
Abstract
Autophagy is an essential degradation pathway in clearing abnormal protein aggregates in mammalian cells and is responsible for protein homeostasis and neuronal health. Several studies have shown that autophagy deficits occurred in early stage of Alzheimer’s disease (AD). Autophagy plays an important role in generation and metabolism of β-amyloid (Aβ), assembling of tau and thus its malfunction may lead to the progress of AD. By considering the above evidences, autophagy may be a new target in developing drugs for AD. So far, a number of mammalian target of rapamycin (mTOR)-dependent and independent autophagy modulators have been identified to have positive effects in AD treatment. In this review, we summarized the latest progress supporting the role for autophagy deficits in AD and the potential therapeutic effects of autophagy modulators in AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
AD, which is the most prevalent neurodegenerative disease, is characterized by deficiency in memory and cognitive functions. The predominant pathological changes of AD are development of Aβ plaques in specific brain areas, neurofibrillary tangles in neurons, and progressive loss of synapses and neurons (Ingelsson et al. 2004). Although the etiology mechanisms underlying these pathological changes are not clear yet, recent studies have shown deficits in the autophagy-lysosome pathway, which is an important system to clear misfolded proteins or damaged organelles, is likely to precede the formation of Aβ plaques or neurofibrillary tangles (Correia et al. 2015). Until now, three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), have been identified in mammalian cells. Among them, macroautophagy is best studied and considered most relevant to AD, CMA has also been thought to play a role in AD. To make it simple, hereon we will refer macroautophagy as “autophagy.” In this review, we begin with the autophagy physiological machinery, evidence of autophagy malfunction in AD, relationship between autophagy dysfunction and AD-related pathology, and finally focus on the therapeutic potential of autophagy modulators.
Autophagy Machinery
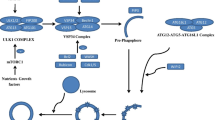
There are several stages for autophagy biogenesis, including phagophore membrane isolation, phagophore elongation and engulf of random cytoplasmic content, and autophagosome maturation and fusion with lysosome (Lee et al. 2013) (Fig. 1). Generally, the autophagy activation is largely depended on cellular starving condition (Kuma et al. 2004, Mizushima et al. 2004), such as low amino acids or glucose concentration. However, recent studies have also shown autophagy could be induced by diverse stimulations, such as reactive oxygen species (ROS) (Scherz-Shouval et al. 2007), hypoxia impairments (Mazure and Pouysségur 2010), subcellular organelle damages (Song et al. 2013), and protein aggregation (Liu et al. 2010). However, in all the regulators of autophagy, the mammalian target of rapamycin complex 1 (mTORC1) has been studied most thoroughly and considered to play a key role in autophagy biogenesis.

Signaling pathway of autophagy
In amino acid depletion environment, mTORC1 complex is de-activated and distributed freely in cytoplasm, where it cannot inhibit autophagy initiation. However, in nutrition-rich condition, the lysosomal amino acid concentration is largely increased, which in turn stimulates v-ATPase on the lysosome membrane. v-ATPase is a lysosome membrane multimeric channel protein majorly functions as ATPase and H+ channel, meanwhile, it also functions as a sensor for lysosomal amino acid concentration. After sensing the increase of amino acid concentration, v-ATPase and its binding partner, Ragulator, would both change their conformation and thus activate downstream signal molecule: RagA/B, by converting its conformation from GDP-bound to GTP-bound. Activated RagA/B then recruit active mTORC1 onto lysosome membrane, on where mTORC1 could be activated by Rheb, a small GTPase attached on lysosome membrane (Zoncu et al. 2011; Jewell et al. 2013; Yadav et al. 2013). After mTORC1 activation, it will negatively control autophagy genesis by blocking Unc-51-like kinase (ULK)1/2 complex, which is an important inducer for autophagy by regulating Beclin1-VPS34 complex (Russell et al. 2013). On the other hand, activated ULK1/2 could active microtubule-associated protein 1 light chain 3 beta (LC3B) by phosphatidylethanolamine (PE) (Fujita et al. 2008). LC3 is important for phagophore elongation and finally form autophagosome (Bernard and Klionsky 2014). Furthermore, activated mTORC1 could also inhibit autophagosome fusion with lysosome by phosphorylating UV radiation resistance-associated gene protein (UVRAG) (Kim et al. 2015) and thus block autophagosome maturation. Therefore, activated mTORC1 could inhibit all autophagy biogenesis required stages, and inhibition of mTOR pathway by either pharmacological compounds such as rapamycin or activation of AMP-activated protein kinase (AMPK) (Wu et al. 2015) could facilitate autophagy process.
Autophagy in Neuronal Cells
Autophagy is an essential homeostatic pathway in neurons (Maday et al. 2012). Similar to other cells, with aging process, neuronal cell will also accumulate intracellular toxicant or damaged organelles such as mitochondria that must to be cleared by autophagy to maintain an appropriate intracellular homeostasis (Mariño et al. 2011). However, unlike other cell types, neurons are post-mitotic cell that can never dilute toxic substance by mitosis. So autophagy-dependent protein/organelles clearance would be more important in neurons.
Neurons are characterized by their highly polarized axons and dendritic compartments, by which they can extend over distances many times larger than their cell soma and thus connect to each other from long distance with minimized cell number. Autophagosomes in neurons majorly formed at axon tip and gradually matured when they are retrograde transporting along axons (Maday et al. 2012). There is also evidence that autophagosome formation can be induced along the axon in order to clear damaged mitochondria (Ashrafi et al. 2014). Furthermore, some research indicated distal biogenesis of autophagosome might be related with synaptic function (Wang et al. 2015a, b). The first step of autophagosome biogenesis at axon tip is recruiting Atg13 and Atg5 to double-FYVE containing protein 1 (DFCP-1), which is a phosphatidylinositol 3-phosphate (PI3P)-enriched omega-shaped ER structure that serves as a platform for autophagosome biogenesis (Maday and Holzbaur 2014). Then in 4–6 min, lipidated LC3 was incorporated into the developing autophagosome (Maday et al. 2012). Autophagy is more efficient in young neurons (Boland et al. 2008), as autophagy-related proteins such as beclin-1, Atg5, and Atg7 will decline with age (Lipinski et al. 2010; Shibata et al. 2006), which potentially contributing to the late onset of many neurodegenerative diseases (Rubinsztein et al. 2011).
Autophagy Malfunction in AD
There is substantial evidence that dysregulation of autophagy occurs in both AD animal models and AD patients. As early as 1967, Suzuki had found a large amount of abnormal aggregated tau protein and subcellular vesicles accumulated in the dystrophic or swollen neurites in AD patient brains (Suzuki and Terry 1967), but the identity of these vesicles was unclear until 2005, when Nixon’s group found immature autophagic vacuoles (AVs) accumulated in dystrophic neuritis in AD brains using immunogold labeling and electron microscopy (Nixon et al. 2005). This is the first direct evidence that autophagy deficiency was involved in AD. In the same year, similar results from PS-1/APP double transgenic mice (Yu et al. 2005) also showed AVs accumulated in neuronal dendrites and soma even before Aβ plaques appeared compared to age-matched controls. In addition to these direct ultramicroscopic results, changes in the expressions of several autophagy-related proteins also indicated autophagy dysfunction in AD. For example, Rubinsztein reported increased expression level of lysosomal protease in the early phase of AD patients (Rubinsztein et al. 2005). A recent study found that expression of the autophagy-related genes: atg1, atg8a, and atg18 in Drosophila melanogaster were down-regulated with aging, and the subsequent reducing of autophagy activity and hyper-generation of Aβ were both considered to correlate with late-onset neuronal dysfunction and AD phenotype (Omata et al. 2014), indicating that age-induced reduction of autophagy-related gene expression is associated with late onset of AD.
There are two types of AD identified: sporadic AD (SAD, also known as late-onset AD), and familial AD (FAD, also known as early-onset AD). In SAD, little is known about the cause of the onset, but it is widely accepted that both genetic and environmental factors contribute to this pathogenesis. Apolipoprotein E4 (apoE4), a major genetic risk factor for SAD, has been found to induce autophagy malfunction. Studies from ApoE4 transgenic mice showed that overexpression of ApoE4 elevated Aβ42 amount in lysosome and finally led to hippocampus neurons death (Belinson et al. 2008). In addition, ApoE4 potentiates lysosomal leakage and apoptosis caused by Aβ peptides in Neuro-2a cells(Ji et al. 2006). Taken together, these studies indicated that ApoE4 and Aβ may work in concert to increase the susceptibility of lysosomal membranes disruption, release of lysosomal enzymes, and hence neuronal degeneration (Ji et al. 2006).
Regarding to FAD, at least three genes, amyloid precursor protein (APP), presenilin-1 and -2(PS-1 and PS-2), have been identified as causative genes so far. FAD is caused by mutation in at least one of the three genes (Tang 2003). Research has found wildtype PS-1, but not mutation forms, is crucial for acidification of lysosome by regulating distribution of v-ATPase, and thus contributes to autophagy degradation in a gamma-secretase-independent way (Lee et al. 2010). Meanwhile, hyper-activation of Glycogen synthase kinase-3 (GSK-3), which is also a high-risk factor for AD, could interfere lysosome acidification via similar mechanism as PS-1 (Avrahami et al. 2013). These investigations suggested that autophagy malfunction is involved in FAD, while the mechanism(s) is still not clear.
Proper formation and degradation of autophagosome is critical for normal autophagic flux. In healthy neurons, low-basal autophagic activity was detected because of the quick subsequent degradation of autophagosome by lysosome. In hippocampus neurons of AD mice, abnormal accumulation of immature AVs in axon was observed before synaptic and neuronal loss (Sanchez-Varo et al. 2012). However, as either autophagosome axonal trafficking deficiency or insufficient lysosome acidification could cause AVs accumulation in axon, the actual mechanisms underlying autophagy dysfunction in AD is still need further investigate. It is also a debate whether dysfunction of autophagy is the cause or result of AD. Reports to date usually show some controversies (Peric and Annaert 2015; Cho et al. 2015; Lee et al. 2014). Many factors may lead to these differences, such as different animal models, cellular models, and experimental paradigms. Also, the different model systems are likely represent of different particular stages of AD pathogenic process.
CMA and AD
Chaperone-mediated autophagy (CMA) is another form of autophagy which functions quite distinct from macroautophagy, as CMA pathway will degrade certain proteins one by one without engulf process, and most of its targeting proteins would be cytoplasm soluble ones. This selective degradation pathway contributes to protein quality control and maintenance of cellular homeostasis particularly under stress conditions (Cuervo 2010; Dice 2007; Kaushik and Cuervo 2012). CMA-mediated degradation needs heat shock cognate protein of 70KD (hsc 70), which is a cytosolic chaperone, to recognize its targets. Most of hsc 70 target proteins share a reserved KFERQ sequence (Chiang et al. 1989). After binding to its target, cytoplasmic hsc70 will then mediate target protein unfolding and deliver it onto lysosome membrane, where lysosomal-associated membrane protein 2A (LAMP-2A) will form a transient multimeric channel to allow them to translocate through lysosome membrane. lysosome-resident form of hsc70 (lys-hsc70) in lysosome luminal side will also assist this process (Cuervo and Dice 1996; Cuervo et al. 1997; Agarraberes et al. 1997). It has been considered that the number of lys-hsc70 positive lysosome would decide the speed of CMA-mediated degradation under physiology condition.
Similar to macroautophagy, CMA is also considered to connect with neurodegeneration diseases such as AD and PD, as CMA plays an important role in both Tau tangles and Aβ plaques generation and its activity is impaired with aging process (Koga and Cuervo 2011). It has been found although Tau bears a CMA recognizing motif in its sequence, trace amount of wildtype Tau is degraded by CMA. Normally, when facing degradation, Tau will be first cleaved in cytoplasm and then remained c-terminal part will translocate into lysosome luminal either through autophagy or CMA, where the second and third cleavage will happen (Wang et al. 2009). However, in certain Tau mutations, the first cleavage will happen in wrong amino acid position. After this cleavage, although remaining c-truncated Tau could still be recognized by hsc70 and recruited onto lysosome membrane, it cannot translocated into lysosome luminal efficiently, thus resulting in accumulation of truncated Tau on lysosome membrane. Some of these products organize as oligomeric structures at the lysosomal membrane and with time, these oligomers can promote disruption of the lysosomal membrane and the subsequent leakage of lysosomal enzymes (Koga and Cuervo 2011).
Another recent study found APP also bears KFERQ motif which could be recognized by hsc70. Deletion of this sequence will keep APP away from lysosome and increase its secretase cleavage products, without influence its ability to bind to hsc70 (Park et al. 2016).
Relationship Between Autophagy Dysfunction and AD-Related Pathology
Aβ, Tau, and Autophagy
As mentioned above, Aβ plaques and neurofibrillary tangles are the two major neuro-pathological changes occurred in AD patients. Emerging evidence has demonstrated complicated interactions among autophagy, Aβ, and tau, which may contribute to the progress of AD.
Autophagy plays an important role in the metabolism of Aβ. First, along with Aβ degradation enzymes (Miners et al. 2008), autophagy is believed to be another major Aβ clearance pathway. Autophagy facilitates the degradation and clearance of APP (Aymloid-β precursor protein) (Zhou et al. 2011) as well as all APP cleavage products including Aβ (Son et al. 2012) and APP-CTFs (amyloid precursor protein-cleaved C-terminal fragment) (Tian et al. 2014). In microglia, Aβ is also degraded by autophagy through the autophagy receptor optineurin (Cho et al. 2014).
Second, autophagy-lysosome system, which is important for Aβ degradation under physiological conditions, is demonstrated to be a novel way for Aβ production under pathological condition or in aging process (Yu et al. 2005). Although Aβ is thought to be produced in lysosome, endoplasmic reticulum, and Golgi apparatus, emerging studies have provided evidence that Aβ generation could be detected in autophagic vacuoles after autophagy activation (Mizushima 2005). Thus, accumulated immature autophagic vacuoles found in AD brains and in APP/PS1 transgenic mice maybe a source of Aβ generation (Nixon et al. 2005). Immunohistochemistry staining showed that Aβ42 were stained in AEL (autophagy–endosomal–lysosomal) vesicles in Drosophila neurons (Ling et al. 2014). Another study found in neuron that although APP and its processing enzyme BACE1 should be separated into distinct vesicles under normal condition, they showed strong co-localization and co-trafficking into autophagy–lysosome pathways under glycine/NMDA-receptor agonist/K+ or GABAA antagonist Picrotoxin (PTX) stimulations (Das et al. 2013). This result indicated under some certain pathological conditions, the convergence of APP and BACE1 in autophagosomes may act as some new Aβ generation places.
Third, autophagy is also involved in the secretion of Aβ. Recent findings support that autophagy is responsible for extracellular release of Aβ. Measurement of extracellular Aβ in autophagy-deficient mice revealed that the Aβ secretion was reduced by 90 %, while restoration of autophagy enhanced Aβ secretion to normal levels (Nilsson and Saido 2014). There is another recent work has found in ATG7 knockdown mice, the secretion Aβ was largely reduced, which was accompanied by a significant intracellular Aβ accumulation (Nilsson, et al. 2015).
On the other side, Aβ could also regulate autophagy. Aβ40 in vascular could induce autophagy in endothelial cell and impair neurovascular regeneration (Hayashi et al. 2009). Further study showed Aβ-induced formation of AVs was regulated through the RAGE-calcium-CaMKKβ-AMPK pathway (Son et al. 2012).
For the case of tau, although ubiquitin–proteasome system (UPS) was considered to be the major pathway for tau turnover, recent studies suggested that autophagy maybe another effective degradation route for tau. A number of studies have demonstrated that dysfunction of autophagy-lysosome system leads to the formation of tau oligomers and insoluble aggregates, while induction of autophagy could alleviate this aggregation (Hamano et al. 2008, Congdon et al. 2012). Moreover, autophagy may affect phosphorylation status of tau. Hyperphosphorylated tau was found to be accumulated in brains of autophagy-deficient mice (Inoue et al. 2012), and this phospho-tau accumulation will be largely reduced after autophagy was restored. Researchers have found autophagic degradation of tau is regulated by nuclear factor erythroid-2-related factor 2 (Nrf2)-mediated activation of the autophagy receptor NDP52 (Jo et al. 2014). Furthermore, Phosphatidylinositol binding clathrin assembly protein (PICALM, also known as CALM) could also regulate Tau degradation by modulating SNAREs (Soluble NSF Attachment protein Receptors) endocytosis, which is critical for autophagy clearance of tau (Moreau et al. 2014). On the other hand, hyperphosphorylation of tau may result in autophagy dysfunction (Lim et al. 2001; Lin et al. 2003). Tau is well identified to facilitate the assembly and stabilization of microtubule, which is critical for autophagosome retrograde trafficking and maturation to fuse with lysosome. On contrast, hyperphosphorylated tau could lead to the instability and disassemble of microtubule cytoskeleton, which could subsequently inhibit autophagosome retrograde trafficking and thus accumulate immature autophagosomes in axons.
Axonal Transfer and Autophagy
Axonal transport is an essential process required to maintain neuronal homeostasis. Impaired axonal transport can lead to axon degeneration and has been found in many neurodegenerative diseases including AD. In mammalian cells, newly formed autophagosomes move along with microtubule tracks (Monastyrska et al. 2009), and during this process, autophagosomes engulf long-lived protein, misfolded protein, or damaged organelles—such as mitochondria—subsequently degrade them after fused with lysosome. Deficits in axonal transport usually result in accumulation of large amounts of autophagosome. Axonal dystrophic neurites arising from neurofibrillary tangles could be easily detected in hippocampus CA3 and CA1regions (Su et al. 1993).
Years of pathological examination in AD brains have yielded many descriptions of abnormal axonal transport in both early and late phase of AD (Bell and Claudio 2006). Meanwhile, the phosphorylated tau level affects axonal transport and degradation (Rodríguez-Martín et al. 2013). These data supported that abnormal protein aggregates disrupt axons, thereby autophagosomes could not transport to cytoplasm and thus failed to fuse with lysosomes. In contrast to this, some other studies gave opposite viewpoints that lysosomal protease abnormalities are the causative factor of axonal degeneration (Xie et al. 2015). However, the mechanism(s) underlying this transport disruption is not clear.
Autophagy and Treatment
Currently, most drugs available for AD treatment are developed based on cholinergic hypothesis. Some Chinese traditional herbs also show potential effects in AD treatment (Yu et al. 2012, Sun et al. 2012). However, these drugs can only alleviate some symptoms of AD. Development of innovative medicines to prevent AD pathogenesis has always been a hotspot field. As AD is caused by abnormal protein aggregation, new strategies that can enhance the degradation of toxic aggregates are essential for effective therapy. Autophagy, which represents a major route for clearance of aggregated proteins and organelles, may serve as an emerging and promising therapeutic target for blocking AD pathogenesis. Although some autophagy inducers showed promising effect in other neurodegeneration disease, research is still underway to identify selective autophagy regulator as potential drugs for AD treatment.
Pharmacological Chemicals Targeting mTOR-Dependent Autophagy Pathway
mTOR is a protein kinase that senses cellular energy availability, and regulates cellular proliferation. mTOR also serves as a major negative regulator in autophagy. In mammalian cells, mTOR could enhance the binding between ULK and ATG13, probably via phosphorylation of ULK. Previous reports have shown that mTOR signaling is hyperactive in selected regions of AD brains (An et al. 2003; Pei and Hugon 2008); genetic reduction of mTOR signaling in the brains of Tg2576 mice resulted in reduced Aβ deposits and rescued memory deficits by increasing autophagy induction and restoring the hippocampus gene expression signature (Caccamo et al. 2014). All these results demonstrated that mTOR hyperactivity could result in Aβ accumulation.
As a major regulator of the autophagy, mTOR is widely considered as a pharmacological target for autophagy regulation (Fig. 2). Moreover, research conducted in recent years revealed a potential role for mTOR-related proteases as anti-AD drug targets. Until now, amounts of chemicals that can regulate mTOR have been identified and some of them may become the potential candidates for AD treatment.

mTOR-dependent autophagy pathways
The beneficial effects of autophagy up-regulation in neurodegenerative diseases first came with the mTORC1 inhibitor rapamycin. Rapamycin is a US Food and Drug Administration (FDA)-approved antifungal antibiotic, anticancer cytostatic agent, and an immunosuppressant. In mammalian cells, rapamycin forms a complex with the immunophilin FKBP12 (FK506-binding protein of 12 kDa), thereby stabilizing raptor (Regulatory-associated protein of mTOR)-mTOR association and inhibiting mTORC1 kinase activity, which could then subsequently induce autophagy initiation. Some studies revealed a potential beneficial effect of inducing autophagy with rapamycin in AD. In two AD mouse models, rapamycin administration both resulted in reduced Aβ generation, lower tau accumulation and better cognitive performance (An et al. 2003; Pei and Hugon 2008). However, although drugs that target proteins involved in mTOR signaling pathway have clinical use potential, mTOR pathway itself involved in many critical cellular processes including cell growth and gene translation, so long-term inhibition of mTOR may cause toxic side effects on patients. Therefore, rapamycin is not considered as an ideal drug for long-term use.
Temsirolimus, which is a newly developed compound approved by FDA and European Medicines Agency for renal cell carcinoma treatment, could enhance Aβ clearance in HEK293-APP695 cells and in brains of APP/PS1 mice in an autophagy-dependent manner (Jiang et al. 2014a). Furthermore, Results showed temsirolimus could enhance autophagic clearance of hyperphosphorylated tau in either okadaic acid-administrated SH-SY5Y cells or P301S transgenic mice brain (Jiang et al. 2014b). Meanwhile, behavioral tests showed temsirolimus improved spatial learning and memory abilities in both APP/PS1 mice and P301S transgenic mice. These results strongly indicated temsirolimus may use as a potential therapeutic strategy for AD treatment (Jiang et al. 2014a, b).
However, there are many issues to be considered and confirmed before these candidate drugs to be applied in clinical therapies. First, whether the beneficial results observed in mice and tissue cultures can be also observed in human remains unknown. Sometimes even chemicals that have beneficial effects in small clinical trials may fail in the subsequent large sample trials. For example, Latrepirdine (Dimebon), which is a mTOR targeted drug, significantly alleviated cognitive deficiency in mild to severe AD patients in a small trial, failed in a phase III trial later (Sweetlove 2012). Second, as we mentioned above, because mTOR is such an important molecule that is involved in many essential cellular functions, treatment by those mTOR inhibitors over a prolonged period might be harmful to patients. Therefore, clinical studies are needed to be conducted carefully to adjust the adverse effects of these kind of chemicals.
Pharmacological Chemicals Targeting mTOR-Independent Autophagy Pathway
In addition to the well-defined mTOR pathway, there are several mTOR-independent signaling pathways that are involved in autophagy initiation (Fig. 3), such as inositol signaling pathway, Ca2+/calpain pathway, cAMP/Epac/Ins(1,4,5)P3 pathway, and JNK1/Beclin-1/PI3KC3 pathway. There are several FDA-approved drugs that have been reported to regulate autophagy through mTOR-independent pathways (Williams et al. 2008). In this section, we will highlight some examples in this category.

mTOR-independent autopahgy pathways
Inositol Signaling Pathway
The first evidence for existing a mTOR-independent pathway came from study that indicated intracellular IP3 levels negatively regulate autophagy. IP3 is a well-known signaling transducer produced by PIP2. IP3 can be hydrolyzed into free inositol that is essential for the inositol signaling pathway. Lowering intracellular inositol or IP3 levels could induce autophagy, while elevation of them could inhibit autophagy by inhibiting autophagosome synthesis.
The most well-studied chemical that can induce autophagy through inositol signaling pathway is lithium, which is a mood stabilizer that can inhibit inosito monophosphatase (IMPase) and lower inositol levels. Although it was used to treat affective disorders for more than a half-century, whether lithium could have positive effects on AD patients is still unclear. Some studies have shown long-term use of lithium is associated with a lower risk of dementia in bipolar disorder patients (Nunes et al. 2007; Kessing et al. 2010). However, contradictory evidences suggested lithium was not useful in the treatment of AD patients (Hampel et al. 2009; Macdonald et al. 2008). Further studies are needed to assess the effect of lithium on AD.
Another autophagy inducer targeting this pathway is scyllo-Inositol (SI), which has shown potential benefit to AD patients in clinical trials. In addition to its inhibitory effect on aggregation and fibril formation of Aβ, SI treatment also decreased the size and the number of accumulated AVs in TgCRND8 mouse model (Lai and McLaurin 2012), suggesting beneficial effect of SI-Aβ interaction may resolve autophagy deficiency in the AD brains. Other inducers include L-690,330, carbamazepine, and sodium valproate, which can enhance the clearance of mutant Huntingtin fragments and attenuate polyglutamine toxicity (Nalivaeva et al. 2009, Sarkar and Rubinsztein 2008). Studies have also shown sodium valproate and carbamazepine could inhibit Aβ aggregates and thus reduce cell toxicity (Nalivaeva et al. 2009, Li et al. 2013). However, further investigations are needed to confirm whether these chemicals have positive effects on AD patients.
Ca2+/Calpain Pathway
Elevation in cytosolic Ca2+ concentration could inhibit autophagy, while decrease of it enhances autophagy. Cytosolic Ca2+ affects autophagy at both the formation and autophagosome–lysosome fusion stages. Several FDA-approved L-type Ca2+ channel antagonists such as verapamil, loperamide, amiodarone, nimodipine, and nitrendipine, have been screened as enhancer of autophagy (Williams et al. 2008), but the effects of these antagonists on AD have not been well-studied. One potential drug is Isradipine, which is a FDA-approved dihydropyridine calcium channel blocker, attenuates Aβ oligomer toxicity by suppressing calcium influx into cytoplasm and suppressing CaV1.2 expression in vitro and in AD animal models, suggesting that isradipine exhibited bio-availability, lowered Aβ plaque burden, and improved autophagy function (Anekonda and Quinn 2011).
Other mTOR-Independent Pathways
There are many other kinds of drugs that target mTOR-independent pathways such as histone acetyltransferase inhibitors (Eisenberg et al. 2009), AMPK, and the protein deacetylase sirtuin 1 enhancers like Cilostazol (Lee et al. 2015). Besides, drugs that regulate transcriptional factor EB (TFEB) (Zhang and Zhao 2015), activate pro-autophagic enzymes, modify the maturation of autophagosome, or inhibit the enzymes antagonizing the formation of membrane structures (Cavieres et al. 2015, Papp et al. 2015) may all have beneficial effects on AD patients. Emerging mTOR-independent autophagy enhancers have been screened, but the precise mechanisms of some enhancers remain unclear (Sarkar et al. 2007b).
Combined Treatment May Have Additional Effects
Since AD is a multi-factor caused disease, the use of combination drug therapy may have more positive effects in AD treatment. Studies have shown that additive therapeutic effects and fewer side effects can be achieved at the same time through simultaneous stimulation of mTOR-dependent and independent routes. The promising effect of combination administration of lithium and rapamycin in Huntington’s disease (HD) treatment may give us some clues. Lithium could activate autophagy through mTOR-independent pathway while rapamycin activate autophagy through mTOR-dependent pathway. Combined administration of lithium and rapamycin results in stronger induction of autophagy, better protective effects on neuron survival, and more efficiency on autophagy substrates clearance such as mutant Huntingtin and α-synuclein compared with single medication alone (Sarkar et al. 2009). Similar effects were also observed in several cell lines from combination use of rapamycin with other mTOR-independent autophagy enhancers, such as trehalose, calpastatin, or SMERs (Sarkar et al. 2007a, b, Williams et al. 2008). In addition to the better effect in autophagy induction, lower doses administration of each chemical reagent may be safer for longer treatment by minimizing dose-related side effects.
When to Deliver Drug is an Important Concern
When designing and evaluating autophagy interventional therapies, timing is always an important factor to be considered as the role of autophagy in different AD stages remains controversial. Early activation of autophagy is supposed to be beneficial, aiding neurons to clear abnormal protein aggregates and organelles. However, in the advanced phase, the activation of autophagy may show less benefit to alleviate AD pathology as more mature and stable aggregates cannot be eliminated efficiently through autophagy. Study has shown that giving rapamycin to 2-month-old 3xTg-AD mice throughout their lifespan induced autophagy and significantly reduced Aβ plaques, Tau tangles, and subsequent cognitive deficits. However, inducing autophagy in 15-month-old 3xTg-AD mice which have established stable plaques, and tangles has no effect on AD-like pathology and cognitive deficits, suggesting autophagy induction exhibited beneficial effects only when they are administrated before the plaques and tangles formed (Spilman et al. 2010; Majumder et al. 2011). Ultimately, understanding the specific steps affected in the autophagy process in AD will be essential for the development of autophagy-based therapeutic drugs.
Gene Therapy for Autophagy Regulation
In addition to pharmacological chemicals, gene therapy is also a selective approach with the advantage of function in a tissue-directed manner. Several studies have shown gene therapy treatment designed to regulate autophagy via lentivirus or adenovirus-associated viral delivery had positive effects in diverse human disease including aging and neurodegenerative diseases. Although most gene therapy studies for AD-targeted NGF, recent work showed that oral vaccination with a recombinant AAV/Aβ vaccine increased the clearance of Aβ from the brain and improved cognitive ability in AD animal models through enhanced autophagy (Wang et al. 2015a, b), suggesting modulating the autophagy pathway by viral delivery may be an important strategy for AD prevention and intervention.
BECN1 gene encoded protein beclin-1, which is a well-known essential inducer of autophagy. Beclin-1 serves as a molecular platform assembling components which regulate the initiation of the autophagosome formation. Activation of Beclin-1 could up-regulate autophagy. In Becn1+/− transgenic mice, significantly reduced Beclin 1 expression lead to a decrease of neuronal autophagy and finally resulted in neurodegeneration (Pickford et al. 2008). Consistent with this, the down-regulation of Beclin1 was also observed in the brains of AD patients especially in those brain regions which were most vulnerable to AD pathology (Pickford et al. 2008). Administration of lentivirus encoding mouse beclin 1 to the frontal cortex and hippocampus of 6-month-old APP transgenic mice for 8 weeks resulted in prominent beclin 1 expression and significant reduced intracellular Aβ immunoreactivity (Pickford et al. 2008), which suggested beclin 1 is a potential target when designing gene therapy for AD.
Up to now, few reports have been released considering enhancing autophagy through gene therapy. Continued investigations seem to hold tremendous potential for successful application of this novel approach.
Concluding Remarks and Future Perspectives
Although the etiology of AD remains unclear, and many factors including genetic mutation, environmental factors, imbalance of energy metabolism, and heavy metal ion (Yu et al. 2010; Li et al. 2010) seem to contribute to the etiology of AD, emerging advances regarding to autophagy have indicated its role as a protective factor in the early phase of AD, but an evil player in the late phase. Autophagy can influence the generation, secretion, and clearance of Aβ, and it will also influence the phosphorylation status and clearance of tau. Thus, chemical modulators of autophagy as well as gene therapy targeting autophagy-related proteins offer great potential for the treatment of AD. A number of mTOR-dependent and independent autophagy modulators have been demonstrated to have positive effects in AD animal models and patients. However, a more throughout understanding of autophagy malfunction in AD, as well as brain pharmacokinetics of autophagy modulators will be critical for designing new experiments with appropriate drug doses in any future clinical trials for AD.
References
Agarraberes FA, Terlecky SR, Dice JF (1997) An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol 137(4):825–834
An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal IG, Winblad B, Pei JJ (2003) Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am J Pathol 163(2):591–607
Anekonda TS, Quinn JF (2011) Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: the case for isradipine. Biochim Biophys Acta 1812(12):1584–1590
Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 206(5):655–670
Avrahami L, Farfara D, Shaham Kol M, Vassar R, Frenkel D, Eldar-Finkelman H (2013) Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem 288:1295–1306
Belinson H, Lev D, Masliah E, Michaelson DM (2008) Activation of the amyloid cascade in apolipoprotein E4 transgenic mice induces lysosomal activation and neurodegeneration resulting in marked cognitive deficits. J Neurosci 28(18):4690–4701
Bell KF, Claudio CA (2006) Altered synaptic function in Alzheimer’s disease. Eur J Pharmacol 545(1):11–21
Bernard A, Klionsky DJ (2014) Defining the membrane precursor supporting the nucleation of the phagophore. Autophagy 10(1):1–2
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci 28(27):6926–6937
Caccamo A, De Pinto V, Messina A, Branca C, Oddo S (2014) Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci 34(23):7988–7998
Cavieres VA, González A, Muñoz VC, Yefi CP, Bustamante HA, Barraza RR, Tapia-Rojas C, Otth C, Barrera MJ, González C, Mardones GA, Inestrosa NC, Burgos PV (2015) Tetrahydrohyperforin inhibits the proteolytic processing of amyloid precursor protein and enhances its degradation by Atg5-dependent autophagy. PLoS ONE 10(8):e0136313
Chiang HL, Terlecky SR, Plant CP, Dice JF (1989) A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246(4928):382–385
Cho MH, Cho K, Kang HJ, Jeon EY, Kim HS, Kwon HJ, Kim HM, Kim DH, Yoon SY (2014) Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 10(10):1761–1775
Cho SJ, Yun SM, Jo C, Lee DH, Choi KJ, Song JC, Park SI, Kim YJ, Koh YH (2015) SUMO1 promotes Aβ production via the modulation of autophagy. Autophagy 11(1):100–112
Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH (2012) Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 8(4):609–622
Correia SC, Resende R, Moreira PI (2015) Pereira CM (2015) Alzheimer’s disease-related misfolded proteins and dysfunctional organelles on autophagy menu. DNA Cell Biol 34(4):261–273
Cuervo AM (2010) Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab 21(3):142–150
Cuervo AM, Dice JF (1996) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273:501–503
Cuervo AM, Dice JF, Knecht E (1997) A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem 272(9):5606–5615
Das U, Scott DA, Ganguly A, Koo EH, Tang Y, Roy S (2013) Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 79(3):447–460
Dice J (2007) Chaperone-mediated autophagy. Autophagy 3:295–299
Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L et al (2009) Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11:1305–1314
Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19(5):2092–2100
Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, Isidoro C, Deture M, Ko LW (2008) Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci 27(5):1119–1130
Hampel H, Ewers M, Bürger K, Annas P, Mörtberg A, Bogstedt A, Frölich L, Schröder J, Schönknecht P, Riepe MW, Kraft I, Gasser T, Leyhe T, Möller HJ, Kurz A, Basun H (2009) Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry 70(6):922–923
Hayashi S, Sato N, Yamamoto A, Ikegame Y, Nakashima S, Ogihara T, Morishita R (2009) Alzheimer disease-associated peptide, amyloid β40, inhibits vascular regeneration with induction of endothelial autophagy. Arterioscler Thromb Vasc Biol 29(11):1909–1915
Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC (2004) Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62(6):925–931
Inoue K, Rispoli J, Kaphzan H, Klann E, Chen EI, Kim J, Komatsu M, Abeliovich A (2012) Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol Neurodegener 7:48
Jewell JL, Russell RC, Guan KL (2013) Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol 14(3):133–139
Ji ZS, Müllendorff K, Cheng IH, Miranda RD, Huang Y, Mahley RW (2006) Reactivity of apolipoprotein E4 and amyloid beta peptide: lysosomal stability and neurodegeneration. J Biol Chem 281(5):2683–2692
Jiang T, Yu JT, Zhu XC, Tan MS, Wang HF, Cao L, Zhang QQ, Shi JQ, Gao L, Qin H, Zhang YD, Tan L (2014a) Temsirolimus promotes autophagic clearance of amyloid-β and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol Res 81:54–63
Jiang T, Yu JT, Zhu XC, Zhang QQ, Cao L, Wang HF, Tan MS, Gao Q, Qin H, Zhang YD, Tan L (2014b) Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 85:121–130
Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV (2014) Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun 5:3496
Kaushik S, Cuervo AM (2012) Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22(8):407–417
Kessing LV, Forman JL, Andersen PK (2010) Does lithium protect against dementia? Bipolar Disord 12(1):87–94
Kim YM, Jung CH, Seo M, Kim EK, Park JM, Bae SS, Kim DH (2015) mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell 57(2):207–218
Koga H, Cuervo AM (2011) Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol Dis 43(1):29–37
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432(7020):1032–1036
Lai AY, McLaurin J (2012) Inhibition of amyloid-beta peptide aggregation rescues the autophagic deficits in the TgCRND8 mouse model of Alzheimer disease. Biochim Biophys Acta 1822(10):1629–1637
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141:1146–1158
Lee KM, Hwang SK, Lee JA (2013) Neuronal autophagy and neurodevelopmental disorders. Exp Neurobiol 22(3):133–142
Lee JK, Jin HK, Park MH, Kim BR, Lee PH, Nakauchi H, Carter JE, He X, Schuchman EH, Bae JS (2014) Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J Exp Med 211(8):1551–1570
Lee HR, Shin HK, Park SY, Kim HY, Bae SS, Lee WS, Rhim BY, Hong KW, Kim CD (2015) Cilostazol upregulates autophagy via SIRT1 activation: reducing amyloid-β peptide and APP-CTFβ levels in neuronal cells. PLoS ONE 10(8):e0134486
Li M, Sun M, Liu Y, Yu J, Yang H, Fan D, Chui D (2010) Copper downregulates neprilysin activity through modulation of neprilysin degradation. J Alzheimer’s Dis 19(1):161–169
Li L, Zhang S, Zhang X, Li T, Tang Y, Liu H, Yang W, Le W (2013) Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-β pathology in a mouse model of Alzheimer’s disease. Curr Alzheimer Res 10:433–441
Lim F, Hernández F, Lucas JJ, Gómez-Ramos P, Morán MA, Avila J (2001) FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and tau filaments in forebrain. Mol Cell Neurosci 18(6):702–714
Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW (2003) Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol 32(9):1091–1105
Ling D, Magallanes M, Salvaterra PM (2014) Accumulation of amyloid-like Aβ1-42 in AEL (autophagy–endosomal–lysosomal) vesicles: potential implications for plaque biogenesis. ASN Neuro 6(2):95–109
Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, Yuan J (2010) Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci USA 107(32):14164–14169
Liu C, Gao Y, Barrett J, Hu B (2010) Autophagy and protein aggregation after brain ischemia. J Neurochem 115(1):68–78
Macdonald A, Briggs K, Poppe M, Higgins A, Velayudhan L, Lovestone S (2008) A feasibility and tolerability study of lithium in Alzheimer’s disease. Int J Geriatr Psychiatry 23(7):704–711
Maday S, Holzbaur EL (2014) Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell 30(1):71–85
Maday S, Wallace KE, Holzbaur EL (2012) Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 196:407–417
Majumder S, Richardson A, Strong R, Oddo S (2011) Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 6(9):e25416
Mariño G, Madeo F, Kroemer G (2011) Autophagy for tissue homeostasis and neuroprotection. Curr Opin Cell Biol 23(2):198–206
Mazure NM, Pouysségur J (2010) Hypoxia-induced autophagy: Cell death or cell survival? Curr Opin Cell Biol 22(2):177–180
Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S (2008) Abeta-degrading enzymes in Alzheimer’s disease. Brain Pathol 18(2):240–252
Mizushima N (2005) A(beta) generation in autophagic vacuoles. J Cell Biol 171(1):15–17
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15(3):1101–1111
Monastyrska I, Rieter E, Klionsky DJ, Reggiori F (2009) Multiple roles of the cytoskeleton in autophagy. Biol Rev Camb Philos Soc 84(3):431–448
Moreau K, Fleming A, Imarisio S, Lopez Ramirez A, Mercer JL, Jimenez-Sanchez M, Bento CF, Puri C, Zavodszky E, Siddiqi F, Lavau CP, Betton M, O’Kane CJ, Wechsler DS, Rubinsztein DC (2014) PICALM modulates autophagy activity and tau accumulation. Nat Commun 5:4998
Nalivaeva NN, Belyaev ND, Turner AJ (2009) Sodium valproate: an old drug with new roles. Trends Pharmacol Sci 30(10):509–514
Nilsson P, Saido TC (2014) Dual roles for autophagy: degradation and secretion of Alzheimer’s disease Aβ peptide. BioEssays 36(6):570–578
Nilsson P, Sekiguchi M, Akagi T, Izumi S, Komori T, Hui K, Sörgjerd K, Tanaka M, Saito T, Iwata N, Saido TC (2015) Autophagy-related protein 7 deficiency in amyloid β (Aβ) precursor protein transgenic mice decreases Aβ in the multivesicular bodies and induces Aβ accumulation in the Golgi. Am J Pathol 185:305–313
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, CataldoA Cuervo AM (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. JNeuropathol Exp Neurol 64:113–122
Nunes PV, Forlenza OV, Gattaz WF (2007) Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br J Psychiatry 190:359–360
Omata Y, Lim YM, Akao Y, Tsuda L (2014) Age-induced reduction of autophagy-related gene expression is associated with onset of Alzheimer’s disease. Am J Neurodegener Dis 3(3):134–142
Papp D, Kovács T, Billes V, Varga M, Tarnóci A, Hackler L Jr, Puskás LG, Liliom H, Tárnok K, Schlett K, Borsy A, Pádár Z, Kovács AL, Hegedűs K, Juhász G, Komlós M, Erdős A, Gulyás B, Vellai T (2015) AUTEN-67, an autophagy-enhancing drug candidate with potent antiaging and neuroprotective effects. Autophagy 27:0
Park JS, Kim DH, Yoon SY (2016) Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep (Epub ahead of print)
Pei JJ, Hugon J (2008) mTOR-dependent signalling in Alzheimer’s disease. J Cell Mol Med 12(6B):2525–2532
Peric A, Annaert W (2015) Early etiology of Alzheimer’s disease: Tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol 129(3):363–381
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest 118(6):2190–2199
Rodríguez-Martín T, Cuchillo-Ibáñez Noble W, Nyenya F, Anderton BH, Hanger DP (2013) Tau phosphorylation affects its axonal transport and degradation. Neurobiol Aging 34(9):2146–2157
Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A (2005) Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 1(1):11–22
Rubinsztein DC, Mariño G, Kroemer G (2011) Autophagy and aging. Cell 146(5):682–695
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15(7):741–750
Sanchez-Varo R, Trujillo-Estrada L, Sanchez-Mejias E, Torres M, Baglietto-Vargas D, Moreno-Gonzalez I, De Castro V, Jimenez S, Ruano D, Vizuete M, Davila JC, Garcia-Verdugo JM, Jimenez AJ, Vitorica J, Gutierrez A (2012) Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol 123(1):53–70
Sarkar S, Rubinsztein DC (2008) Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS J 275(17):4263–4270
Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC (2007a) Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 282(8):5641–5652
Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O’Kane CJ, Schreiber SL, Rubinsztein DC (2007b) Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol 3(6):331–338
Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC (2009) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 16(1):46–56
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26(7):1749–1760
Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J (2006) Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 281(20):14474–14485
Son SM, Jung ES, Shin HJ, Byun J, Mook-Jung I (2012) Aβ-induced formation of autophagosomes is mediated by RAGE-CaMKKβ-AMPK signaling. Neurobiol Aging 33(5):1006.e11–1006.e23
Song Z, Zhao D, Yang L (2013) Molecular mechanisms of neurodegeneration mediated by dysfunctional subcellular organelles in transmissible spongiform encephalopathies. Acta Biochim Biophys Sin (Shanghai) 45(6):452–464
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V (2010) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 5(4):e9979
Su JH, Cummings BJ, Cotman CW (1993) Identification and distribution of axonal dystrophic neuritis in Alzheimer’s disease. Brain Res 625(2):228–237
Sun M, Zhou T, Zhou L, Chen Q, Yu Y, Yang H, Zhong K, Zhang X, Xu F, Cai S, Yu A, Zhang H, Xiao R, Xiao D, Chui D (2012) Formononetin protects neurons against hypoxia-induced cytotoxicity through upregulation of ADAM10 and sAβPPα. J Alzheimer’s Dis 28(4):795–808
Suzuki K, Terry RD (1967) Fine structural localization of acid phosphatase in senile plaques in Alzheimer’s presenile dementia. Acta Neuropathol 8:276–284
Sweetlove M (2012) Phase III CONCERT trial of latrepirdine. Negative results. Pharm Med 26(2):113–115
Tang YP (2003) Genetic studies in Alzheimer’s disease. Dialog Clin Neurosci 5(1):17–26
Tian Y, Chang JC, Greengard P, Flajolet M (2014) The convergence of endosomal and autophagosomal pathways: implications for APP-CTF degradation. Autophagy 10(4):694–696
Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18(21):4153–4170
Wang HC, Zhang T, Kuerban B, Jin YL, Le W, Hara H, Fan DS, Wang YJ, Tabira T, Chui DH (2015a) Autophagy is involved in oral rAAV/Aβ vaccine-induced Aβ clearance in APP/PS1 transgenic mice. Neurosci Bull 31(4):491–504
Wang T, Martin S, Papadopulos A, Harper CB, Mavlyutov TA, Niranjan D, Glass NR, Cooper-White JJ, Sibarita JB, Choquet D, Davletov B, Meunier FA (2015b) Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type a. J Neurosci 35(15):6179–6194
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O’Kane CJ, Floto RA, Rubinsztein DC (2008) Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol 4:295–305
Wu T, Wang MC, Jing L, Liu ZY, Guo H, Liu Y, Bai YY, Cheng YZ, Nan KJ, Liang X (2015) Autophagy facilitates lung adenocarcinoma resistance to cisplatin treatment by activation of AMPK/mTOR signaling pathway. Drug Des Devel Ther 9:6421–6431
Xie Y, Zhou B, Lin MY, Sheng ZH (2015) Progressive endolysosomal deficits impair autophagic clearance beginning at early asymptomatic stages in fALS mice. Autophagy 11(10):1934–1936
Yadav RB, Burgos P, Parker AW, Iadevaia V, Proud CG, Allen RA, O’Connell JP, Jeshtadi A, Stubbs CD, Botchway SW (2013) mTOR direct interactions with Rheb-GTPase and raptor: sub-cellular localization using fluorescence lifetime imaging. BMC Cell Biol 14:3
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, LeeJH Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Näslund J, Mathews PM, Cataldo AM, Nixon RA (2005) Macroautophagy: a novel beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 171:87–98
Yu J, Sun M, Chen Z, Lu J, Liu Y, Zhou L, Xu X, Fan D, Chui D (2010) Magnesium modulates amyloid-beta protein precursor trafficking and processing. J Alzheimers Dis 20(4):1091–1106
Yu Y, Zhou L, Sun M, Zhou T, Zhong K, Wang H, Liu Y, Liu X, Xiao R, Ge J, Tu P, Fan DS, Lan Y, Hui C, Chui D (2012) Xylocoside G reduces amyloid-β induced neurotoxicity by inhibiting NF-κB signaling pathway in neuronal cells. J Alzheimer’s Dis 30(2):263–275
Zhang YD, Zhao JJ (2015) TFEB participates in the Aβ-induced pathogenesis of Alzheimer’s disease by regulating the autophagy-lysosome pathway. DNA Cell Biol 34(11):661–668
Zhou F, van Laar T, Huang H, Zhang L (2011) APP and APLP1 are degraded through autophagy in response to proteasome inhibition in neuronal cells. Protein Cell 2(5):377–383
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334(6056):678–683
Author information
Authors and Affiliations
Corresponding author
Additional information
Qian Li and Yi Liu have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Li, Q., Liu, Y. & Sun, M. Autophagy and Alzheimer’s Disease. Cell Mol Neurobiol 37, 377–388 (2017). https://doi.org/10.1007/s10571-016-0386-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-016-0386-8