
Abstract
Inhibition of ionotropic glutamate receptors (iGluRs) is a potential target of therapy for ischemic stroke. Perampanel is a potent noncompetitive α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor (AMPAR) antagonist with good oral bioavailability and favorable pharmacokinetic properties. Here, we investigated the potential protective effects of perampanel against focal cerebral ischemia in a middle cerebral artery occlusion (MCAO) model in rats. Oral administration with perampanel significantly reduced MCAO-induced brain edema, brain infarct volume, and neuronal apoptosis. These protective effects were associated with improved functional outcomes, as measured by foot-fault test, adhesive removal test, and modified neurological severity score (mNSS) test. Importantly, perampanel was effective even when the administration was delayed to 1 h after reperfusion. The results of enzyme-linked immunosorbent assay (ELISA) showed that perampanel significantly decreased the expression of pro-inflammatory cytokines IL-1β and TNF-α, whereas it increased the levels of anti-inflammatory cytokines IL-10 and TGF-β1 after MCAO. In addition, perampanel treatment markedly decreased the expression of inducible nitric oxide synthase (iNOS) and neuronal nitric oxide synthase (nNOS), and also inhibited nitric oxide (NO) generation in MCAO-injured rats at 24 and 72 h after reperfusion. In conclusion, this study demonstrated that the orally active AMPAR antagonist perampanel protects against experimental ischemic stroke via regulating inflammatory cytokines and NOS pathways.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke, which results in the obstruction of brain arteries and loss of nutrients and oxygen to the brain, is still one of the leading causes of death and adult disability worldwide (Mozaffarian et al. 2016). Brain ischemia is known to trigger a series of complex events, such as excessive release of excitatory amino acid, intracellular calcium overload, reactive oxygen species (ROS) generation, and dysfunction of the blood–brain barrier, but the exact mechanism has not been determined (Pandya et al. 2011). Although strategies for the reperfusion of blood flow to brain tissue are effective in the early stage, no neuroprotective agents have been successfully used in clinical practice for ischemic stroke patients (Appireddy et al. 2015; Lin and Sanossian 2015).
Excessive release of glutamate, a major excitatory neurotransmitter in the mammalian CNS, has been shown to contribute to neurodegeneration in many neurological diseases. Through interacting with ionotropic glutamate receptors (iGluRs), such as N-methyl-d-aspartic acid receptor (NMDAR) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR), glutamate mediates fast synaptic transmission, and overactivation of these receptors is involved in neuronal injury following brain ischemia (Arundine and Tymianski 2004; Chen et al. 2012a, b). Blocking AMPAR activation by antagonists has been investigated for anti-ischemic activity in both in vitro and in vivo experiments with mixed success. The selective competitive AMPAR antagonist NBQX was shown to induce robust neuroprotective effects in models of focal and global ischemia (Smith and Meldrum 1993; Sheardown et al. 1990). Talampanel, a recently developed noncompetitive AMPA receptor antagonist, was shown to improve the functional deficit after transient focal cerebral ischemia in rats (Erdo et al. 2006). However, these compounds exert many shortcomings, such as poor solubility, short half-life, and precipitation in the kidney, which limit their utility in the clinical setting (Langan et al. 2003; Hanada et al. 2011). Perampanel [2-(2-oxo-1-phenyl-5-pyridin-2-yl-1,2 -dihydropyridin-3-yl) benzonitrile; E2007] is a potent noncompetitive AMPAR antagonist with good oral bioavailability and favorable pharmacokinetic properties (Rogawski and Hanada 2013). Here, we investigated the neuroprotective effect of perampanel against brain ischemia and the potential underlying mechanisms with focus on inflammatory cytokines and nitric oxide synthase (NOS) pathway.
Materials and Methods
Middle Cerebral Artery Occlusion (MCAO) Model
Brain ischemia was induced by MCAO for 60 min in Sprague Dawley (SD) rats. Briefly, animals were anesthetized using 2% isoflurane in oxygen and placed in the stereotaxic frame. A midline incision was performed to isolate right common carotid artery (CCA) and the internal carotid artery (ICA). A 6–0 nylon monofilament coated with silicone was used to achieve occlusion at 2–3 mm from the origin of the MCA. The occluding filament was subsequently withdrawn at 60 min for reperfusion. Sham animals were subjected to the same surgical procedures except the occlusion. We maintained temperature (37.0 ± 0.5 °C) using a thermostatically controlled heating blanket connected to a thermometer probe in the rectum. All experimental protocols and animal handling procedures were performed in accordance with the National Institutes of Health (NIH) guidelines for the use of experimental animals and approved by the Institutional Animal Care and Use Committee of the Peking University. Perampanel was dissolved in 0.1% dimethyl sulfoxide (DMSO), and the dose of perampanel was selected according to previously published data (Hanada et al. 2011). The animals in perampanel group were orally administrated with 10 mg/kg perampanel as a one-time dose 1 h prior to MCAO. The animals in vehicle group were orally administrated with 0.1% DMSO and subjected to MCAO.
Brain Water Content Measurement
Brain edema was determined with the wet–dry method 24 h after MCAO. Briefly, rats were sacrificed by decapitation under deep anesthesia, and the brain was quickly removed. Tissue samples from the right hemispheres were dissected and weighed immediately to obtain wet weight. Dry weight was determined after heating the tissue for 48 h at 100 °C. Brain water content was then calculated using the following formula: % H2O = (1 − dry weight/wet weight) × 100%.
Infarct Volume Assessment
Brain infarct area was evaluated using 2,3,5-triphenyltetrazolium chloride (TTC) staining. Briefly, the rats were sacrificed and the brains were sectioned into 2-mm-thick coronal slices. Coronal brain slices were stained in 2% TTC at 37 °C for 15 min in the dark and then photographed. The infarct tissue areas were measured using Image-Pro Plus software by an investigator blinded to the study groups. To account for edema, the infarcted area was estimated by subtracting the uninfarcted region in the ipsilateral hemisphere from the contralateral hemisphere, and the infarct volume was expressed as a percentage of the contralateral hemisphere.
TUNEL Staining
Neuronal apoptosis was measured by TUNEL staining. In brief, brain sections of 4 μm thickness were cut and treated with proteinase K solution (20 μg/ml) for 10 min at room temperature to permeabilize the tissues. TUNEL staining was performed by labeling the tissues with a fluorescein TUNEL reagent mixture for 60 min at 37 °C according to the manufacturer’s suggested protocol, and the tissues were examined under fluorescence microscopy. The number of TUNEL-positive cells was counted by an investigator blinded to the grouping.
Functional Tests
The functional outcomes were measured by foot-fault test, adhesive removal test, and modified neurological severity score (mNSS) test according to previously published methods at 2, 24, and 48 h after MCAO (Ning et al. 2014).
Inflammatory Cytokine Measurement
To detect the expression of inflammation-related cytokines, rats were sacrificed at 6, 12, and 24 h after ischemia and the brain tissue homogenates were obtained from the whole right cerebral hemisphere. The concentrations of IL-1β, TNF-α, IL-10, and TGF-β1 were measured using specific ELISA kits according to the manufacturer's instructions (Boster Biological Technology, Wuhan, China).
Nitrite Assay
Nitrite is generated by the rapid oxidation of NO. To assay nitrite, 100-ml aliquots of the samples obtained above were mixed with 100 ml of equal volumes of Griess reagent mixture in a 96-well microtiter plate. After 10 min of incubation at room temperature, the absorbance at a wavelength of 540 nm was measured in a microplate reader. A range of twofold dilutions of sodium nitrite (0–128 μM) in PBS were run in each assay to generate a standard curve.
Western Blot Analysis
The homogenates obtained above were also used for Western blot analysis. Forty micrograms of protein was resolved on 10% SDS-PAGE gel and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% nonfat milk and incubated with the iNOS, nNOS, or β-actin primary antibodies. The membranes were then washed and incubated for 1 h at room temperature with secondary antibodies. The ImageJ software was used to quantify the optical density of each band.
Statistical Analysis
Statistical analysis was performed using SPSS 16.0, a statistical software package. In each experiment, six animals were used, and six values were randomly selected from the results of each animal. Statistical evaluation of the data was performed by Mann–Whitney U test. The neurologic function and expression of inflammatory cytokines were analyzed by repeated measures ANOVA. A value of p < 0.05 was considered statistically significant.
Results
Perampanel Attenuates Focal Cerebral Ischemic Injury in Rats
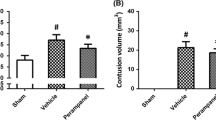
MCAO operation was used to model focal cerebral ischemia, and 10 mg/kg perampanel was administered orally 1 h before MCAO onset. The results of brain water content showed that perampanel significantly inhibited brain edema after ischemia (Fig. 1a). Infarct volume corrected for brain edema in cerebral hemisphere was reduced by 38.1% on 7 days (Fig. 1b). Subanalysis showed that infarct volume in both cortex and striatum was reduced by perampanel. In addition, TUNEL staining was used to detect apoptosis in brain sections (Fig. 1c), and the results showed that the number of TUNEL-positive cells in perampanel-treated group was lower than that in the vehicle group (Fig. 1d). As shown in Fig. 1e, no significant difference in body weight was observed.

Perampanel attenuates focal cerebral ischemic injury in rats. Animals were orally administered with 10 mg/kg perampanel or saline water 1 h prior to MCAO. Brain edema at 24 h was assayed by measuring brain water content (a), and infarct volume in ipsilateral cortex, striatum, and hemisphere at 7 days was determined by TTC staining (b). Apoptosis in brain sections was detected by TUNEL staining (c, d), and body weight was measured in each group (E). Data are expressed as mean ± SEM (n = 6). *p < 0.05 versus vehicle
Perampanel Improves Functional Outcome After Cerebral Ischemia
To test whether perampanel-induced reduction in infarct volume was associated with behavioral improvements, a battery of functional outcomes were tested. The results showed that perampanel significantly improved functional outcome after ischemia, as evidenced by reduced scores in mNSS test (Fig. 2a), decreased time in adhesive removal test (Fig. 2b), as well as reduced percentage of foot faults (Fig. 2c).

Perampanel improves functional outcome after cerebral ischemia. Animals were orally administered with 10 mg/kg perampanel or saline water 1 h prior to MCAO. The functional outcome was measured by mNSS test (a), adhesive removal test (b), and foot-fault test (c) 2, 24, and 48 h later. Data are expressed as mean ± SEM (n = 6). *p < 0.05 versus vehicle
Therapeutic Time Window of Perampanel in Rats
We then sought to determine whether perampanel could still exert protective effects when it was administered after MCAO onset. The results of brain water content showed that remarkable brain edema reductions were observed when perampanel was administered 0 and 1 h after reperfusion, but not when it was delayed to 3 and 6 h later (Fig. 3a). Accordingly, a significant reduction in hemisphere infarct volume, as measured by TTC staining, was detected when perampanel treatment was started at 0 or 1 h after reperfusion (Fig. 3c).

Therapeutic time window of perampanel in rats. Animals were orally administered with 10 mg/kg perampanel or saline water at 0, 1, 3, or 6 h after reperfusion. The brain water content at 24 h (a) and infarct volume in ipsilateral hemisphere at 7 days (b) were measured. Data are expressed as mean ± SEM (n = 6). *p < 0.05 versus vehicle
Perampanel Differentially Regulates Inflammatory Cytokines After Cerebral Ischemia
To investigate the potential anti-inflammatory activity of perampanel treatment, we measured the expression of inflammatory cytokines at 0, 6, 12, and 24 h after ischemia. As shown in Fig. 4a, the expression of IL-1β significantly increased after MCAO operation, and perampanel partially reversed this increase at 6, 12, and 24 h. A similar result was also observed for TNF-α (Fig. 4b). We also determined the content of two anti-inflammatory cytokines, IL-10 and TGF-β1. The results of ELISA showed that the concentrations of IL-10 and TGF-β1 were markedly higher in perampanel-treated group than those in the vehicle group (Fig. 4c, d).

Perampanel differently regulates inflammatory cytokines after cerebral ischemia. Animals were orally administered with 10 mg/kg perampanel or saline water 1 h prior to MCAO. The expression levels of IL-1β (a), TNF-α (b), IL-10 (c), and TGF-β1 (d) in brain homogenate were measured 6, 12, and 24 h later. Data are expressed as mean ± SEM (n = 6). *p < 0.05 versus vehicle
Perampanel Inhibits NO Expression Through iNOS and nNOS
We performed Western blot to detect the expression of iNOS and nNOS proteins in the ipsilateral hemispheres after ischemia. The iNOS protein expression in the vehicle group was up to 3.8-fold and 3.3-fold higher than that in Sham group at 24 and 72 h after reperfusion, respectively (Fig. 5a). However, the iNOS expression in the perampanel-treated group was significantly lower compared to that in the vehicle group at both 24 and 72 h. As shown in Fig. 5b, a similar result on nNOS protein expression was also observed. In addition, we also indirectly determined the NO concentration of the ipsilateral hemisphere by measuring nitrite concentration in each group (Fig. 5c). The results showed that ischemia significantly increased the concentration of NO at both 24 and 72 h, which was prevented by perampanel treatment.

Perampanel inhibits NO expression through iNOS and nNOS. Animals were orally administered with 10 mg/kg perampanel or saline water 1 h prior to MCAO. The expression of iNOS (a) and nNOS (b) was detected by Western blot. Nitrite production in the ipsilateral hemisphere was measured (c). Data are expressed as mean ± SEM (n = 6). #p < 0.05 versus Sham. *p < 0.05 versus vehicle
Discussion
Excessive release of glutamate plays an important role in mediating ischemic neuronal injury, and dysfunction of glutamate receptors, especially iGluRs, has been implicated as a key mechanism underlying ischemia-induced toxicity in neurons (Arundine and Tymianski 2004). In this study, we demonstrated the neuroprotective effects of perampanel, an orally active, noncompetitive AMPAR antagonist, against focal cerebral ischemic injury in rats. The protection afforded by perampanel is accompanied by improved functional outcome and sustained for at least 7 days after MCAO operation. Importantly, perampanel treatment is still effective even if the administration was delayed to 1 h after reperfusion. In addition, perampanel differentially regulated the expression of inflammatory cytokines following ischemia and inhibited NO generation via NOS pathways.
As a recently developed noncompetitive AMPAR antagonist, perampanel has unique advantages. Perampanel was shown to inhibit AMPAR-mediated responses during neurotransmission with an IC50 of 93–230 nM, with no obvious inhibition on kainate- and NMDAR-mediated responses (Ceolin et al. 2012; Hanada et al. 2011). After oral administration, perampanel is rapidly absorbed from the gastrointestinal tract with 100% bioavailability, and its peak plasma concentrations occur 0.5–2.5 h later (Patsalos 2015). Perampanel is mainly metabolized in the liver, and none of its metabolites is pharmacologically active (Traynor 2012; Krauss et al. 2012; Kwan et al. 2015). Unlike many competitive AMPAR antagonists that failed to penetrate the blood–brain barrier (BBB), perampanel was shown to abrogate brain endothelial cell permeability in response to ischemia through regulating claudin-5 (Lv et al. 2015). In addition, perampanel has been shown to exert anticonvulsant activity in animal models of epilepsy (Hanada 2014), and it is clinically available as round, biconvex, film-coated tablets for patients (Patsalos 2015). A recent study using controlled cortical impact (CCI) model showed that perampanel protects against traumatic brain injury (TBI) via anti-oxidative and anti-inflammatory activities (Chen et al. 2016). Our present results extended the neuroprotective effects of perampanel into in vivo stroke models. All these data indicate that perampanel is a selective, negative allosteric modulator of AMPAR with good oral bioavailability and favorable pharmacological properties, making it an ideal candidate for brain ischemia treatment.
Previous studies on neuroprotective agents have achieved great success in animal models, but no clinical trial has succeeded in demonstrating their clinical efficacy in patients (Ginsberg 2008). As previously pointed out, a well-accepted view of neuroprotective research is that “everything works in animals but nothing works in people” (O’Collins et al. 2006). One key issue is that most neuroprotective agents were effective only when administrated prior to ischemia, which might be problematic in clinical conditions (Menon and Zahed 2009). One major finding of the present study was the time window of therapeutic opportunity that is available with perampanel administration. Our results showed that perampanel is still effective in alleviating brain edema and reducing infarct volume when the administration was delayed to 1 h after reperfusion, which is more relevant to clinical practice. However, it is worth noting that there is practically no evidence that neuroprotection for acute ischemic stroke is possible with any agent beyond 6 h (Ginsberg 2008). Thus, some effective strategies, such as thrombolysis should be considered as auxiliary treatment methods for perampanel treatment.
Neuroinflammation is a hallmark of stroke pathology. It is well known that altered production and release of inflammatory cytokines is involved in CNS-confined inflammatory responses under neurological conditions, including ischemic stroke. IL-1β and TNF-α are two potent inflammatory cytokines that are shown to be able to modulate the size of ischemic damage in experimental stroke in many animal models (McCoy and Tansey 2008; Simi et al. 2007). These cytokines can directly act on the ischemic neurons and therefore promote the evolution of the infarct. Evidence that the levels of inflammatory cytokines influence stroke infarct is also supported by the findings of reduced infarct volume in IL-1β- and TNF-α-deficient mice (Lambertsen et al. 2012). In addition, another component of the inflammatory process is the anti-inflammatory phase, which is characterized by the activation of several anti-inflammatory cytokines (Hamilton et al. 2002). Previous reports have shown that both exogenous administration and gene transfer of IL-10 and TGF-β1 mediated neuroprotection after brain injury (Ooboshi et al. 2005; Pang et al. 2001; Frenkel et al. 2003). In the present study, the increased expression of all these inflammatory cytokines was confirmed in MCAO model, which was consistent with previous studies (Zhang et al. 2013, 2012). Recently, perampanel was shown to attenuate traumatic brain injury through anti-oxidative and anti-inflammatory activities (Chen et al. 2016). Similar results of the modulation induced by perampanel on inflammatory cytokines levels were observed in our present study, indicating the involvement of anti-inflammatory activity in perampanel-induced protection against experimental ischemic stroke.
NO is a highly reactive chemical that can interact with oxygen to form reactive nitrogen species (RNS). In central nervous system, NO is mainly synthesized by neuronal NO synthase (nNOS) in neuronal cells and by inducible NO synthase (iNOS) that is expressed in glia only during inflammatory conditions (Brown 2007). Previous studies demonstrated that reactive NO significantly increases after brain ischemia, and over-generated NO leads to a reduction in energy production and promotes neuroinflammation (Liu et al. 2002). NO can stimulate superoxide and hydrogen dioxide generation in mitochondria, induce Bax-associated apoptosis through p53 activation, and also can promote necrosis via inhibiting respiration and glycolysis (Brown and Borutaite 2006; Yung et al. 2004). Therefore, inhibiting NO generation via targeting iNOS and nNOS activity has been considered to be a neuroprotective strategy in brain ischemia. nNOS mutant mice showed smaller infarcts and attenuated apparent diffusion coefficient changes in the peri-infarct zone after experimental stroke (Zaharchuk et al. 1997). A study in rat cortex after MCAO showed that the “core” of damage in the striatum and the “penumbra” of damage in the fronto-parietal cortex were affected by nNOS and iNOS expression during the acute phase of ischemia (Vannucchi et al. 2007). In addition, iNOS inhibitors have been demonstrated to exert protective effects in both in vitro and in vivo ischemic models, and many neuroprotective agents were shown to be protective against neuronal ischemia via inhibiting iNOS activity (del Zoppo et al. 2000; Licinio et al. 1999). In the present study, significant increases in iNOS and nNOS expression were observed at both 24 and 72 h after MCAO, and all these changes were partially prevented by perampanel treatment. It was speculated that perampanel-induced inhibition on calcium overload, the main downstream signaling cascade of AMPAR, might contribute to the reduction of nNOS- and iNOS-mediated NO generation, which needs to be further determined.
One limitation of the present study is that the functional outcome in rats was only measured within 48 h after MCAO operation, and more long-term functional outcomes after ischemia warrant further investigation. In addition, the molecular mechanisms underlying perampanel-induced inhibition on iNOS and nNOS pathway needs to be further determined. Also, besides glutamate-induced toxicity, many more signaling cascades are also involved in neuronal injury after stroke, such as adenosine receptors and endocannabinoid system (Dias et al. 2012; Chen et al. 2014), and more experiments on these pathways after perampanel treatment need to be performed.
In conclusion, our data demonstrated, for the first time, that perampanel treatment significantly reduced the infarct size and neurological deficits in MCAO-injured rats. These effects of perampanel were associated with the modulation of inflammatory cytokines and NOS pathways.
References
Appireddy RM, Demchuk AM, Goyal M, Menon BK, Eesa M, Choi P, Hill MD (2015) Endovascular therapy for ischemic stroke. J Clin Neurol 11(1):1–8. doi:10.3988/jcn.2015.11.1.1
Arundine M, Tymianski M (2004) Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci 61(6):657–668. doi:10.1007/s00018-003-3319-x
Brown GC (2007) Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans 35(Pt 5):1119–1121. doi:10.1042/BST0351119
Brown GC, Borutaite V (2006) Interactions between nitric oxide, oxygen, reactive oxygen species and reactive nitrogen species. Biochem Soc Trans 34(Pt 5):953–956. doi:10.1042/BST0340953
Ceolin L, Bortolotto ZA, Bannister N, Collingridge GL, Lodge D, Volianskis A (2012) A novel anti-epileptic agent, perampanel, selectively inhibits AMPA receptor-mediated synaptic transmission in the hippocampus. Neurochem Int 61(4):517–522. doi:10.1016/j.neuint.2012.02.035
Chen T, Cao L, Dong W, Luo P, Liu W, Qu Y, Fei Z (2012a) Protective effects of mGluR5 positive modulators against traumatic neuronal injury through PKC-dependent activation of MEK/ERK pathway. Neurochem Res 37(5):983–990. doi:10.1007/s11064-011-0691-z
Chen T, Zhang L, Qu Y, Huo K, Jiang X, Fei Z (2012b) The selective mGluR5 agonist CHPG protects against traumatic brain injury in vitro and in vivo via ERK and Akt pathway. Int J Mol Med 29(4):630–636. doi:10.3892/ijmm.2011.870
Chen Z, Xiong C, Pancyr C, Stockwell J, Walz W, Cayabyab FS (2014) Prolonged adenosine A1 receptor activation in hypoxia and pial vessel disruption focal cortical ischemia facilitates clathrin-mediated AMPA receptor endocytosis and long-lasting synaptic inhibition in rat hippocampal CA3-CA1 synapses: differential regulation of GluA2 and GluA1 subunits by p38 MAPK and JNK. J Neurosci 34(29):9621–9643. doi:10.1523/JNEUROSCI.3991-13.2014
Chen T, Dai SH, Jiang ZQ, Luo P, Jiang XF, Fei Z, Gui SB, Qi YL (2016) The AMPAR antagonist perampanel attenuates traumatic brain injury through anti-oxidative and anti-inflammatory activity. Cell Mol Neurobiol. doi:10.1007/s10571-016-0341-8
del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ (2000) Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol 10(1):95–112
Dias RB, Ribeiro JA, Sebastiao AM (2012) Enhancement of AMPA currents and GluR1 membrane expression through PKA-coupled adenosine A(2A) receptors. Hippocampus 22(2):276–291. doi:10.1002/hipo.20894
Erdo F, Berzsenyi P, Nemet L, Andrasi F (2006) Talampanel improves the functional deficit after transient focal cerebral ischemia in rats. A 30-day follow up study. Brain Res Bull 68(4):269–276. doi:10.1016/j.brainresbull.2005.08.018
Frenkel D, Huang Z, Maron R, Koldzic DN, Hancock WW, Moskowitz MA, Weiner HL (2003) Nasal vaccination with myelin oligodendrocyte glycoprotein reduces stroke size by inducing IL-10-producing CD4+ T cells. J Immunol 171(12):6549–6555
Ginsberg MD (2008) Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology 55(3):363–389. doi:10.1016/j.neuropharm.2007.12.007
Hamilton TA, Ohmori Y, Tebo J (2002) Regulation of chemokine expression by antiinflammatory cytokines. Immunol Res 25(3):229–245. doi:10.1385/IR:25:3:229
Hanada T (2014) The discovery and development of perampanel for the treatment of epilepsy. Expert Opin Drug Discov 9(4):449–458. doi:10.1517/17460441.2014.891580
Hanada T, Hashizume Y, Tokuhara N, Takenaka O, Kohmura N, Ogasawara A, Hatakeyama S, Ohgoh M, Ueno M, Nishizawa Y (2011) Perampanel: a novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia 52(7):1331–1340. doi:10.1111/j.1528-1167.2011.03109.x
Krauss GL, Bar M, Biton V, Klapper JA, Rektor I, Vaiciene-Magistris N, Squillacote D, Kumar D (2012) Tolerability and safety of perampanel: two randomized dose-escalation studies. Acta Neurol Scand 125(1):8–15. doi:10.1111/j.1600-0404.2011.01588.x
Kwan P, Brodie MJ, Laurenza A, FitzGibbon H, Gidal BE (2015) Analysis of pooled phase III trials of adjunctive perampanel for epilepsy: impact of mechanism of action and pharmacokinetics on clinical outcomes. Epilepsy Res 117:117–124. doi:10.1016/j.eplepsyres.2015.09.002
Lambertsen KL, Biber K, Finsen B (2012) Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab 32(9):1677–1698. doi:10.1038/jcbfm.2012.88
Langan YM, Lucas R, Jewell H, Toublanc N, Schaefer H, Sander JW, Patsalos PN (2003) Talampanel, a new antiepileptic drug: single- and multiple-dose pharmacokinetics and initial 1-week experience in patients with chronic intractable epilepsy. Epilepsia 44(1):46–53
Licinio J, Prolo P, McCann SM, Wong ML (1999) Brain iNOS: current understanding and clinical implications. Mol Med Today 5(5):225–232. doi:10.1016/S1357-4310(99)01453-7
Lin MP, Sanossian N (2015) Reperfusion therapy in the acute management of ischemic stroke. Cardiol Clin 33(1):99–109. doi:10.1016/j.ccl.2014.09.009
Liu PK, Robertson CS, Valadka A (2002) The association between neuronal nitric oxide synthase and neuronal sensitivity in the brain after brain injury. Ann N Y Acad Sci 962:226–241
Lv JM, Guo XM, Chen B, Lei Q, Pan YJ, Yang Q (2015) The noncompetitive AMPAR antagonist perampanel abrogates brain endothelial cell permeability in response to ischemia: involvement of claudin-5. Cell Mol Neurobiol. doi:10.1007/s10571-015-0257-8
McCoy MK, Tansey MG (2008) TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation 5:45. doi:10.1186/1742-2094-5-45
Menon DK, Zahed C (2009) Prediction of outcome in severe traumatic brain injury. Curr Opin Crit Care 15(5):437–441. doi:10.1097/MCC.0b013e3283307a26
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB (2016) Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation 133(4):e38–e360. doi:10.1161/CIR.0000000000000350
Ning R, Chopp M, Zacharek A, Yan T, Zhang C, Roberts C, Lu M, Chen J (2014) Neamine induces neuroprotection after acute ischemic stroke in type one diabetic rats. Neuroscience 257:76–85. doi:10.1016/j.neuroscience.2013.10.071
O’Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW (2006) 1,026 experimental treatments in acute stroke. Ann Neurol 59(3):467–477. doi:10.1002/ana.20741
Ooboshi H, Ibayashi S, Shichita T, Kumai Y, Takada J, Ago T, Arakawa S, Sugimori H, Kamouchi M, Kitazono T, Iida M (2005) Postischemic gene transfer of interleukin-10 protects against both focal and global brain ischemia. Circulation 111(7):913–919. doi:10.1161/01.CIR.0000155622.68580.DC
Pandya RS, Mao L, Zhou H, Zhou S, Zeng J, Popp AJ, Wang X (2011) Central nervous system agents for ischemic stroke: neuroprotection mechanisms. Cent Nerv Syst Agents Med Chem 11(2):81–97
Pang L, Ye W, Che XM, Roessler BJ, Betz AL, Yang GY (2001) Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-ss1 expression. Stroke 32(2):544–552
Patsalos PN (2015) The clinical pharmacology profile of the new antiepileptic drug perampanel: a novel noncompetitive AMPA receptor antagonist. Epilepsia 56(1):12–27. doi:10.1111/epi.12865
Rogawski MA, Hanada T (2013) Preclinical pharmacology of perampanel, a selective non-competitive AMPA receptor antagonist. Acta Neurol Scand 127(Suppl 197):19–24. doi:10.1111/ane.12100
Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honore T (1990) 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science 247(4942):571–574
Simi A, Tsakiri N, Wang P, Rothwell NJ (2007) Interleukin-1 and inflammatory neurodegeneration. Biochem Soc Trans 35(Pt 5):1122–1126. doi:10.1042/BST0351122
Smith SE, Meldrum BS (1993) Cerebroprotective effect of a non-N-methyl-D-aspartate antagonist, NBQX, after focal ischaemia in the rat. Funct Neurol 8(1):43–48
Traynor K (2012) Perampanel approved for epilepsy. Am J Health Syst Pharm 69(23):2024. doi:10.2146/news120080
Vannucchi MG, Bizzoco E, Corsani L, Gianfriddo M, Pedata F, Faussone-Pellegrini MS (2007) Relationships between neurons expressing neuronal nitric oxide synthase, degree of microglia activation and animal survival. A study in the rat cortex after transient ischemia. Brain Res 1132(1):218–227. doi:10.1016/j.brainres.2006.11.029
Yung HW, Bal-Price AK, Brown GC, Tolkovsky AM (2004) Nitric oxide-induced cell death of cerebrocortical murine astrocytes is mediated through p53- and Bax-dependent pathways. J Neurochem 89(4):812–821. doi:10.1111/j.1471-4159.2004.02395.x
Zaharchuk G, Hara H, Huang PL, Fishman MC, Moskowitz MA, Jenkins BG, Rosen BR (1997) Neuronal nitric oxide synthase mutant mice show smaller infarcts and attenuated apparent diffusion coefficient changes in the peri-infarct zone during focal cerebral ischemia. Magn Reson Med 37(2):170–175
Zhang Y, Zhang FG, Meng C, Tian SY, Wang YX, Zhao W, Chen J, Zhang XS, Liang Y, Zhang SD, Xing YJ (2012) Inhibition of sevoflurane postconditioning against cerebral ischemia reperfusion-induced oxidative injury in rats. Molecules 17(1):341–354. doi:10.3390/molecules17010341
Zhang BJ, Men XJ, Lu ZQ, Li HY, Qiu W, Hu XQ (2013) Splenectomy protects experimental rats from cerebral damage after stroke due to anti-inflammatory effects. Chin Med J 126(12):2354–2360
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There is no conflict of interest.
Rights and permissions
About this article

Cite this article
Niu, HX., Wang, JZ., Wang, DL. et al. The Orally Active Noncompetitive AMPAR Antagonist Perampanel Attenuates Focal Cerebral Ischemia Injury in Rats. Cell Mol Neurobiol 38, 459–466 (2018). https://doi.org/10.1007/s10571-017-0489-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-017-0489-x