HUVEC Cells in Dose- and Time-Dependent Manner
Luciferase reporter gene assay is a common method for high throughput drug screening. Based on this method (Fig. 1a), we constructed a HUVEC cell line that stably expressed luciferase reporter gene driven by MALAT1 promoter (− 2100 ~ + 105) and used it to screen our in-house natural product library to seek for MALAT1 regulators. We found that PD could significantly up-regulate luciferase activity driven by MALAT1 promoter (Fig. 1b). MTT and LDH assays were used to detect the effect of PD on HUVEC and rBMVEC cell viability. The results showed that PD has no cytotoxicity in both cell cultures (Fig. S1A, B). In order to detect the effect of PD on the expression of endogenous MALAT1 in HUVEC and rBMVEC cells, we used the RT-PCR method to detect the changes of MALAT1 expression in both cells. The results showed that PD could up-regulate the expression of endogenous MALAT1 in the time- and dose-dependent manners in both endothelial cell cultures (Fig. 1c, d).

Polydatin, a novel MALAT1 up-regulator, time-dependently and dose-dependently increases MALAT1 expression in endothelial cells. a A schematic drawing for screening compounds that may regulate MALAT1 expression based on the MALAT1 promoter (− 2100 ~ + 105) luciferase reporter gene assay in HUVEC cells. b Polydatin (PD) time-dependently and dose-dependently increased luciferase activity of MALAT1 promoter-luciferase in HUVEC. Cells were incubated with PD (20 µM) for different times or with various concentrations of PD for 36 h. c PD time-dependently and dose-dependently increases the expression of MALAT1 in HUVEC Cells. Cells were incubated with various concentrations of PD for 24 h or with PD (20 µM) for different times. d PD time-dependently and dose-dependently increases the expression of MALAT1 in rBMVEC cells by RT-PCR. Cells were incubated with PD (20 µM) for different times or with various concentrations of PD for 24 h. Data are shown as the mean ± SEM (n = 3) after normalization to the control (DMSO). One-way ANOVA with Bonferroni post test. *p < 0.05, **p < 0.01, ***p < 0.001 versus control (CN)
PD, a New MALAT1 Regulator, Significantly Attenuates OGD-Induced Endothelial Cell Injury
We submitted HUVEC and rBMVEC cultures to OGD, mimicking in vivo ischemia conditions to verify whether PD could prevent endothelial cell injury induced by ischemic stroke (Fig. 2a). PD significantly enhanced the expression of MALAT1 and reduced cellular death in endothelial cells submitted to OGD (Figs. 2b, c, S2A, B). Furthermore, in order to verify whether the endothelial protection by PD is associated with up-regulation of MALAT1, GapmeR-MALAT1 was used to silence the expression of MALAT1 in endothelial cells (Fig. 2d). When MALAT1 was knockdown by GapmeR-MALAT1 in rBMVEC cells, the death of endothelial cells was increased and the endothelial protection of PD was significantly attenuated (Fig. 2e). Similar results are also shown in HUVEC cells (Fig. S2C, D). These data indicate that MALAT1 plays an important role for endothelial protection in ischemia and is involved in the PD-mediated endothelial protection.


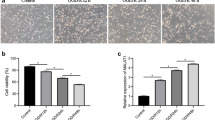
Polydatin protects against OGD-induced endothelial cell injury through regulating MALAT1 expression in rBMVEC cells. a A schematic diagram of PD inhibition of OGD-induced endothelial cell damage by up-regulating MALAT1 expression. The rBMVEC cells were incubated with PD (10, 20 µM) for 24 h, then cultured under OGD conditions for another 12 h. The MALAT1 expression was measured by RT-PCR (b), and the cell viability was determined by LDH assay (c). d The expression of MALAT1 was significantly inhibited by GapmeR-MALAT1 transfected into rBMVEC cells for 24 h. GapmeR-MALAT1-mediated MALAT1 silencing abolished the PD prevention of cell death induced by OGD (e). f, g MALAT1 knockdown also reversed the PD inhibition of mRNA expressions of pro-inflammatory factors (TNF-α, IL-6, COX-2) and attenuated the PD enhancement of mRNA expressions of BBB markers (Claudin-5, ZO-1, Occludin) and protein expressions of ZO-1 and Occludin in rBMVEC cells. h Immunofluorescence method determined that PD protected OGD-induced reduction of ZO-1 expression, but MALAT1 silencing reversed this effect. i PD significantly inhibited the apoptosis of rBMVEC cells induced by OGD for 12 h, determined by TUNEL assay, and this effect was reversed by MALAT1 silencing. Data are shown as the mean ± SEM (n = 3) after normalization to the control (CN). One-way ANOVA with Bonferroni post test. ***p < 0.001 versus CN group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus GapmeR-NC group; $p < 0.05, $$p < 0.01, $$$p < 0.001 versus GapmeR-NC + PD group
To further study the effects of PD on endothelial inflammation and vascular integrity under OGD conditions, we examined the expression changes of endothelial pro-inflammatory cytokines and BBB markers. PD significantly inhibited the OGD-induced gene expression of endothelial pro-inflammatory factors (TNF-α, IL-6, COX-2) and enhanced the gene expression of BBB markers (Claudin-5, Occludin, ZO-1) in rBMVEC cells (Fig. 2f). PD treatment also enhanced the protein expressions of Occludin and ZO-1 (Fig. 2g). In addition, the knockdown of MALAT1 mediated by GapmeR-MALAT1 reversed these effects of PD in endothelial cells (Fig. 2f, g).
The immunostaining data also showed that PD had a significant inhibitory effect on OGD-induced reduction of the ZO-1 protein expression, whereas knockdown of MALAT1 by GapmeR-MALAT1 reversed this effect of PD (Fig. 2h).
Apoptosis of cerebral vascular endothelial cells induced by ischemia is a common phenomenon in the early stage of ischemic stroke. In order to evaluate whether PD can protect endothelial cells from apoptosis caused by OGD, we detected the apoptosis rate of endothelial cells by TUNEL assay and found that PD reduced the apoptosis rate of rBMVEC cells induced by OGD from 62.75 to 24%, an effect reversed by GapmeR-MALAT1 with MALAT1 knockdown (Fig. 2i).
The above results indicated that the MALAT1 up-regulator PD could prevent OGD-induced endothelial cell injury by inhibiting the endothelial inflammation and enhancing the expression of BBB markers in rBMVEC cells, which was mediated through enhanced MALAT1 expression.
C/EBPβ is Responsible for the MALAT1 Up-Regulation by PD
In order to further investigate which transcription factor mediates the up-regulation of MALAT1 expression by PD, we constructed luciferase reporter gene systems with three truncated MALAT1 promoter fragments to estimate the binding location of possible transcription factors acting on MALAT1 promoters (Fig. 3a). The results showed that PD significantly up-regulated the activity of the reporter gene driven by the − 195 ~ + 105 promoter region of MALAT1 (Fig. 3b), indicating that the transcription factor may act on the − 195 ~ + 105 promoter region of MALAT1 to regulate MALAT1 expression by PD. PROMO database analysis shows that one of the transcription factors, C/EBPβ may bind to the − 195 ~ + 105 promoter region of MALAT1 with higher possibility. To verify whether PD regulates the expression of MALAT1 by C/EBPβ, we knockdown and overexpressed C/EBPβ in the HEK-293T cells (Fig. 3c) to test whether C/EBPβ has an effect on the reporter gene activity of MALAT1. The results showed that knockdown of C/EBPβ significantly suppressed the reporter gene activity driven by the − 195 ~ + 105 promoter of MALAT1, whereas overexpression of C/EBPβ enhanced this reporter gene activity (Fig. 3d). Furthermore, changes in the expression of C/EBPβ significantly affected this reporter gene activity driven by PD (Fig. 3e). These results indicate that C/EBPβ is an important transcription factor for PD-mediated MALAT1 expression.

C/EBPβ is responsible for the MALAT1 up-regulation by polydatin. a A schematic diagram of luciferase reporter gene constructs containing different lengths of the MALAT1 promoter fragment. b Polydatin (PD) significantly increased the activity of luciferase reporter genes driven by three truncated MALAT1 promoter fragments in HEK-293T cells. c The expression of C/EBPβ protein was knockdown by the C/EBPβ siRNA transfection (siRNA-C/EBPβ) and was overexpressed by the pcDNA3.1-C/EBPβ plasmid transfection for 24 h in HEK-293T cells. d The C/EBPβ silencing significantly decreased the activity of the reporter gene driven by MALAT1 promoter (− 195 ~ + 105), while the overexpression of C/EBPβ remarkably increased its activity. e The C/EBPβ silencing significantly reversed the PD enhancement of the reporter gene driven by the MALAT1 promoter (− 195 ~ + 105), while the overexpression of C/EBPβ remarkably increased the PD effect. f The wild type and mutant base sequences of C/EBPβ binding site in MALAT1 promoter (− 195 ~ + 105) region. g C/EBPβ knockdown or overexpression had no effect on the activity of the reporter gene driven by the MALAT1 promoter (− 195 ~ + 105) region containing mutant bases of possible C/EBPβ binding site (Mut). h PD had no effect on the activity of the Mut. i, j PD time-dependently and dose-dependently increased the mRNA and protein expressions of C/EBPβ in rBMVEC cells. Data are shown as the mean ± SEM (n = 3) after normalization to the control. One-way ANOVA with Bonferroni post test. *p < 0.05, **p < 0.01, ***p < 0.001 versus control (CN, 0 µM or 0 h)
To further prove that C/EBPβ may directly regulate the expression of MALAT1, we mutated the possible binding base sequence of C/EBPβ in the MALAT1 promoter region to confirm the interaction between C/EBPβ and MALAT1 promoter (Fig. 3f). The results showed that the change of C/EBPβ expression could not affect the activity of the reporter gene driven by the MALAT1 promoter containing mutant possible binding sequence (Fig. 3g), and PD could not regulate the activity of this mutant reporter gene (Fig. 3h), suggesting that C/EBPβ may bind to the − 195 ~ + 105 promoter region of MALAT1 to mediate the PD regulation of MALAT1 expression. In primary rBMVEC and HUVEC cells, PD treatment increased the expression of C/EBPβ gene and protein in a time- and dose-dependent manner (Figs. 3i, j, S2A–D), indicating that C/EBPβ is a key transcription factor that regulates the expression of MALAT1 by PD in endothelial cells.
PD Effects are Mediated by the CREB-PGC-1α-PPARγ Pathway as Downstream of MALAT1 in Endothelial Cells
We knockdown and overexpressed MALAT1 in HEK-293T cells in order to determine downstream signaling pathways of MALAT1 after PD treatment (Fig. 4a). Peroxisome proliferator-activated receptor γ (PPARγ) is a ligand-activated nuclear receptor, playing an active role in the treatment of ischemic stroke (Liu and Wang 2017). Pioglitazone, the small molecular agonist of PPARγ could significantly reduce the cerebral infarction volume and the release of inflammatory factors in ischemic rats (Medhi et al. 2010), and significantly reduced the risk of ischemic stroke in clinical trials (Kernan et al. 2016). We determined whether MALAT1 influences PPARγ activity in HEK-293T cells, using the PPRE-driven reporter gene assay system to detect PPARγ activity (Fig. 4b). We found that the knockdown of MALAT1 by siRNA significantly inhibited PPARγ activity. Conversely the overexpression of MALAT1 by pEGFP-C1-MALAT1 enhanced PPARγ activity (Fig. 4c).



The CREB-PGC-1α-PPARγ pathway as downstream of MALAT1 is involved in the PD effects. a RT-PCR analysis of MALAT1 expression mediated by siRNA-MALAT1 and pEGFP-C1-MALAT1 in HEK-293T cells. b A schematic diagram of the system of PPRE-driven luciferase reporter gene expression for PPARγ activity. c MALAT1 silencing reduced the PPARγ activity, while MALAT1 overexpression increased the PPARγ activity in HEK-293T cells. d, e RT-PCR analysis of the gene expression of PPARγ, CD36, ABCG1, and PGC-1α. f The PGC-1α inhibitor SR-18292 blocked the MALAT1 overexpression-enhanced PPARγ activity. g Polydatin (PD) time-dependently and dose-dependently increased the PPARγ activity. h MALAT1 silencing attenuated PD-induced PPARγ activity, while MALAT1 overexpression enhanced the PD effect. i The PGC-1α inhibitor SR-18292totally inhibited PD-induced PPARγ activity. j C/EBPβ silencing attenuated PD-induced PPARγ activity, while C/EBPβ overexpression enhanced PD effect. k A schematic diagram of the system of CRE-driven luciferase reporter gene expression for CREB activity. l MALAT1 silencing reduced the CREB transcriptional activity, while MALAT1 overexpression increased the CREB activity in HEK-293T cells. m, n The CREB inhibitor 666-15 (1 µM) attenuated the MALAT1 overexpression-induced the PGC-1α gene expression and the PPARγ activity. o PD time-dependently and dose-dependently increased the CREB transcriptional activity in HEK-293T cells. p, q The CREB inhibitor 666-15 (1 µM) totally blocked the PD-induced PGC-1α gene expression in HEK-293T and rBMVEC cells. r The CREB inhibitor 666-15 (1 µM) totally blocked the PD-induced PPARγ activity in HEK-293T cells. s C/EBPβ silencing reduced the CREB transcriptional activity, while C/EBPβ overexpression increased the CREB activity in HEK-293T cells. t A schematic diagram of the upstream signaling pathway of PPARγ activity induced by PD involving C/EBPβ, MALAT1, CREB and PGC-1α proteins. u, v PD time-dependently and dose-dependently increased the phosphorylated CREB (p-CREB) and PGC-1α protein levels but not PPARγ in rBMVEC cells. w The MALAT1 silencing by GapmeR-MALAT1 in rBMVEC cells reduced the protein expression levels of the phosphorylated CREB and PGC-1α but not PPARγ. x The MALAT1 silencing by GapmeR-MALAT1 attenuated the PD effects on the enhancement of protein expression levels of the phosphorylated CREB and PGC-1α in rBMVEC cells under OGD condition. Data are shown as the mean ± SEM (n = 3) after normalization to the control (DMSO, CN). One-way ANOVA with Bonferroni post test. *p < 0.05, **p < 0.01, ***p < 0.001 versus CN or siRNA-NC group; #p < 0.05, ###p < 0.001 versus GapmeR-NC group; $$$p < 0.001 versus GapmeR-NC + PD group
We determined gene expressions of PPARγ and that of its downstream target genes, CD36 and ABCG1. The results show that MALAT1 knockdown reduced ABCG1 gene expression but the gene expressions of CD36 and ABCG1 were increased by overexpression of MALAT1. Since modulation of MALAT1 did not affect PPARγ gene expression (Fig. 4d), our results suggest that MALAT1 may regulate PPARγ activity through its co-factors.
We examined the effect of MALAT1 on the expression of PGC-1α, an important coactivator regulating PPARγ activity. We found that MALAT1 knockdown inhibited PGC-1α gene expression and conversely, the overexpression of MALAT1 enhanced PGC-1α gene expression (Fig. 4e). In addition, the PPARγ activity increased by overexpression of MALAT1 was significantly reversed by the PGC-1α inhibitor SR-18292 (Fig. 4f) (Sharabi et al. 2017). This suggests that MALAT1 may enhance the expression of PGC-1α to increase PPARγ activity.
PD treatment time and dose dependently increased PPARγ activity in endothelial cells (Fig. 4g). This effect was reversed by MALAT1 knockdown and conversely enhanced by MALAT1 overexpression (Fig. 4h). The role of PD in enhancing PPARγ activity can be blocked by the PGC-1α inhibitor SR-18292 (Fig. 4i). Taken together, our results suggest that PD may increase PPARγ activity through the stimulation of the MALAT1/PGC-1α pathway.
In addition, the MALAT1 upstream transcription factor C/EBPβ also increased PPARγ activity (Fig. 4j) and was also involved in the enhanced PPARγ activity as a result of PD treatment (Fig. 4j).
CREB is a transcription factor that has been reported to regulate the expression of PGC-1α (Singh et al. 2015). MALAT1 could bind to phosphorylated CREB (p-CREB) to maintain the CREB activity (Yao et al. 2016). We proposed that MALAT1 may regulate PGC-1α expression through CREB activation. To test this hypothesis, we used a reporter gene driven by CRE to detect the transcriptional activity of CREB (Xue et al. 2011) (Fig. 4k). We found that the knockdown of MALAT1 significantly reduced the transcriptional activity of CREB, while the overexpression of MALAT1 enhanced CREB activity (Fig. 4l). In addition, 666-15, an inhibitor of CREB, can significantly reverse the upregulation of PGC-1α and PPARγ activity induced by overexpression of MALAT1, but not the expression of PPARγ (Fig. 4m, n). This suggests that MALAT1 promotes PGC-1α expression and PPARγ activity by increasing CREB transcriptional activity.
In HEK-293T cells, PD enhanced CREB transcriptional activity in a dose- and time-dependent manner (Fig. 4o). PD-induced enhancement of PPARγ activity and PGC-1α expression can be blocked by the CREB inhibitor 666-15 in HEK-293T and rBMVEC cells (Fig. 4p–r), suggesting that PD may increase the PGC-1α expression and PPARγ activity through modulating CREB transcriptional activity.
C/EBPβ, as a MALAT1 upstream transcription factor, also enhanced the activity of CREB (Fig. 4s), further supporting that PD effects involved the C/EBPβ-MALAT1-CREB-PGC-1α-PPARγ pathway (Fig. 4t).
In rBMVEC cells, PD increased the expressions of phosphorylated CREB and PGC-1α protein in a dose- and time-dependent manner, but PD had no effect on the protein expression of PPARγ (Fig. 4u, v).
In order to further confirm CREB-PGC-1α as downstream signaling pathway of MALAT1, we detected the expressions of phosphorylated CREB, PGC-1α, and PPARγ in rBMVEC cells after the MALAT1 knockdown by GapmeR-MALAT1. MALAT1 knockdown significantly down-regulated the protein expressions of phosphorylated CREB and PGC-1α, but had no effect on the protein expression of PPARγ (Fig. 4w). When rBMVEC cells pretreated with PD were submitted to OGD, PD significantly enhanced the protein expressions of phosphorylated CREB and PGC-1α, but had no effect on the PPARγ protein expression, while knockdown of MALAT1 reversed these effects (Fig. 4x).
PD Protects Against OGD-Induced Cerebrovascular Endothelial Cell Injury Through the Regulation of the PGC-1α/PPARγ Pathway
In order to confirm whether PGC-1α-PPARγ pathway plays an important role for PD protection of OGD-induced endothelial injury, we used two PPARγ antagonists, GW9662 (Lisa et al. 2002) and T0070907 (Lee et al. 2002) in rBMVEC cells. The effect of PD on protection against endothelial cell death induced by OGD was significantly reversed by GW9662 and T0070907 (Fig. 5a). Furthermore, the PD inhibition of OGD-induced endothelial inflammatory cytokines (the TNF-α and IL-6 gene expression) and BBB markers (Claudin-5, Occludin and ZO-1 gene expression, and the Occludin and ZO-1 proteins) was also reversed by GW9662 and T0070907 (Fig. 5b–d). In addition, the immunostaining data show that GW9662 also reversed the PD enhancement of ZO-1 protein expression in rBMVEC cells submitted to OGD (Fig. 5e). Additionally, the PD inhibition of OGD-induced endothelial cell apoptosis was significantly attenuated by GW9662 (Fig. 5f).


Polydatin protects against OGD-induced endothelial cell injury through the PGC-1α/PPARγ pathway in rBMVEC cells. a–c The specific PPARγ inhibitors GW9662 (GW) or T0070907 (T007) reversed the PD reduction of OGD-induced cell death and inflammatory cytokines (TNF-α, IL-6) gene expression, and also reversed the PD enhancement of the gene expression of tight junction protein (Occludin, Claudin-5, ZO-1). d, e GW or T007 remarkably attenuated the PD enhancement of the protein expression of Occludin and ZO-1 by Western blotting or immunostaining. f TUNEL staining showed that GW reversed the PD inhibition of the OGD-caused endothelial cell apoptosis in rBMVEC cells. g The knockdown of PGC-1α protein expression in rBMVEC cells was mediated by PGC-1α siRNA (siRNA-PGC-1α). LDH assay show that the PGC-1α knockdown significantly reversed the PD inhibition of OGD-induced cell death (h) and the inflammatory cytokines (TNF-1α and IL-6) gene expression (i), and also reversed the PD enhancement of the gene expression of tight junction protein (Occludin, Claudin-5, ZO-1) in rBMVEC cells (j). k The PGC-1α silencing totally attenuated the PD enhancement of the protein expression of ZO-1 and Occludin in rBMVEC cells under OGD condition. Data are shown as the mean ± SEM (n = 3) after normalization to the control (DMSO). One-way ANOVA followed by Bonferroni post-hoc test (a–f, h–k) and unpaired two-tailed T test (G). *p < 0.05, **p < 0.01, ***p < 0.001 versus control (CN); #p < 0.05, ##p < 0.01, ###p < 0.001 versus OGD or OGD + siRNA-NC; $p < 0.05, $$p < 0.01, $$$p < 0.001 versus PD + OGD or siRNA-NC + PD + OGD
Similarly, PGC-1α gene silencing by PGC-1α siRNA (Fig. 5g) reversed the PD protection of OGD-induced endothelial cell injury (Fig. 5h) and the PD inhibition of OGD-induced endothelial inflammatory cytokines (TNF-α and IL-6 gene expression) and BBB markers (Claudin-5, Occludin and ZO-1 gene expression, and the Occludin and ZO-1 proteins) expressions (Fig. 5i–k). These results indicate that PD protected cerebrovascular endothelial cell injury against OGD mainly through the PGC-1α/PPARγ pathway regulation.
PD Reduces Cerebral Infarct Volume, Protects Cerebrovascular Endothelial Cells and BBB Integrity in Rats Subject to tMCAO
To confirm the beneficial effects of PD on ischemia in vivo, rats were subjected to tMCAO and treated by PD. The experiment design is shown in Fig. 6a. The results show that PD administration significantly reduced the infarct volume and improved neurological deficit in ischemic rats (Fig. 6b). In order to evaluate the protective effect of PD on BBB integrity, Evans blue staining was used to evaluate the permeability of BBB. The results show that the permeability of BBB was significantly increased in the vehicle group at 24 h post ischemia, while PD administration remarkably improved BBB integrity, indicating that PD could protect the BBB integrity in ischemic rats (Fig. 6c).

Polydatin ameliorates brain ischemic injury and protects the blood–brain barrier integrity in rats subject to tMCAO. a Experimental design of the tMCAO model. Rats were intravenously injected with polydatin (PD, 30 mg/kg) 10 min before ischemia. The rat brains were removed to perform histological analysis 24 h after reperfusion. b PD reduced cerebral infarct volume as assessed by TTC staining and improved sensorimotor recovery as evaluated by the Longa test. c PD significantly reduced ischemia-induced blood–brain barrier (BBB) disruption, which was tested by Evans blue extravasation assay. d–f The brain microvessels from rats with ischemia were isolated to test the expressions of MALAT1, C/EBPβ, PGC-1α, and PPARγ. RT-PCR and Western blotting results showed that PD markedly enhanced the MALAT1 expression and the mRNA and protein expression of C/EBPβ and PGC-1α but not PPARγ. PD also increased the protein expression of phosphorylated CREB (f). g, h Ischemia reduced the gene and protein expressions of tight junction proteins (Occludin, Claudin-5, ZO-1), while PD administration significantly enhanced the expressions of tight junction proteins (Occludin, Claudin-5, ZO-1). i, j PD administration reduced the gene expression of inflammatory factors (TNF-α, IL-1β, IL-6, COX-2, ICAM-1, VCAM-1, MCP-1) and the protein expression of COX-2, ICAM-1 and VCAM-1 in brain microvessels from rats subjected to tMCAO. Data are shown as the mean ± SEM (n = 8–12) after normalization to the control (Sham). One-way ANOVA with Bonferroni post test. *p < 0.05, **p < 0.01, ***p < 0.001 versus Sham group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus vehicle group
In order to study the molecular mechanisms for above PD effects in vivo, we extracted cerebrovascular tissue of rats from the sham, vehicle and PD-treated groups and determined gene and protein expression. The results show that PD significantly enhanced MALAT1 expression (Fig. 6d), and markedly increased the gene and protein expression of C/EBPβ and PGC-1α, but not PPARγ (Fig. 6e, f). PD also enhanced the gene and protein expression levels of the BBB markers, Claudin-5, Occludin and ZO-1 (Fig. 6g, h). In addition, PD administration significantly inhibited ischemia-induced the gene expression levels of inflammatory factors, TNF-α, IL-1β, IL-6, COX-2, ICAM-1, VCAM-1 and MCP-1, and also reduced ischemia-induced the protein expression levels of COX-2, ICAM-1 and VCAM-1 (Fig. 6i, j).
Discussion
LncRNAs have been reported to regulate physiological and pathological responses (Valluri et al. 2017; Heward and Lindsay 2014). The lncRNA MALAT1 contributes to regulate endothelial cell function, and was proposed to be involved in the mechanisms of vascular endothelial cell damage caused by ischemic stroke (Zhang et al. 2016, 2017). It was proposed that MALAT1 could protect endothelial cells against inflammatory response and apoptosis (Puthanveetil et al. 2015). Thus, promoting the expression of MALAT1 may ameliorate brain damage caused by ischemia, and a small molecular compound that up-regulates the expression of endogenous MALAT1 may be a promising therapeutic strategy for ischemic stroke.
In this study, we established a reporter gene system driven by − 2100 ~ + 105 promoter region of MALAT1 in HUVEC cells. Our in-house compound library containing 1200 natural products were screened and a component of Polygonum cuspidatum, PD, was shown to remarkably up-regulate the expression of MALAT1 in HUVEC and rBMVEC cells in a dose- and time-dependent manner. In addition, our in vivo results indicate that PD could enhance MALAT1 expression in cerebrovascular endothelial cells to exhibit beneficial effects in rats with ischemia. We studied the molecular mechanisms involved in the reported PD neuroprotection against ischemic stroke (Gao et al. 2016; Cheng et al. 2006).
In the present study, we demonstrated that PD reduced cerebral infarct volume and improved blood–brain barrier damage in rats subject to tMCAO. Our data indicate that the neuroprotection of PD may be mediated by maintaining the BBB integrity of rats with ischemia. Cerebrovascular endothelial cells, the main components of BBB structure, are first damaged after ischemia and produce variety of pro-inflammatory factors, which further exacerbate ischemic brain damage. Our results showed that PD could improve cell viability loss and inhibit apoptosis in rBMVEC cells after OGD injury, and significantly inhibited the expression of pro-inflammatory factors (TNF-α, IL-6, COX-2) and the degradation of tight junction proteins (Claudin-5, Occludin, ZO-1). We found that the protective effect of PD against endothelial injury was reversed by MALAT1 knockdown, indicating that PD protection against ischemic stroke involved the regulation of MALAT1 expression.
Although the crucial role of MALAT1 in regulating endothelial cell function has been well recognized, the upstream factors mediating MALAT1 expression are still unclear. C/EBPβ, a transcription factor, is involved in the control of cellular growth, proliferation and pro-inflammatory process and participates in pathological responses in endothelial cells (Manea et al. 2013; Chuang et al. 2014). We discovered that C/EBPβ may bind to the − 195 ~ + 105 promoter region of MALAT1 to regulate MALAT1 expression by the experiment of luciferase reporter gene driven by three truncated human MALAT1 promoter segments. And the C/EBPβ binding base sequence can be found in the MALAT1 − 195 ~ + 105 promoter region of human and rat origins, so we think that C/EBPβ is a key transcription factor regulating the expression of MALAT1. We also found that PD increased C/EBPβ gene expression and the effect of PD on increase in MALAT1 expression was abolished by C/EBPβ gene silencing. PD up-regulation of C/EBPβ gene expression was also observed in cerebrovascular endothelial cells isolated from the brain of rats with ischemia. These results indicate that C/EBPβ may be a key transcription factor responsible for regulating MALAT1 expression by PD.
Our study further elucidated the downstream mechanisms of PD-induced MALAT1 up-regulation in cerebrovascular endothelial cells. PPARγ has been proved to exert a vital role in the treatment for endothelial dysfunction and ischemic stroke (Xu et al. 2017) and C/EBPβ could regulate the PPARγ activity (Wu et al. 2018). This indicates that MALAT1 may be an important mediator for C/EBPβ-regulated PPARγ activity. In this study, we uncovered that MALAT1 significantly increased PPARγ activity, without influencing its protein expression. A recent study has shown that MALAT1 binding to CREB maintains CREB phosphorylation by inhibiting PP2A-mediated dephosphorylation, which leads to continuous CREB signaling activation (Yao et al. 2016). Moreover, CREB is an important transcription factor regulating PGC-1α expression (Singh et al. 2015), a transcriptional coactivator interacting with PPARγ. This suggests that the CREB-PGC-1α-PPARγ pathway may act downstream of MALAT1.
In the present study, we confirmed that MALAT1 regulated CREB phosphorylation to maintain its transcriptional activity. Furthermore, our data showed that the effect of MALAT1 on regulating PGC-1α was attenuated by the inhibition of CREB activity, indicating that MALAT1 possibly binds with CREB to maintain the phosphorylation of CREB to further activate the CREB signaling pathway, thus promoting PGC-1α expression. We further confirmed that PD could significantly up-regulate CREB phosphorylation, the expression of PGC-1α and the activity of PPARγ, suggesting that PD might activate MALAT1/CREB/PGC-1α/PPARγ signaling pathway to protect endothelial cells against ischemia.
Based on the understanding of mechanism underlying PD-regulated MALAT1 expression, we further proved that the activation of PGC-1α/PPARγ signaling pathway is essential for the protective effect of PD against ischemia-induced endothelial injury. It has been reported that PPARγ could directly bind to the subunit P65/P50 of NF-κB, forming a transcriptional repression complex and reducing the binding activity of NF-κB to DNA (Su et al. 2000), thus inhibiting its transcriptional activity and suppressing the production of downstream inflammatory factors. Our data showed that the effects of PD on inhibition of OGD-induced endothelial cell injury and enhancement of the tight junction proteins expressions were reversed by PPARγ inhibition or PGC-1α gene silencing. Altogether, our results demonstrate that PD plays a protective role in ischemia-induced endothelial injury by activating the PGC-1α/PPARγ pathway via MALAT1.
One limitation of our present study is that the mechanism of PD regulating C/EBPβ expression in rBMVEC cells has not been clarified. Microscale thermophoresis (MST) or surface plasmon resonance (SPR) technology should be used to further detect whether PD directly binds to C/EBPβ protein to regulate its transcriptional activity or only regulates its expression. Previous studies showed that PD enhanced Nrf2-ARE pathway by activating Sirt1 to exert anti-oxidative activities. MALAT1 has been particularly shown to activate Nrf2 signaling and regulate the deacetylation activity of Sirt1 (Ye et al. 2017; Huang et al. 2015). Thus, whether Sirt1- or Nrf2-mediated anti-oxidative pathway participates in the protective role of PD in endothelial injury caused by ischemia would be further studied.
Local brain inflammation is one of the pathological markers of ischemic stroke damage. Post-ischemic inflammation is mainly caused by activation of microglia and macrophages and peripheral leukocytes into brain parenchyma. A few minutes after ischemic stroke, microglia are activated to produce a series of inflammatory mediators that migrate from the surrounding tissues of the lesion to the lesion. At the same time, endothelial cells activate and express a variety of adhesion molecules, leading to destruction of the blood–brain barrier and leukocyte adhesion and exudation (Wang et al. 2007; Doyle et al. 2008). Leukocytes exuded in the lesion together with activated multiple glial cells release various harmful molecules such as protease, glutamate, cytokines, chemokines, free radicals, prostaglandins, and nitric oxide. These inflammatory molecules can aggravate the adhesion and exudation of inflammatory cells and cause a vicious cycle of inflammation, and can damage cells, blood vessels and extracellular matrix, resulting in blood–brain barrier destruction, brain edema and cell death (Zoppo 2009). In our study, we only studied that PD improves ischemic stroke which is associated with inhibition of endothelial cell damage. As for whether PD affects the inflammatory response of immune cells, we are not clear. From the published literature, PD can indeed inhibit the inflammatory response of immune cells (Lou et al. 2015; Zhao et al. 2017), but whether this effect is involved in the PD ameliorating ischemic stroke has not been determined. We will address this issue in the subsequent studies.
We selected male οSD rats for our in vivo experiments with the tMACO model to avoid the effect of reproductive hormones during the rat estrous cycle. In future experiments, we will continue to study the effects of PD on brain damage in female tMCAO rats. In addition, we only studied the effects of PD treatment on brain injury in tMCAO rats 10 min before surgery. In the subsequent experiments, we will determine the therapeutic window of PD amelioration of brain damage caused by ischemia. In behavioral tests, we used Longa test to assess the brain protection of PD in the acute phase of ischemic stroke, and the results suggest that PD can improve behavioral deficits. However, in the subsequent recovery of long-term behavior, we do not know the effect of PD. In the acute phase of cerebral ischemia, BBB damage leads to impaired brain parenchyma and thus loss of behavior. Longa test can be used to evaluate the efficacy of the compound from a behavioral point of view (Longa et al. 1989; Wang et al. 2016). But in the late stage of cerebral ischemia, damaged tissues are repaired and behavioral changes are slowly recovered. In order to evaluate the efficacy of compounds comprehensively, it is necessary to examine their effects on long-term behavioral recovery. In the published literature (Akinrinmade et al. 2017; Shi et al. 2017; Belayev et al. 2018), long-term behavioral recovery has been regarded as an important indicator for evaluating the protective effects of compounds on ischemic stroke. So in the follow-up study, we will determine the effect of PD on long-term behavioral recovery after ischemic stroke.
Conclusion
In summary, our study, for the first time, discovered that PD, a novel MALAT1 regulator, protected against ischemia-induced endothelial injury in vitro and in vivo through modulating C/EBPβ/MALAT1/CREB/PGC-1α/PPARγ pathway (Fig. 7). These findings may promote the study of MALAT1 up-regulators such as PD to be further developed as therapeutic agents for ischemic stroke.

Schematic diagram for the proposed mechanisms of beneficial effects of polydatin on ischemic stroke. Polydatin promotes the expression of MALAT1 by up-regulating C/EBPβ transcription factor, and then MALAT1 may bind to the phosphorylated CREB to maintain its phosphorylation, resulting in promoting the expression of PGC-1α. PGC-1α acts as a transcriptional coactivator to enhance the activity of PPARγ, resulting in expression of PPARγ target genes, which protect against the cerebrovascular endothelial cells injury and further blood–brain barrier (BBB) damage caused by ischemia. Therefore, PD, as a MALAT1 regulator with beneficial effects on brain ischemic stroke, may inhibit cerebrovascular endothelial cell damage through C/EBPβ-MALAT1-CREB-PGC-1α-PPARγ signaling pathway, thereby maintaining the BBB integrity and reducing brain damage caused by ischemia
Abbreviations
- ABCG1:
-
ATP-binding cassette sub-family G member 1
- BBB:
-
Blood–brain barrier
- CD36:
-
Cluster of differentiation 36
- C/EBPβ:
-
CCAAT/enhancer-binding proteinβ
- COX-2:
-
Cyclooxygenase-2
- CREB:
-
cAMP response element binding
- DMEM:
-
Dulbecco’s modified eagle’s medium
- DMSO:
-
Dimethyl sulfoxide
- FBS:
-
Fetal bovine serum
- HUVEC:
-
Human umbilical vein endothelial cells
- IL-6:
-
Interleukin-6
- LDH:
-
Lactate dehydrogenase
- LncRNA:
-
Long non-coding RNA
- MALAT1:
-
Metastasis-associated lung adenocarcinoma transcript 1
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OGD:
-
Oxygen and glucose deprivation
- PBS:
-
Phosphate-buffered saline
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma co-activator 1α
- PPARγ:
-
Peroxisome proliferative activated receptorγ
- rBMVEC:
-
Rat brain microvascular endothelial cell
- SD rats:
-
Sprague–Dawley rats
- tMCAO:
-
Transient middle cerebral artery occlusion
- TNF-α:
-
Tumor necrosis factor-α
- TTC:
-
2,3,5-Triphenyltetrazolium chloride
- ZO-1:
-
Zonula occludens-1
References
Akinrinmade O, Omoruyi S, Dietrich D, Ekpo O (2017) Long-term consumption of fermented rooibos herbal tea offers neuroprotection against ischemic brain injury in rats. Acta Neurobiol Exp 77(1):94–105
Belayev L, Hong SH, Menghani H, Marcell SJ, Obenaus A, Freitas RS, Khoutorova L, Balaszczuk V, Jun B, Oriá RB, Bazan NG (2018) Docosanoids promote neurogenesis and angiogenesis, blood-brain barrier integrity, penumbra protection, and neurobehavioral recovery after experimental ischemic stroke. Mol Neurobiol 55(8):7090-7106
Chang S, Ruan WC, Xu YZ, Wang YJ, Pang J, Zhang LY et al (2017) The natural product 4,10-aromadendranediol induces neuritogenesis in neuronal cells in vitro through activation of the ERK pathway. Acta Pharmacol Sin 38(1):29–40
Cheng Y, Zhang HT, Sun L, Guo S, Ouyang S, Zhang Y et al (2006) Involvement of cell adhesion molecules in polydatin protection of brain tissues from ischemia-reperfusion injury. Brain Res 1110(1):193–200
Chuang YF, Yang HY, Ko TL, Hsu YF, Sheu JR, Ou G et al (2014) Valproic acid suppresses lipopolysaccharide-induced cyclooxygenase-2 expression via MKP-1 in murine brain microvascular endothelial cells. Biochem Pharmacol 88(3):372–383
del Zoppo GJ (2006) Stroke and neurovascular protection. N Engl J Med 354(6):553–555
del Zoppo GJ (2009) Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 158(3):972–982
Doyle KP, Simon RP, Stenzelpoore MP (2008) Mechanisms of ischemic brain damage. Neuropharmacology 55(3):310–318
Fisher M (2008) Injuries to the vascular endothelium: vascular wall and endothelial dysfunction. Rev Neurol Dis 5(Suppl 1):S4–S11
Gao Y, Chen T, Lei X, Li Y, Dai X, Cao Y et al (2016) Neuroprotective effects of polydatin against mitochondrial-dependent apoptosis in the rat cerebral cortex following ischemia/reperfusion injury. Mol Med Rep 14(6):5481–5488
Guo W, Liu W, Chen Z, Gu Y, Peng S, Shen L et al (2017) Tyrosine phosphatase SHP2 negatively regulates NLRP3 inflammasome activation via ANT1-dependent mitochondrial homeostasis. Nat Commun 8(1):2168
Hawkins BT, Davis TP (2005) The blood–brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57(2):173–185
Heward JA, Lindsay MA (2014) Long non-coding RNAs in the regulation of the immune response. Trends Immunol 35(9):408–419
Huang J, Upadhyay UM, Tamargo RJ (2006) Inflammation in stroke and focal cerebral ischemia. Surg Neurol 66(3):232–245
Huang K, Chen C, Hao J, Huang J, Wang S, Liu P et al (2015) Polydatin promotes Nrf2-ARE anti-oxidative pathway through activating Sirt1 to resist AGEs-induced upregulation of fibronetin and transforming growth factor-β1 in rat glomerular messangial cells. Mol Cell Endocrinol 399:178–189
Jinmo K, Miyeon K, Kanghyun L, Sangkwan M, Jamakattel-Pandit N, Hoyoung C et al (2010) Key compound groups for the neuroprotective effect of roots of Polygonum cuspidatum on transient middle cerebral artery occlusion in Sprague-Dawley rats. Nat Prod Res 24(13):1214–1226
Kenneth J, Thomas D (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2–∆∆CT method. Methods A 25(4):402–408
Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M et al (2016) Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med 64(1):260
Lee G, Elwood F, Mcnally J, Weiszmann J, Lindstrom M, Amaral K et al (2002) T0070907, a selective ligand for peroxisome proliferator-activated receptor γ, functions as an antagonist of biochemical and cellular activities. J Biol Chem 277(22):19649–19657
Lisa ML, Derek JP, Randy KB, Jeff EC, Jon LC, Thomas GC et al (2002) Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry 41(21):6640–6650
Liu J, Wang LN (2017) Peroxisome proliferator-activated receptor gamma agonists for preventing recurrent stroke and other vascular events in people with stroke or transient ischaemic attack. Cochrane Database Syst Rev 12:CD010693
Liu JY, Yao J, Li XM, Song YC, Wang XQ, Li YJ et al (2014) Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis 5:e1506
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20(1):84–91
Lou T, Jiang W, Xu D, Chen T, Fu Y (2015) Inhibitory effects of polydatin on lipopolysaccharide-stimulated RAW 264.7 cells. Inflammation 38(3):1213–1220
Manea SA, Todirita A, Manea A (2013) High glucose-induced increased expression of endothelin-1 in human endothelial cells is mediated by activated CCAAT/enhancer-binding proteins. PLoS ONE 8(12):e84170
Medhi B, Aggarwal R, Chakrabarti A (2010) Neuroprotective effect of pioglitazone on acute phase changes induced by partial global cerebral ischemia in mice. Indian J Exp Biol 48(8):793–799
Michalik KM, You X, Manavski Y, Doddaballapur A, Zörnig M, Braun T et al (2014) Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res 114(9):1389–1397
Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S (2015) Long non-coding RNA malat1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med 19(6):1418–1425
Qureshi IA, Mehler MF (2012) Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci 13(8):528–541
Ruan W, Zhao F, Zhao S, Zhang L, Shi L, Pang T (2018) Knockdown of long noncoding RNA MEG3 impairs VEGF-stimulated endothelial sprouting angiogenesis via, modulating VEGFR2 expression in human umbilical vein endothelial cells. Gene 649:32–39
Ruetzler CA, Furuya K, Takeda H, Hallenbeck JM (2001) Brain vessels normally undergo cyclic activation and inactivation: evidence from tumor necrosis factor-alpha, heme oxygenase-1, and manganese superoxide dismutase immunostaining of vessels and perivascular brain cells. J Cereb Blood Flow Metab 21(3):244–252
Sandoval KE, Witt KA (2008) Blood–brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 32(2):200–219
Schellinger PD, Kaste M, Hacke W (2004) An update on thrombolytic therapy for acute stroke. Curr Opin Neurol 17(1):69–77
Sharabi K, Hua L, Tavares CDJ, Dominy JE, Camporez JP, Perry RJ et al (2017) Selective chemical inhibition of PGC-1α gluconeogenic activity ameliorates type 2 diabetes. Cell 169(1):148–160
Shi Y, Jiang X, Zhang L, Pu H, Hu X, Zhang W et al (2017) Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood–brain barrier disruption after ischemic brain injury. Proc Natl Acad Sci USA 114(7):E1243–E1252
Singh S, Simpson RL, Bennett RG (2015) Relaxin activates peroxisome proliferator-activated receptor γ (PPARγ) through a pathway involving PPARγ coactivator 1α (PGC1α). J Biol Chem 290(2):950–959
Su WC, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G et al (2000) Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-γ and nuclear factor-κB. J Biol Chem 275(42):32681–32687
Valluri S, Rupam G, Srividya S, George C (2017) Long non-coding rnas regulating immunity in insects. Noncoding RNA 3(1):14
Wang Q, Tang XN, Yenari MA (2007) The inflammatory response in stroke. J Neuroimmunol 184(1–2):53–68
Wang J, Li C, Chen T, Fang Y, Shi X, Pang T et al (2016) Nafamostat mesilate protects against acute cerebral ischemia via blood–brain barrier protection. Neuropharmacology 105:398–410
Wang W, Jiang B, Sun H, Ru X, Sun D, Wang L et al (2017) Prevalence, incidence and mortality of stroke in China: results from a nationwide population-based survey of 480,687 adults. Circulation 135(8):759–771
Wang Y, Ruan W, Mi J, Xu J, Wang H, Cao Z et al (2018a) Balasubramide derivative 3C modulates microglia activation via CaMKKβ-dependent AMPK/PGC-1α pathway in neuroinflammatory conditions. Brain Behav Immun 67:101–117
Wang Y, Huang Y, Xu Y, Ruan W, Wang H, Zhang Y et al (2018b) A Dual AMPK/Nrf2 activator reduces brain inflammation after stroke by enhancing microglia M2 polarization. Antioxid Redox Signal 28(2):141–163
Wu JS, Kao MH, Tsai HD, Cheung WM, Chen JJ, Ong WY et al (2018) Clinacanthus nutans mitigates neuronal apoptosis and ischemic brain damage through augmenting the C/EBPβ-driven PPAR-γ transcription. Mol Neurobiol 55(7):5425–5438
Xie F, Li BX, Kassenbrock A, Xue C, Wang X, Qian DZ et al (2015) Identification of a potent inhibitor of CREB-mediated gene transcription with efficacious in vivo anticancer activity. J Med Chem 58(12):5075–5087
Xu Y, Xu Y, Wang Y, Wang Y, He L, Jiang Z et al (2015) Telmisartan prevention of LPS-induced microglia activation involves M2 microglia polarization via CaMKKβ-dependent AMPK activation. Brain Behav Immun 50:298–313
Xu M, Yang X, Zeng Q, He H, Lu P, Huang G (2017) Birc5 is a novel target of peroxisome proliferator-activated receptor γ in brain microvascular endothelium cells during cerebral ischemia. Mol Med Rep 16(6):8882–8890
Xue H, Qiao Y, Ni P, Wang J, Chen C, Huang G (2011) A CRE that binds CREB and contributes to PKA-dependent regulation of the proximal promoter of human RAB25 gene. Int J Biochem Cell Biol 43(3):348–357
Yao J, Wang XQ, Li YJ, Shan K, Yang H, Wang YN et al (2016) Long non-coding RNA MALAT1 regulates retinal neurodegeneration through CREB signaling. EMBO Mol Med 8(4):346–362
Ye J, Piao H, Jiang J, Jin G, Zheng M, Yang J et al (2017) Polydatin inhibits mast cell-mediated allergic inflammation by targeting PI3K/Akt, MAPK, NF-κB and Nrf2/HO-1 pathways. Sci Rep 7(1):11895
Yin KJ, Hamblin M, Chen YE (2014) Non-coding RNAs in cerebral endothelial pathophysiology: emerging roles in stroke. Neurochem Int 77:9–16
Zhang JH, Badaut J, Tang J, Obenaus A, Hartman R, Pearce WJ (2012) The vascular neural network-a new paradigm in stroke pathophysiology. Nat Rev Neurol 8(12):711–716
Zhang J, Yuan L, Zhang X, Hamblin MH, Zhu T, Meng F et al (2016) Altered long non-coding RNA transcriptomic profiles in brain microvascular endothelium after cerebral ischemia. Exp Neurol 277:162–170
Zhang X, Tang X, Liu K, Hamblin MH, Yin KJ (2017) Long non-coding RNA Malat1 regulates cerebrovascular pathologies in ischemic stroke. J Neurosci 37(7):1797–1806
Zhao G, Jiang K, Wu H, Qiu C, Deng G, Peng X (2017) Polydatin reduces Staphylococcus aureus lipoteichoic acid-induced injury by attenuating reactive oxygen species generation and TLR2-NFkB signalling. J Cell Mol Med 21(11):2796–2808
Zilleßen P, Celner J, Kretschmann A, Pfeifer A, Racké K, Mayer P et al (2016) Metabolic role of dipeptidyl peptidase 4 (DPP4) in primary human (pre)adipocytes. Sci Rep 6:23074
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81570236, 81870360, 81402385, 81571134, 81773995, 81320108029), the Natural Science Foundation of Jiangsu Province (BK20160032), “Double First-Class” University Project (CPU2018GY06, CPU2018GY20), the Six Talent Peaks Project of Jiangsu Province (T.P.), Shanghai Key Laboratory of Psychotic Disorders (13dz2260500), the Postgraduate Research & Practice Innovation Program of Jiangsu Province. We would like to acknowledge Dr. Xiujun Li of the Affiliated Drum Tower Hospital of Nanjing University Medical School, for providing pEGFP-C1-MALAT1 and control plasmids; Dr. Shanshan Guo of Ningxia Medical University for providing the C/EBPβ overexpression plasmid pcDNA3.1-C/EBPβ; Prof. Qin Jiang of Nanjing Medical University for generously providing us with the rat MALAT1 primer sequence. We also gratefully acknowledge the excellent technical assistance of Haojie Wang, Chenglong Gao, Tailin He and Xin Guan in the animal experiments.
Author information
Authors and Affiliations
Contributions
All authors listed contributed immensely to this study. WR and JL performed the experiments and wrote the paper. YX, YW, FZ, and XY performed the animal experiments and analyzed the data. HJ, LZ, JMS, LS, TP, as experts in molecular pharmacology provided technical supports and designed the research.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ruan, W., Li, J., Xu, Y. et al. MALAT1 Up-Regulator Polydatin Protects Brain Microvascular Integrity and Ameliorates Stroke Through C/EBPβ/MALAT1/CREB/PGC-1α/PPARγ Pathway. Cell Mol Neurobiol 39, 265–286 (2019). https://doi.org/10.1007/s10571-018-00646-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-018-00646-4