
Abstract
There is accumulating evidence suggesting that changes in brain perfusion are present long before the clinical symptoms of Alzheimer’s disease (AD), perhaps even before amyloid-β accumulation or brain atrophy. This evidence, consistent with the vascular hypothesis of AD, implicates cerebral blood flow (CBF) in the pathogenesis of AD and suggests its utility as a biomarker of preclinical AD. The extended preclinical phase of AD holds particular significance for disease modification, as treatment would likely be most effective in this early asymptomatic stage of disease. This highlights the importance of identifying reliable and accurate biomarkers of AD that can differentiate normal aging from preclinical AD prior to clinical symptom manifestation. Cerebral perfusion, as measured by arterial spin labeling magnetic resonance imaging (ASL-MRI), has been shown to distinguish between normal controls and adults with AD. In addition to demonstrating diagnostic utility, CBF has shown usefulness as a tool for identifying those who are at risk for AD and for predicting subtle cognitive decline and conversion to mild cognitive impairment and AD. Taken together, this evidence not only implicates CBF as a useful biomarker for tracking disease severity and progression, but also suggests that ASL-measured CBF may be useful for identifying candidates for future AD treatment trials, especially in the preclinical, asymptomatic phases of the disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD), characterized by progressive cognitive decline, is the most common form of dementia (Alzheimer’s Association 2014). With a global prevalence of 5 million that is expected to quadruple by 2040, it is clear that AD is a growing world-wide health concern (Hebert et al. 2013). Recent advances in AD research have shown that neuropathological changes begin years, if not decades, before the clinical manifestation of this disease becomes evident (Morris 2005). This extended preclinical phase may hold particular significance for disease modification, as potential therapies are likely to be most effective in the early, asymptomatic stages of AD. As such, increased research efforts have been aimed at identifying reliable and accurate biomarkers of AD that can differentiate normal aging from preclinical AD prior to clinical symptom manifestation.
By definition, a biomarker is a characteristic that is objectively measured and evaluated as an indicator of pathologic processes, and can be used as a diagnostic tool for identifying patients, staging a disease, indicating disease prognosis, or for predicting and monitoring clinical response to an intervention (Biomarkers Definitions Working Group 2001). Within the context of AD, it has been proposed that an ideal biomarker should detect a fundamental feature of AD neuropathology, be validated in neuropathologically confirmed cases, and have high diagnostic sensitivity and specificity (<80 %). Moreover, a useful biological marker of AD should be reliable, reproducible, non-invasive, simple to perform, and inexpensive (Growdon et al. 1998; Frank et al. 2003). Our ability to identify effective biomarkers in AD is largely dependent on our knowledge of its pathogenesis. Although AD etiology has become increasingly clear, the exact cause of this disease remains unknown. While a number of hypotheses exist, much of the literature favors either the ‘amyloid cascade hypothesis’ or the ‘vascular hypothesis’ (Kelleher and Soiza 2013). The amyloid cascade hypothesis describes a sequence of biomarker changes that occur along a continuum from normal aging to dementia. Within this model, amyloid biomarkers are implicated as the primary cause of AD and the first to become abnormal, followed much later by tau-related markers of neural injury and neurodegeneration, leading to structural brain changes, subtle cognitive decline, mild cognitive impairment (MCI), and eventually dementia (Jack et al. 2010). However, evidence suggesting that amyloid-β may be necessary but not sufficient for the development of AD, and its questionable association with cognitive decline has led to increasing criticism of the amyloid cascade hypothesis. This model also appears incomplete, as it does not consider vascular dysfunction in the etiology of AD, despite accumulating evidence supporting its contribution. Within the vascular model, pathology results from cerebral hypoperfusion, leading to glial and neuronal damage, and eventually resulting in neurodegeneration (e.g., deposition of amyloid-β and tau), cognitive decline, and dementia (Kelleher and Soiza 2013). Although currently accepted biomarkers of AD include those related to amyloid, tau, cerebral metabolism, cerebral blood flow (CBF) or perfusion, and brain atrophy, etiological studies of AD suggest that amyloid and perfusion markers may hold the most promise in very early disease detection. The recent evidence supporting a strong relationship between cerebral perfusion and disease progression further implicates CBF as a useful biomarker of preclinical AD (Binnewijzend et al. 2013; Wang 2014; Wierenga et al. 2014; Binnewijzend et al. 2015).
The term perfusion is generally used to describe the process by which arterial blood is delivered to a capillary bed in the tissue. Similarly, CBF, is used to describe the “rate of delivery of arterial blood to the capillary bed of a particular mass of tissue” and it is measured in milliliters of blood per 100 g of tissue per minute (Buxton 2009). Average CBF values in humans approximate 50 mL/100 g per minute, with gray matter blood flow being approximately three times higher than white matter flow (Buxton 2009). The building blocks of cellular energy needed by the brain, oxygen and glucose, are delivered by a constant supply of CBF, since neither of them is stored in appreciable amounts as reserve (Golanov and Reis 1997). The connection between neural activity, energy metabolism, and blood flow is the foundation of functional neuroimaging measures. CBF can be measured by a variety of methods, the most common being positron emissions tomography (PET), single-photon emission computed tomography (SPECT), and arterial spin labeling (ASL) magnetic resonance imaging (MRI). Over the past decades, neuroimaging studies have demonstrated that CBF is altered in AD and AD risk (Wierenga et al. 2014).
The following review discusses the utility of CBF as a tool to differentiate AD from normal aging and to predict cognitive decline and conversion to MCI. When available, we focus on studies assessing cerebral perfusion with ASL, a non-invasive MRI technique that magnetically labels arterial water and uses it as an endogenous tracer to measure CBF. Due to its ability to quantify CBF, ASL-MRI has the potential to accurately estimate the magnitude and location of neural function (Liu and Brown 2007) and it is a non-invasive and easily repeatable method with good reliability and reproducibility (Parkes et al. 2004). This review also examines whether behavioral strategies can modify CBF to prevent or delay the development of AD and concludes by discussing the future of ASL-MRI as a biomarker of preclinical AD.
CBF in Aging
Resting CBF in Aging
Advancing age is the greatest risk factor for developing AD, with most patients diagnosed at age 65 or older (Alzheimer’s Association 2014). There is also a well-established association between elevated blood pressure and reduced CBF with age, with an overall reduction in CBF of between 18 and 28 % in older adults compared to young adults (Singh et al. 2010; Popa-Wagner et al. 2013). Evidence suggests that this decline in CBF is rapid rather than gradual, with decreases of approximately 40 % in adults compared to children (Biagi et al. 2007), and cognitively healthy adults (20–67 years) showing global cerebral perfusion reductions of 0.45 % per year (Parkes et al. 2004). Hypoperfusion tends to be widespread, affecting frontal, temporal (e.g., superior temporal cortex, hippocampus, parahippocampal gyrus), parietal (e.g., precuneus), and subcortical regions (e.g., thalamus, caudate) (Parkes et al. 2004; Bangen et al. 2009; Lee et al. 2009; Wierenga et al. 2013). However, Lee et al. (2009) report significant variability of CBF within a sample of cognitively intact older adults, with individuals showing relative increases and decreases compared to the group average in multiple regions, including the anterior and posterior cingulate, precuneus, caudate, and superior temporal cortex, suggesting these regions may be particularly sensitive to vascular changes in aging. These regions overlap with the default mode network, a network of brain regions active during rest and intrinsic processing and inhibited during cognitive involvement, that has demonstrated altered metabolic processing in aging and AD (Greicius et al. 2004), supporting the notion that vascular function may contribute to these early functional changes.
Functional CBF in Aging
Functional CBF, or the change in CBF from baseline as a result of cognitive task performance, may be a more sensitive measure of early functional brain changes given some advantages over the more typically used blood oxygen level dependent (BOLD) signal. These advantages include the direct assessment of cerebrovascular function, measurement of a well-defined physiological quantity (e.g., units of mL of blood per 100 g of tissue per minute), and better localization of functional activity to the arterial side of the vascular tree, as the ASL perfusion signal is well localized to the capillary bed (Lee et al. 2001; Liu and Brown 2007). For example, cognitively intact older adults showed increased percent change (%Δ) in CBF in the medial temporal lobe (MTL) during a picture encoding task compared to their younger counterparts, in the context of lower resting CBF at baseline (Restom et al. 2007; Bangen et al. 2009). Performance on primary measures of learning and memory did not differ between the groups, suggesting that the increase in %Δ in CBF may reflect age-related compensatory mechanisms to accommodate for lower resting perfusion (Bangen et al. 2009). Moreover, memory abilities were associated with %Δ in CBF in the MTL during picture encoding (Bangen et al. 2009), but not with the simultaneously collected %Δ in BOLD, suggesting that CBF may be more tightly linked to cognition than the BOLD response in older adults.
Resting CBF in Genetic Risk for AD
The apolipoprotein (APOE) gene, thought to play a role in cerebrovascular integrity (Tiraboschi et al. 2004; Yip et al. 2005), is considered the single most important genetic factor in AD (Coon et al. 2007). Possession of the APOE ε4 allele increases AD risk 3–8 fold, in a dose dependent fashion, and significantly lowers the age of disease onset (Saunders et al. 1993; Corder et al. 1993; Coon et al. 2007). Cognitively normal APOE ε4 carriers demonstrate both increased and decreased CBF relative to normal controls. Across studies, perfusion decreases have been observed in the MTL, left lingual gyrus, and the precuneus, whereas increases have been reported in the frontal, parietal, and temporal cortex, as well as the anterior/posterior cingulate cortex and cerebellum (Bangen et al. 2009; Thambisetty et al. 2010; Wierenga et al. 2013). Interestingly, the effect of APOE genotype on CBF appears to be mediated by age, with older adults displaying more areas of hypoperfusion and younger adults displaying more areas of hyperperfusion (Wierenga et al. 2013). Moreover, in young APOE ε4 carriers, increased CBF appears to be associated with better executive functioning, perhaps suggesting an early compensatory mechanism (Wierenga et al. 2013). Similarly, the finding that older adults at risk for AD demonstrate abnormally low CBF may indicate a breakdown of this early compensatory mechanism. We have suggested that the interaction between age and APOE genotype reflects the differential impact of the genotype on cerebrovascular function across the lifespan. There is also evidence that disease severity may mediate the relationship between APOE and CBF, with differing patterns of CBF alteration in ε4 carriers with AD and those with MCI, thought to reflect the prodromal stage of AD. Specifically, APOE ε4 carriers with MCI showed regional hyperperfusion in the parahippocampal and bilateral cingulate gyri, whereas APOE ε4 carriers with AD displayed hypoperfusion in the right frontal and occipital lobes (Kim et al. 2013). Taken together, the evidence presented here suggests that CBF is altered in genetic risk for AD. Moreover, the early hyperperfusion and later hypoperfusion observed in carriers of the ε4 allele suggests the presence of a vascular regulatory mechanism, likely in response to an increased need for oxygen and glucose or alterations in brain metabolism.
Resting CBF in Familial Risk
First degree familial history of AD has been shown to increase risk of developing AD by as much as 10-fold (Silverman et al. 2005; Jarvik et al. 2008). Furthermore, this risk factor is associated with alterations in brain perfusion, as cognitively normal middle-aged adults with a familial history of AD demonstrate decreased CBF in right superior and middle frontal cortices, compared to those without a familial history (Okonkwo et al. 2014). When looking specifically at maternal history of AD, there appears to be a particular risk of hypoperfusion in midlife, as evidenced by reduced CBF in the hippocampus and parietofrontal regions in those who have a mother with AD, compared to those with either no history or only a paternal history of AD (Okonkwo et al. 2014). Interestingly, the effect of genetic and familial risk factors appears to be additive, as APOE ε4 carriers who also have a familial history of AD display diminished perfusion in frontal regions, supramarginal gyri, and hippocampi compared to those with no risk factors for AD (Fleisher et al. 2009; Okonkwo et al. 2014). Overall, studies of CBF in those with a familial history of AD implicate CBF as an early marker of brain changes in those at risk for developing AD.
Resting CBF in Vascular Risk
Elevated vascular risk burden, or the presence of vascular risk factors (e.g., diabetes, hypertension, heart disease, and current smoking), is associated with an increased risk of cognitive decline and AD (Luchsinger et al. 2005; Austin et al. 2011; Gorelick et al. 2011; Bangen et al. 2014). Moreover, AD risk increases with the number of coexisting vascular risk factors present (Luchsinger et al. 2005). Studies investigating the link between vascular risk and CBF suggest that increased vascular risk factors are associated with a decrease in resting CBF over time, and with greater %Δ CBF in response to functional tasks (Bangen et al. 2009; Muller et al. 2012). Recent evidence also suggests that vascular risk burden might moderate the relationship between increasing age and reduced CBF, with a relationship between advancing age and reduced CBF only observed among those possessing multiple vascular risk factors. Moreover, only those with elevated vascular risk showed an association between reduced CBF and poorer cognitive performance (Bangen et al. 2014). Taken together, these studies demonstrate that CBF may be the mechanism by which elevated vascular risk burden impacts brain health and cognition, suggesting its utility in identifying those at risk for AD.
CBF in Cognitive Risk
Resting CBF and Cognitive Performance
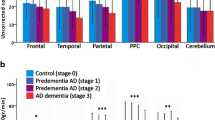
An examination of the relationship between CBF and cognitive performance may further elucidate the role of CBF as a biomarker of preclinical AD. Consistent with theories of neural efficiency, lower temporal and parietal CBF has been associated with better attention in younger adults, and lower CBF in cognitively healthy older adults has been related to better tonic alertness (Bertsch et al. 2009). Other studies have shown that increased CBF in the posterior cingulate, precuneus, and superior parietal lobe in normal aging is associated with improved immediate memory and working memory function (Okonkwo et al. 2014), and increased CBF in the hippocampus is correlated with improved spatial memory only in older adults (Heo et al. 2010). Similarly, in MCI, increased MTL CBF has been shown to be correlated with better memory performance, whereas decreased CBF was linked to visuospatial and general cognitive dysfunction (Bangen et al. 2012; Yoon et al. 2012). Our group found that improved verbal memory was correlated with an increase in resting CBF in the medial frontal gyrus in MCI APOE ε4 carriers and elevated CBF in the parahippocampal gyrus in APOE ε4 cognitively normal adults (Fig. 1). These results highlight the role of CBF in memory function and are largely consistent with a compensatory response that may shift from posterior to anterior cortices as the disease progresses to overcome pathologic encroachments (Wierenga et al. 2012). Furthermore, there is evidence that resting perfusion in AD patients correlates negatively with disease severity, as measured by the MMSE (Sandson et al. 1996; Alsop et al. 2008). Together, these findings suggest that CBF plays a role in maintaining cognitive function and that the relationship between CBF and cognition may be modified by age and genotype.

Modified from Wierenga et al. (2012). Analysis of variance results demonstrating an interaction of cognitive status (cognitively normal, MCI) and APOE genotype (ε3, ε4) for CBF with corresponding graphical presentation of significant CBF differences. Increased CBF in the left parahippocampal and fusiform gyrus was correlated only with verbal memory for cognitively normal (CN) ε4 adults and increased CBF in the left medial frontal gyrus was only correlated with verbal memory for MCI ε4 adults. This suggests a compensatory response that may shift from posterior to anterior cortices to overcome pathologic encroachments as the disease progresses. Results are thresholded and clustered (protecting a whole-brain voxel-wise P < 0.05; red P < 0.05, orange P < 0.025, yellow P < 0.01). Error bars represent the standard error of the mean. Results are overlaid onto sagittal slices of a high-resolution anatomical image averaged across all participants (L left, R right, C cluster, PHG/FG parahippocampal gyrus/fusiform gyrus) (Color figure online)
Resting CBF in Mild Cognitive Impairment
Mild cognitive impairment (MCI), or the presence of cognitive impairment in the absence of significant functional impairment, is arguably the most well-characterized risk factor for AD (Marra et al. 2011). Across studies, MCI is associated with both increases and decreases in CBF, with increases in the MTL, anterior cingulate, insula, putamen, the left hippocampus, right amygdala, ventral striatum, and the basal ganglia (Dai et al. 2009; Alexopoulos et al. 2012; Wolk and Detre 2012; Wierenga et al. 2014), and decreases that reflect a lateral temporo-parietal-frontal pattern, extending to the MTL, posterior cingulate, and precuneus (Johnson et al. 2005; Xu et al. 2007; Dai et al. 2009; Bangen et al. 2012; Alexopoulos et al. 2012; Wolk and Detre 2012; Wierenga et al. 2014). The evidence of both increased and decreased perfusion in those with MCI is suggestive of early neurodegeneration resulting in CBF dysregulation that may reflect changing metabolic demands to maintain cognitive function (Ostergaard et al. 2013). Overall, the consistent presence of regional perfusion changes in MCI suggests that CBF can be used to identify those at cognitive risk for AD.
In addition to being able to identify those with concurrent MCI, there is evidence supporting the utility of CBF in predicting who is likely to convert to MCI. Evidence from a 2013 PET study suggests that those who later develop MCI demonstrate a greater increase in perfusion over time in orbitofrontal, medial frontal, and anterior cingulate regions, compared to those who do not develop MCI, and that MCI converters show greater CBF decreases in parietal, temporal, and thalamic regions (Beason-Held et al. 2013). A 2015 study also found that, compared to cognitively stable participants, those who showed subtle cognitive decline at an 18-month follow-up demonstrated reduced CBF at baseline, most notably in the posterior cingulate cortex, an area commonly associated with AD. Furthermore, the observed pattern of reduced CBF was similar to that of patients who already had MCI at baseline (Xekardaki et al. 2015) (Fig. 2). The authors suggest that normal cognitive function despite reduced CBF may reflect neurocognitive reserve. More specifically, cognitively normal controls who later show subtle cognitive decline likely already have perfusion alterations at baseline comparable to those with MCI, but they appear cognitively normal due to cognitive reserve that has not yet been depleted (Xekardaki et al. 2015). Together, this evidence suggests that CBF alteration may take place years before the onset of cognitive symptoms in those who eventually convert to MCI. Furthermore, it demonstrates that CBF can be used to identify those with MCI and predict cognitive decline and conversion to MCI.

Receiver operating characteristic curve (ROC) demonstrating the ability of CBF to differentiate cognitive decliners from those who remained cognitively stable. ASL relative CBF in posterior cingulate cortex (PCC) enabled discrimination of deteriorated cognitive function (dCON) (P < .001) from stable cognitive function (sCON); however, there was no difference between deteriorated cognitive function and mild cognitive impairment (not shown). Published in: Xekardaki et al. 2015
Functional CBF in Mild Cognitive Impairment
Very few studies have investigated CBF in response to functional tasks among those at risk for AD, and existing results are mixed. One study showed that in adults with MCI, APOE ε4 carriers demonstrated greater %Δ in CBF in the right MTL compared to non-carriers during a memory encoding task (Bangen et al. 2012). Other studies have found decreased response in the MTL in response to memory encoding tasks in adults with MCI, with these reductions ranging from 22 to 30 %, compared to controls (Xu et al. 2007; Fleisher et al. 2009). It has been suggested that this decreased response is due to differences in resting state, given that both high- and low-risk groups tended to show similar levels of perfusion when accounting for resting state CBF levels (Fleisher et al. 2009). Taken together, these results suggest that the ability to modulate CBF in response to memory tasks may be reduced in individuals at risk for AD. They also implicate the measurement of functional CBF response as a possible early marker of brain changes in adults at cognitive risk for developing AD.
CBF in Preclinical AD Biomarker Risk
Well-validated AD biomarkers include elevated amyloid-β and/or tau-related proteins in the brain, cerebral hypometabolism, measures of brain atrophy, and measures of cognitive functioning. This multitude of AD biomarkers and their inter-relationships has made it difficult to determine the etiological importance of each independent biomarker. Furthermore, it has led to difficulties in establishing both additive and putative weight for these markers of AD. With this in mind, clinicians and researchers tend to rely on a combination of biomarkers in the detection of AD, MCI, and preclinical AD, rather than any one marker in isolation. The National Institutes of Aging–Alzheimer’s Association (NIA-AA) workgroup recently proposed a hypothetical staging framework for the classification of preclinical AD, defined as clinically normal individuals who are thought to be in the early asymptomatic stages of AD. Within this staging framework, classification of preclinical AD is largely based on biomarkers associated with the amyloid cascade hypothesis, including markers of amyloid-β accumulation, neuronal injury, and cognitive functioning (Sperling et al. 2011). Stage 1 can be operationally defined as asymptomatic amyloidosis, Stage 2 as asymptomatic amyloidosis + “downstream” neurodegeneration as evidenced by positive cerebrospinal fluid (CSF) amyloid-β and tau with no evidence of subtle cognitive change, and Stage 3 as amyloidosis + neuronal injury + subtle cognitive/behavioral decline not meeting the criteria for MCI. Although changes in brain perfusion are not included in this staging criteria, the authors acknowledge that other biomarkers of AD may show changes prior to those utilized within their model, especially in those at genetic risk for AD by way of the APOE gene (Sperling et al. 2011). Exploring CBF alterations within this newly defined preclinical stage of AD would likely provide further evidence for its usefulness as a preclinical biomarker and could argue for its inclusion in the current NIA-AA staging framework. Unfortunately, such studies are lacking, with AD-related perfusion studies largely restricted to genetic and cognitive risk factors. To date, we are aware of no published studies directly exploring CBF in preclinical AD, as defined by this new set of diagnostic criteria. One study examined CBF changes in relation to the NIA-AA-based stages of AD; however, the authors collapsed the MCI and preclinical AD categories into one “predementia” stage, making interpretation within the NIA-AA-defined stages difficult. Although results showed an association between decreased CBF and advancing stages of AD, across the continuum from normal aging to AD, they showed no CBF differences between predementia patients and controls (Binnewijzend et al. 2015). While these results support CBF as a biomarker of AD progression, they suggest that CBF changes are not directly related to amyloid-β deposition. Further CBF research distinguishing between the NIA-AA-based preclinical and MCI groups is needed to confirm these findings.
Understanding how CBF relates to the NIA-AA based biomarkers of AD may also provide information as to its usefulness as a preclinical marker of AD. While we are aware of no studies exploring the relationship between ASL and MRI measured CBF and CSF biomarkers of AD, a 2012 SPECT study showed that high levels of CSF phosphorylated tau (P-tau) and total tau (T-tau) were associated with decreased CBF in the right superior posterior medial frontal lobe, whereas high levels of P-tau were also correlated with CBF increases in the left frontotemporal border zone area (Stomrud et al. 2012). Although this study found no relationship between CBF and CSF β-amyloid1–42 (Aβ42), results from both human and animal PET studies consistently demonstrate that higher PET measured amyloid-β load is related to both increases and decreases in regional CBF (Sojkova et al. 2008; Mattsson et al. 2014; Maier et al. 2014). Across studies, amyloid-related CBF decreases have been found in the precuneus, cingulate, supramarginal gyrus, thalamus, entorhinal cortex, hippocampus, midbrain, and inferior temporal cortex, while amyloid-related CBF increases have been found in the medial and inferior frontal, precuneus, and inferior parietal regions (Sojkova et al. 2008, 2011; McLaren et al. 2013; Mattsson et al. 2014). Consistent with studies examining amyloid-related cross sectional differences in CBF, those measuring longitudinal change in CBF show both greater decreases and increases in perfusion over time in non-demented older adults with high amyloid-β deposition compared to those with low deposition (Sojkova et al. 2008). A more recent study showed that elevated amyloid-β deposition was associated with hypoperfusion in several regions, and that this relationship was independent of diagnostic group (i.e., MCI, AD). Furthermore, results showed that, compared to normal controls, amyloid-β positive diagnostic groups demonstrated regional CBF decreases, with precuneus, entorhinal cortex, and hippocampus hypoperfusion in dementia, inferior parietal cortex hypoperfusion in late MCI and dementia, and inferior temporal cortex hypoperfusion in early and late MCI, whereas amyloid-β negative groups did not (Mattsson et al. 2014). Consistent with these findings, a 2015 study found a negative correlation between regional CBF and age that was only present in those with elevated amyloid-β deposition (Gietl et al. 2015). Taken together, these results suggest that preserved CBF in the absence of amyloid pathology could reflect healthy aging, whereas CBF reductions with age, in the presence of amyloid deposition, may reflect AD. Further, these results are also consistent with evidence of increased CBF at earlier stages of AD, followed by a phase of reduced CBF (Thambisetty et al. 2010; Ostergaard et al. 2013).
Overall, research exploring cerebral perfusion as it relates to amyloid and tau markers, suggests that CBF is sensitive to these biomarkers of preclinical AD and AD risk. Moreover, it has led to the proposal that perfusion changes may occur even before amyloid-β accumulation or other pathological signs are evident, and that early alterations in CBF should be worked into current staging frameworks of AD biomarker pathology (Smith et al. 1999; Bookheimer et al. 2000; Iadecola 2004; Ruitenberg et al. 2005; Knopman and Roberts 2010; Sheline et al. 2010; Wierenga et al. 2014) (Fig. 3). However, studies exploring ASL-MRI-measured CBF in relation to CSF biomarkers of AD are needed to confirm these findings and to better understand the relationship between changes in cerebral perfusion and specific biomarkers of preclinical AD.

Hypothetical model of the temporal ordering of physiological biomarkers of AD. This figure was modified by Wierenga et al. (2014) from Jack at al. (2010) to include early alterations in CBF in the sequence of biomarkers across the continuum from normal aging to MCI to AD. Direction of CBF alteration is not specified because, as reviewed here, both hyper and hypoperfusion reflect abnormality in different stages of cognitive decline
Can Behavioral Interventions Modify CBF to Improve Cognition and Prevent or Delay the Development of AD?
Given that general cognitive function and AD risk and progression are associated with CBF, the question remains as to whether CBF can be modified to optimize CBF levels. If so, does CBF modification positively impact the neurodegenerative process? Evidence shows that physical activity in cognitively normal older adults is correlated with healthy cognitive aging (Kramer and Erickson 2007; Prakash et al. 2015) and increases in CBF (Chapman et al. 2013; Maass et al. 2015), suggesting that physical activity might be a promising intervention for AD prevention (Rolland et al. 2010; Smith et al. 2010). For example, life-long aerobic exercise preserved baseline CBF in areas associated with AD and aging (Thomas et al. 2013), and a recent randomized controlled trial of exercise with cognitively normal older adults found that, compared to the control group, adults in the exercise group showed increased CBF in the anterior cingulate cortex with improved cognitive performance after 12 weeks (Chapman et al. 2013). Furthermore, there is evidence that the association between CBF and physical activity patterns may be modified by AD risk. We recently reported a relationship between sedentary time and CBF that was modified by genetic risk in the left hippocampus, such that APOE ε4 carriers showed higher CBF as a function of longer sedentary time relative to non-carriers. We believe this suggests that sedentary time may act as a behavioral risk factor possibly exacerbating the regulatory increases in CBF usually seen in APOE ε4 carriers (Zlatar et al. 2014). Preliminary work currently being collected by our research group suggests that there is a strong positive association between higher sedentary time (measured objectively via accelerometry) and higher levels of CSF tau. Similarly, work by Head and colleagues found that cognitively normal sedentary APOE ε4 carriers may be at augmented risk of cerebral amyloid deposition as measured by PET PIB (Head et al. 2012). These results highlight the importance of understanding how behavioral lifestyle factors may interact with genetic vulnerability for AD and possibly affect disease pathology and course.
Not only has physical activity been shown to change CBF, but cognitive enhancement interventions may also be a key behavioral tool to maintain CBF levels in older adults. Mozolic and colleagues demonstrated that cognitive training increased resting CBF in the prefrontal cortex of older adults, which has been implicated in age-related functional compensatory recruitment (Mozolic et al. 2010, 2011). This suggests that cognitive training might extend the utility of compensatory mechanisms, and perhaps preserve healthy cognition in adults at risk for AD (Cabeza 2002; Park and Reuter-Lorenz 2009; Mozolic et al. 2011). More intervention research studies are needed to determine if increased physical activity and/or cognitive training can modify CBF and in turn prevent conversion to MCI or AD.
Future Directions and Emerging Biomarkers
Calibrated fMRI estimates functional changes in cerebral metabolic rate of oxygen (CMRO2) by simultaneously acquiring BOLD and CBF data in response to a cognitive and hypercapnic challenge (Davis et al. 1998; Liu and Wong 2005). This is a promising technique with the potential to reduce some of the confounds of cerebrovascular dysfunction in the measurement of neural function. Recent applications of calibrated fMRI to aging generally conclude that the BOLD response can provide misleading reflections of underlying physiological changes [since the BOLD signal reflects local changes in deoxyhemoglobin content, which in turn exhibits a complex dependence on changes in CBF, cerebral blood volume (CBV), and (CMR02) (Buxton 2009)] and that a calibrated approach provides a more quantitative reflection of underlying metabolic changes than the BOLD response alone (Ances et al. 2009; Hutchison et al. 2013).
Measuring resting CBF in conjunction with hypercapnia may provide a more comprehensive assessment of vascular function. For example, a measure of cerebrovascular reactivity (CVRh), or the amount of vessel restriction and relaxation, can be obtained by combining a baseline resting CBF scan with end tidal CO2 produced by conditions of hypercapnia, and is a physiological parameter showing great clinical potential as a new hemodynamic marker and indicator of cerebrovascular health (Bulte et al. 2009; Hajjar et al. 2010; Villien et al. 2013). Like CBF, CVRh is able to predict cognitive decline and to distinguish between different stages of the AD process. Furthermore, reduced CVRh is associated with hypertension (Hajjar et al. 2010), cognitive decline (Pfefferkorn et al. 2001; Silvestrini et al. 2006; Vicenzini et al. 2007; Cantin et al. 2011; Zavoreo et al. 2013), and is more sensitive to vascular burden as measured by the Framingham Stroke Risk Profile than resting CBF or brain volume (Glodzik et al. 2011). In fact, results from a 2015 study suggest that APOE ε4 carriers have lower CVRh than non-carriers and that the cognitive decline associated with the ε4 allele is attributable to lower CVRh (Hajjar et al. 2015).
Another measure showing predictive value in AD is CBF velocity (CBFv), which may distinguish between normal controls, individuals at genetic risk, adults with MCI, and AD patients (Maalikjy Akkawi et al. 2003, 2005; Sun et al. 2007; Claassen et al. 2009). Similarly, research looking at flow territory asymmetry, using vessel encoding ASL (VE-ASL), suggests that it is a promising predictor of cognitive decline, and should be the focus of further investigation (Donahue et al. 2014). Recent studies of acute changes in blood pressure and CBF in AD patients, using transcranial doppler ultrasound, have found that AD is associated with elevated global cerebrovascular resistance (CVR) and autoregulatory dysfunction (Pantano et al. 1984). We have also shown that a cerebrovascular resistance index (CVRi), calculated as the ratio of mean arterial blood pressure and regional CBF, is sensitive to the AD process and warrants further exploration (Nation et al. 2013). Specifically, we found elevated CVRi in very old cognitively normal, MCI and AD groups which was associated with poorer cognitive performance in regions that are vulnerable to cerebral small vessel disease or neurodegeneration in AD (Nation et al. 2013; Clark et al. 2015). Notably, CVRi can be derived from ASL-based perfusion estimates to assess regional differences in autoregulation, further highlighting the utility of this non-invasive technique. Taken together, these considerations point toward the importance of integrating CBF and hemodynamic vascular measures in studies examining AD risk.
Discussion and Conclusions
In summary, changes in cerebral perfusion are present long before the development of the clinical symptoms of AD. Evidence from this review is consistent with the vascular hypothesis of AD and supports the notion that vascular dysfunction plays a crucial role in the pathogenesis of AD and that changes in CBF are present even before other AD pathology is evident. This highlights the potential utility of ASL-measured CBF in helping to detect the preclinical phase of AD in conjunction with other biomarkers (Lee et al. 2011; Ostergaard et al. 2013). ASL-MRI is consistent with previous PET and SPECT studies measuring brain perfusion, yet it offers the advantage of being non-invasive, cost-effective, highly reliable, reproducible, and easily repeatable (Parkes et al. 2004). This suggests that ASL-MRI is the most promising tool to non-invasively study changes in CBF as they relate to normal and pathological aging, as well as to investigate differences in perfusion before and after behavioral interventions developed to target CBF as a protective mechanism to delay AD (Uchihashi et al. 2011; Chen et al. 2011; Takahashi et al. 2013; Heijtel et al. 2014). Despite its clear advantages, ASL-MRI has several methodological limitations related to its labeling sequences that may confine its widespread use.
The main labeling sequences used in ASL-MRI are continuous ASL, pulsed ASL, and pseudo-continuous ASL. Continuous ASL applies a constant gradient in the direction of flow, pulsed ASL uses a single radiofrequency pulse or a short train of pulses, and pseudo-continuous ASL employs both radiofrequency and gradient pulses that are applied as a train of short pulses. While continuous ASL shows a considerable advantage to pulsed ASL in signal-to-noise ratio, it suffers from magnetization transfer effects. Similarly, pseudo-continuous ASL improves both signal-to-noise ratio and magnetization effects, yet it demonstrates reduces tagging efficiency. Further limitations related to ASL labeling sequences include sensitivity to transit time effects, limited brain coverage, low spatial resolution, and less sensitivity to white matter CBF (Buxton 2009; Wong 2013). Taken together, these limitations may explain some of the seemingly contradictory findings in AD risk and AD-related ASL studies, including inconsistencies regarding hyperperfusion and hypoperfusion, and regional differences in CBF alteration between studies. Other possible explanations for disparate findings in AD-related CBF research include patient demographics (i.e., age and vascular risk burden (Bangen et al. 2014)), disease severity, and diagnostic criteria.
Given that the NIA-AA-based stages of preclinical AD are currently defined by the presence of other pathophysiological abnormalities, one could argue against the usefulness of CBF as a biomarker, as it could be viewed as a marker of a construct that is currently defined by the presence of other biomarker abnormalities. As discussed earlier, the NIA-AA acknowledges that there may be other biomarkers that show changes prior to the markers currently used in their staging framework. This notion is supported by research showing evidence of perfusion changes in at-risk individuals prior to the accumulation of amyloid-β or brain atrophy and suggests that CBF should be included in the NIA-AA-based stages of AD. When evaluating the potential usefulness of CBF as a biomarker of preclinical AD, it is also important to consider the fact that decreased CBF and related vascular dysfunction are not specific to AD, as they are also observed in other neurological conditions (Schuff et al. 2009; Firbank et al. 2011). Although this may limit the specificity of CBF and caution against its use in isolation, it does not preclude its usefulness in conjunction with other, more established biomarkers of AD. For example, amyloid-β and tau accumulation, brain atrophy, and cognitive dysfunction are also non-specific to the AD process, but they are considered to be AD-related biomarkers. This ultimately highlights the importance of establishing the specificity of CBF to distinguish between AD pathology and other vascular etiologies through research using consistent ASL protocols with different populations.
With the recently revised criteria put forth by the NIA for diagnosis of AD (McKhann et al. 2011), MCI (Albert et al. 2011), and preclinical AD (Sperling et al. 2011), it is clear that research will increasingly focus on the role of biomarkers in disease detection, diagnosis, and clinical outcome. Although the utility of CBF as a biomarker of AD risk and disease conversion is promising, future studies following the framework of the NIA-AA diagnostic criteria for preclinical AD (Sperling et al. 2011) are needed to firmly establish the role of CBF in the cascade of pathophysiological events that precipitate AD. An essential next step in this line of research will be to generate a microvascular biomarker profile and pathological signature of AD using multimodal neuropsychological and neuroimaging-derived assessments to determine its usefulness in predicting AD pathology.
References
Albert MS, DeKosky ST, Dickson D et al (2011) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 7:270–279. doi:10.1016/j.jalz.2011.03.008
Alexopoulos P, Sorg C, Förschler A et al (2012) Perfusion abnormalities in mild cognitive impairment and mild dementia in Alzheimer’s disease measured by pulsed arterial spin labeling MRI. Eur Arch Psychiatry Clin Neurosci 262:69–77. doi:10.1007/s00406-011-0226-2
Alsop DC, Casement M, de Bazelaire C et al (2008) Hippocampal hyperperfusion in Alzheimer’s disease. NeuroImage 42:1267–1274. doi:10.1016/j.neuroimage.2008.06.006
Alzheimer’s Association (2014) 2014 Alzheimer’s disease facts and figures. Alzheimers Dement 10:e47–e92. doi:10.1016/j.jalz.2014.02.001
Ances BM, Liang CL, Leontiev O et al (2009) Effects of aging on cerebral blood flow, oxygen metabolism, and blood oxygenation level dependent responses to visual stimulation. Hum Brain Mapp 30:1120–1132. doi:10.1002/hbm.20574
Austin BP, Nair VA, Meier TB et al (2011) Effects of hypoperfusion in Alzheimer’s disease. J Alzheimers Dis 26:123–133. doi:10.3233/JAD-2011-0010
Bangen KJ, Restom K, Liu TT et al (2009) Differential age effects on cerebral blood flow and BOLD response to encoding: associations with cognition and stroke risk. Neurobiol Aging 30:1276–1287. doi:10.1016/j.neurobiolaging.2007.11.012
Bangen KJ, Restom K, Liu TT et al (2012) Assessment of Alzheimer’s disease risk with functional magnetic resonance imaging: an arterial spin labeling study. J Alzheimers Dis JAD 31(Suppl 3):S59–S74. doi:10.3233/JAD-2012-120292
Bangen K, Nation D, Clark L et al (2014) Interactive effects of vascular risk burden and advanced age on cerebral blood flow. Front Aging Neurosci 6:159. doi:10.3389/fnagi.2014.00159
Beason-Held LL, Goh JO, An Y et al (2013) Changes in brain function occur years before the onset of cognitive impairment. J Neurosci Off J Soc Neurosci 33:18008–18014. doi:10.1523/JNEUROSCI.1402-13.2013
Bertsch K, Hagemann D, Hermes M et al (2009) Resting cerebral blood flow, attention, and aging. Brain Res 1267:77–88. doi:10.1016/j.brainres.2009.02.053
Biagi L, Abbruzzese A, Bianchi MC et al (2007) Age dependence of cerebral perfusion assessed by magnetic resonance continuous arterial spin labeling. J Magn Reson Imaging 25:696–702. doi:10.1002/jmri.20839
Binnewijzend MAA, Kuijer JPA, Benedictus MR et al (2013) Cerebral blood flow measured with 3D pseudocontinuous arterial spin-labeling MR imaging in Alzheimer disease and mild cognitive impairment: a marker for disease severity. Radiology 267:221–230. doi:10.1148/radiol.12120928
Binnewijzend MAA, Benedictus MR, Kuijer JPA et al (2015) Cerebral perfusion in the predementia stages of Alzheimer’s disease. Eur Radiol. doi:10.1007/s00330-015-3834-9
Biomarkers Definitions Working Group (2001) Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 69:89–95. doi:10.1067/mcp.2001.113989
Bookheimer SY, Strojwas MH, Cohen MS et al (2000) Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med 343:450–456. doi:10.1056/NEJM200008173430701
Bulte DP, Drescher K, Jezzard P (2009) Comparison of hypercapnia-based calibration techniques for measurement of cerebral oxygen metabolism with MRI. Magn Reson Med Off J Soc Magn Reson Med Soc Magn Reson Med 61:391–398. doi:10.1002/mrm.21862
Buxton RB (2009) Introduction to functional magnetic resonance imaging: principles and techniques, 2nd edn. Cambridge University Press, New York
Cabeza R (2002) Hemispheric asymmetry reduction in older adults: the HAROLD model. Psychol Aging 17:85–100
Cantin S, Villien M, Moreaud O et al (2011) Impaired cerebral vasoreactivity to CO2 in Alzheimer’s disease using BOLD fMRI. NeuroImage 58:579–587. doi:10.1016/j.neuroimage.2011.06.070
Chapman SB, Aslan S, Spence JS et al (2013) Shorter term aerobic exercise improves brain, cognition, and cardiovascular fitness in aging. Front Aging Neurosci 5:75. doi:10.3389/fnagi.2013.00075
Chen Y, Wolk DA, Reddin JS et al (2011) Voxel-level comparison of arterial spin-labeled perfusion MRI and FDG-PET in Alzheimer disease. Neurology 77:1977–1985. doi:10.1212/WNL.0b013e31823a0ef7
Claassen JAHR, Diaz-Arrastia R, Martin-Cook K et al (2009) Altered cerebral hemodynamics in early alzheimer disease: a pilot study using transcranial doppler. J Alzheimers Dis JAD 17:621–629. doi:10.3233/JAD-2009-1079
Clark LR, Nation DA, Wierenga CE et al (2015) Elevated cerebrovascular resistance index is associated with cognitive dysfunction in the very-old. Alzheimers Res Ther 7:3. doi:10.1186/s13195-014-0080-3
Coon KD, Myers AJ, Craig DW et al (2007) A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 68:613–618
Corder EH, Saunders AM, Strittmatter WJ et al (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923
Dai W, Lopez OL, Carmichael OT et al (2009) Mild cognitive impairment and alzheimers disease: patterns of altered cerebral blood flow at MR imaging. Radiology 250:856–866. doi:10.1148/radiol.2503080751
Davis TL, Kwong KK, Weisskoff RM, Rosen BR (1998) Calibrated functional MRI: mapping the dynamics of oxidative metabolism. Proc Natl Acad Sci USA 95:1834–1839
Donahue MJ, Hussey E, Rane S et al (2014) Vessel-encoded arterial spin labeling (VE-ASL) reveals elevated flow territory asymmetry in older adults with substandard verbal memory performance. J Magn Reson Imaging JMRI 39:377–386. doi:10.1002/jmri.24150
Firbank MJ, He J, Blamire AM et al (2011) Cerebral blood flow by arterial spin labeling in poststroke dementia. Neurology 76:1478–1484. doi:10.1212/WNL.0b013e318217e76a
Fleisher AS, Podraza KM, Bangen KJ et al (2009) Cerebral perfusion and oxygenation differences in Alzheimer’s disease risk. Neurobiol Aging 30:1737–1748. doi:10.1016/j.neurobiolaging.2008.01.012
Frank RA, Galasko D, Hampel H et al (2003) Biological markers for therapeutic trials in Alzheimer’s disease. In: Proceedings of the biological markers working group; NIA initiative on neuroimaging in Alzheimer’s disease. Neurobiol Aging 24:521–536
Gietl AF, Warnock G, Riese F et al (2015) Regional cerebral blood flow estimated by early PiB uptake is reduced in mild cognitive impairment and associated with age in an amyloid-dependent manner. Neurobiol Aging 36:1619–1628. doi:10.1016/j.neurobiolaging.2014.12.036
Glodzik L, Rusinek H, Brys M et al (2011) Framingham cardiovascular risk profile correlates with impaired hippocampal and cortical vasoreactivity to hypercapnia. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 31:671–679. doi:10.1038/jcbfm.2010.145
Golanov E, Reis D (1997) Oxygen and cerebral blood flow. In: Welch KMA, Caplan LR, Reis DJ, Siesjo BK, Weir B (eds) Primer on cerebrovascular diseases. Academic Press, San Diego, pp 58–60
Gorelick PB, Scuteri A, Black SE et al (2011) Vascular contributions to cognitive impairment and dementia. Stroke J Cereb Circ 42:2672–2713. doi:10.1161/STR.0b013e3182299496
Greicius MD, Srivastava G, Reiss AL, Menon V (2004) Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci 101:4637–4642. doi:10.1073/pnas.0308627101
Growdon JH, Selkoe DJ, Roses A et al (1998) Consensus report of the working group on biological markers of Alzheimer’s disease.[Ronald and Nancy Reagan Institute of the Alzheimer’s Association and National Institute on Aging Working Group on Biological Markers of Alzheimer’s Disease. Neurobiol Aging 19:109–116
Hajjar I, Zhao P, Alsop D, Novak V (2010) Hypertension and cerebral vasoreactivity: a continuous arterial spin labeling magnetic resonance imaging study. Hypertension 56:859–864. doi:10.1161/HYPERTENSIONAHA.110.160002
Hajjar I, Sorond F, Lipsitz LA (2015) Apolipoprotein E, carbon dioxide vasoreactivity, and cognition in older adults: effect of hypertension. J Am Geriatr Soc 63:276–281. doi:10.1111/jgs.13235
Head D, Bugg JM, Goate AM et al (2012) Exercise engagement as a moderator of the effects of APOE genotype on amyloid deposition. Arch Neurol 69:636–643. doi:10.1001/archneurol.2011.845
Hebert LE, Weuve J, Scherr PA, Evans DA (2013) Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80:1778–1783. doi:10.1212/WNL.0b013e31828726f5
Heijtel DFR, Mutsaerts HJMM, Bakker E et al (2014) Accuracy and precision of pseudo-continuous arterial spin labeling perfusion during baseline and hypercapnia: a head-to-head comparison with (15)O H2O positron emission tomography. NeuroImage 92C:182–192. doi:10.1016/j.neuroimage.2014.02.011
Heo S, Prakash RS, Voss MW et al (2010) Resting hippocampal blood flow, spatial memory and aging. Brain Res 1315:119–127. doi:10.1016/j.brainres.2009.12.020
Hutchison JL, Lu H, Rypma B (2013) Neural mechanisms of age-related slowing: the ΔCBF/ΔCMRO2 ratio mediates age-differences in BOLD signal and human performance. Cereb Cortex NY N 1991 23:2337–2346. doi:10.1093/cercor/bhs233
Iadecola C (2004) Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5:347–360. doi:10.1038/nrn1387
Jack CR Jr, Knopman DS, Jagust WJ et al (2010) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9:119–128. doi:10.1016/S1474-4422(09)70299-6
Jarvik L, LaRue A, Blacker D et al (2008) Children of persons with Alzheimer disease: what does the future hold? Alzheimer Dis Assoc Disord 22:6–20. doi:10.1097/WAD.0b013e31816653ac
Johnson NA, Jahng G-H, Weiner MW et al (2005) Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 234:851–859. doi:10.1148/radiol.2343040197
Kelleher RJ, Soiza RL (2013) Evidence of endothelial dysfunction in the development of Alzheimer’s disease: is Alzheimer’s a vascular disorder? Am J Cardiovasc Dis 3:197–226
Kim SM, Kim MJ, Rhee HY et al (2013) Regional cerebral perfusion in patients with Alzheimer’s disease and mild cognitive impairment: effect of APOE epsilon4 allele. Neuroradiology 55:25–34. doi:10.1007/s00234-012-1077-x
Knopman DS, Roberts R (2010) Vascular risk factors: imaging and neuropathologic correlates. J Alzheimers Dis JAD 20:699–709. doi:10.3233/JAD-2010-091555
Kramer AF, Erickson KI (2007) Capitalizing on cortical plasticity: influence of physical activity on cognition and brain function. Trends Cogn Sci 11:342–348. doi:10.1016/j.tics.2007.06.009
Lee SP, Duong TQ, Yang G et al (2001) Relative changes of cerebral arterial and venous blood volumes during increased cerebral blood flow: implications for BOLD fMRI. Magn Reson Med 45:791–800
Lee C, Lopez OL, Becker JT et al (2009) Imaging cerebral blood flow in the cognitively normal aging brain with arterial spin labeling: implications for imaging of neurodegenerative disease. J Neuroimaging Off J Am Soc Neuroimaging 19:344–352. doi:10.1111/j.1552-6569.2008.00277.x
Lee JS, Im DS, An Y-S et al (2011) Chronic cerebral hypoperfusion in a mouse model of Alzheimer’s disease: an additional contributing factor of cognitive impairment. Neurosci Lett 489:84–88. doi:10.1016/j.neulet.2010.11.071
Liu TT, Brown GG (2007) Measurement of cerebral perfusion with arterial spin labeling: Part 1. Methods. J Int Neuropsychol Soc JINS 13:517–525. doi:10.1017/S1355617707070646
Liu TT, Wong EC (2005) A signal processing model for arterial spin labeling functional MRI. NeuroImage 24:207–215. doi:10.1016/j.neuroimage.2004.09.047
Luchsinger J, Reitz C, Honig MDLS et al (2005) Aggregation of vascular risk factors and risk of incident Alzheimer’s disease. Neurology 65:545–551. doi:10.1212/01.wnl.0000172914.08967.dc
Maalikjy Akkawi N, Borroni B, Agosti C et al (2003) Volume reduction in cerebral blood flow in patients with Alzheimer’s disease: a sonographic study. Dement Geriatr Cogn Disord 16:163–169. doi:10.1159/000071005
Maalikjy Akkawi N, Borroni B, Agosti C et al (2005) Volume cerebral blood flow reduction in pre-clinical stage of Alzheimer disease: evidence from an ultrasonographic study. J Neurol 252:559–563. doi:10.1007/s00415-005-0689-z
Maass A, Düzel S, Goerke M et al (2015) Vascular hippocampal plasticity after aerobic exercise in older adults. Mol Psychiatry 20:585–593. doi:10.1038/mp.2014.114
Maier FC, Wehrl HF, Schmid AM et al (2014) Longitudinal PET-MRI reveals β-amyloid deposition and rCBF dynamics and connects vascular amyloidosis to quantitative loss of perfusion. Nat Med 20:1485–1492. doi:10.1038/nm.3734
Marra C, Ferraccioli M, Vita MG et al (2011) Patterns of cognitive decline and rates of conversion to dementia in patients with degenerative and vascular forms of MCI. Curr Alzheimer Res 8:24–31
Mattsson N, Tosun D, Insel PS et al (2014) Association of brain amyloid-β with cerebral perfusion and structure in Alzheimer’s disease and mild cognitive impairment. Brain J Neurol 137:1550–1561. doi:10.1093/brain/awu043
McKhann GM, Knopman DS, Chertkow H et al (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 7:263–269. doi:10.1016/j.jalz.2011.03.005
McLaren, Schultz, Chhatwal (2013) ApoE e4 and amyloid burden independently increase cerebral blood flow in cognitively normal older adults. In: Seventh Human Amyloid Imaging Conference, Miami
Morris JC (2005) Early-stage and preclinical Alzheimer disease. Alzheimer Dis Assoc Disord 19:163–165
Mozolic JL, Hayasaka S, Laurienti PJ (2010) A Cognitive training intervention increases resting cerebral blood flow in healthy older adults. Front Hum Neurosci. doi:10.3389/neuro.09.016.2010
Mozolic JL, Long AB, Morgan AR et al (2011) A cognitive training intervention improves modality-specific attention in a randomized controlled trial of healthy older adults. Neurobiol Aging 32:655–668. doi:10.1016/j.neurobiolaging.2009.04.013
Muller M, van der Graaf Y, Visseren FL et al (2012) Hypertension and longitudinal changes in cerebral blood flow: the SMART-MR study. Ann Neurol 71:825–833. doi:10.1002/ana.23554
Nation DA, Wierenga CE, Clark LR et al (2013) Cortical and subcortical cerebrovascular resistance index in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis JAD 36:689–698. doi:10.3233/JAD-130086
Okonkwo OC, Xu G, Oh JM et al (2014) Cerebral blood flow is diminished in asymptomatic middle-aged adults with maternal history of Alzheimer’s disease. Cereb Cortex N Y N 1991 24:978–988. doi:10.1093/cercor/bhs381
Ostergaard L, Aamand R, Gutierrez-Jimenez E et al (2013) The capillary dysfunction hypothesis of Alzheimer’s disease. Neurobiol Aging 34:1018–1031
Pantano P, Baron JC, Lebrun-Grandié P et al (1984) Regional cerebral blood flow and oxygen consumption in human aging. Stroke J Cereb Circ 15:635–641
Park DC, Reuter-Lorenz P (2009) The adaptive brain: aging and neurocognitive scaffolding. Annu Rev Psychol 60:173–196. doi:10.1146/annurev.psych.59.103006.093656
Parkes LM, Rashid W, Chard DT, Tofts PS (2004) Normal cerebral perfusion measurements using arterial spin labeling: reproducibility, stability, and age and gender effects. Magn Reson Med 51:736–743. doi:10.1002/mrm.20023
Pfefferkorn T, von Stuckrad-Barre S, Herzog J et al (2001) Reduced cerebrovascular CO(2) reactivity in CADASIL: a transcranial Doppler sonography study. Stroke J Cereb Circ 32:17–21
Popa-Wagner A, Buga A-M, Popescu B, Muresanu D (2013) Vascular cognitive impairment, dementia, aging and energy demand. A vicious cycle. J Neural Transm Vienna Austria 1996. doi:10.1007/s00702-013-1129-3
Prakash RS, Voss MW, Erickson KI, Kramer AF (2015) Physical activity and cognitive vitality. Annu Rev Psychol 66:769–797. doi:10.1146/annurev-psych-010814-015249
Restom K, Bangen KJ, Bondi MW et al (2007) Cerebral blood flow and BOLD responses to a memory encoding task: a comparison between healthy young and elderly adults. NeuroImage 37:430–439. doi:10.1016/j.neuroimage.2007.05.024
Rolland Y, Abellan van Kan G, Vellas B (2010) Healthy brain aging: role of exercise and physical activity. Clin Geriatr Med 26:75–87. doi:10.1016/j.cger.2009.11.002
Ruitenberg A, den Heijer T, Bakker SLM et al (2005) Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam study. Ann Neurol 57:789–794. doi:10.1002/ana.20493
Sandson TA, O’Connor M, Sperling RA et al (1996) Noninvasive perfusion MRI in Alzheimer’s disease: a preliminary report. Neurol Novemb 47:1339–1342
Saunders AM, Strittmatter WJ, Schmechel D et al (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43:1467–1472
Schuff N, Matsumoto S, Kmiecik J et al (2009) Cerebral blood flow in ischemic vascular dementia and Alzheimer’s disease, measured by arterial spin-labeling magnetic resonance imaging. Alzheimers Dement J Alzheimers Assoc 5:454–462. doi:10.1016/j.jalz.2009.04.1233
Sheline YI, Morris JC, Snyder AZ et al (2010) APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Aβ42. J Neurosci Off J Soc Neurosci 30:17035–17040. doi:10.1523/JNEUROSCI.3987-10.2010
Silverman JM, Ciresi G, Smith CJ et al (2005) Variability of familial risk of Alzheimer disease across the late life span. Arch Gen Psychiatry 62:565–573. doi:10.1001/archpsyc.62.5.565
Silvestrini M, Pasqualetti P, Baruffaldi R et al (2006) Cerebrovascular reactivity and cognitive decline in patients with Alzheimer disease. Stroke J Cereb Circ 37:1010–1015. doi:10.1161/01.STR.0000206439.62025.97
Singh M, Mensah GA, Bakris G (2010) Pathogenesis and clinical physiology of hypertension. Cardiol Clin 28:545–559. doi:10.1016/j.ccl.2010.07.001
Smith CD, Andersen AH, Kryscio RJ et al (1999) Altered brain activation in cognitively intact individuals at high risk for Alzheimer’s disease. Neurology 53:1391–1396
Smith JC, Paulson ES, Cook DB et al (2010) Detecting changes in human cerebral blood flow after acute exercise using arterial spin labeling: implications for fMRI. J Neurosci Methods 191:258–262. doi:10.1016/j.jneumeth.2010.06.028
Sojkova J, Beason-Held L, Zhou Y et al (2008) Longitudinal cerebral blood flow and amyloid deposition: an emerging pattern? J Nucl Med 49:1465–1471. doi:10.2967/jnumed.108.051946
Sojkova, Goh, Beason-Held (2011) Increased beta-amyloid deposition is related to regional cerebral blood flow in nondemented older adults. In: Fifth Human Amyloid Imaging Conference, Miami
Sperling RA, Aisen PS, Beckett LA et al (2011) Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 7:280–292. doi:10.1016/j.jalz.2011.03.003
Stomrud E, Forsberg A, Hägerström D et al (2012) CSF biomarkers correlate with cerebral blood flow on SPECT in healthy elderly. Dement Geriatr Cogn Disord 33:156–163. doi:10.1159/000338185
Sun Z-W, Zhu Y-X, Liu H-Y et al (2007) Decreased cerebral blood flow velocity in apolipoprotein E epsilon4 allele carriers with mild cognitive impairment. Eur J Neurol 14:150–155. doi:10.1111/j.1468-1331.2006.01579.x
Takahashi H, Ishii K, Hosokawa C et al (2013) Clinical application of 3D arterial spin-labeled brain perfusion imaging for Alzheimer disease: comparison with brain perfusion SPECT. AJNR Am J Neuroradiol. doi:10.3174/ajnr.A3780
Thambisetty M, Beason-Held L, An Y et al (2010) APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol 67:93–98. doi:10.1001/archneurol.2009.913
Thomas BP, Yezhuvath US, Tseng BY et al (2013) Life-long aerobic exercise preserved baseline cerebral blood flow but reduced vascular reactivity to CO2. J Magn Reson Imaging JMRI 38:1177–1183. doi:10.1002/jmri.24090
Tiraboschi P, Hansen LA, Masliah E et al (2004) Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 62:1977–1983
Uchihashi Y, Hosoda K, Zimine I et al (2011) Clinical application of arterial spin-labeling MR imaging in patients with carotid stenosis: quantitative comparative study with single-photon emission CT. AJNR Am J Neuroradiol 32:1545–1551. doi:10.3174/ajnr.A2525
Vicenzini E, Ricciardi MC, Altieri M et al (2007) Cerebrovascular reactivity in degenerative and vascular dementia: a transcranial Doppler study. Eur Neurol 58:84–89. doi:10.1159/000103642
Villien M, Bouzat P, Rupp T et al (2013) Changes in cerebral blood flow and vasoreactivity to CO2 measured by arterial spin labeling after 6 days at 4350m. NeuroImage 72:272–279. doi:10.1016/j.neuroimage.2013.01.066
Wang Z (2014) Characterizing early Alzheimer’s disease and disease progression using hippocampal volume and arterial spin labeling perfusion MRI. J Alzheimers Dis JAD 42(Suppl 4):S495–S502. doi:10.3233/JAD-141419
Wierenga CE, Dev SI, Shin DD et al (2012) Effect of mild cognitive impairment and APOE genotype on resting cerebral blood flow and its association with cognition. J Cereb Blood Flow Metab 32:1589–1599. doi:10.1038/jcbfm.2012.58
Wierenga CE, Clark LR, Dev SI et al (2013) Interaction of age and APOE genotype on cerebral blood flow at rest. J Alzheimers Dis JAD 34:921–935. doi:10.3233/JAD-121897
Wierenga CE, Hays CC, Zlatar ZZ (2014) Cerebral blood flow measured by arterial spin labeling MRI as a preclinical marker of Alzheimer’s disease. J Alzheimers Dis JAD. doi:10.3233/JAD-141467
Wolk DA, Detre JA (2012) Arterial spin labeling MRI: an emerging biomarker for Alzheimer’s disease and other neurodegenerative conditions. Curr Opin Neurol 25:421–428. doi:10.1097/WCO.0b013e328354ff0a
Wong EC (2013) New developments in arterial spin labeling pulse sequences. NMR Biomed 26:887–891. doi:10.1002/nbm.2954
Xekardaki A, Rodriguez C, Montandon M-L et al (2015) Arterial spin labeling may contribute to the prediction of cognitive deterioration in healthy elderly individuals. Radiology 274:490–499. doi:10.1148/radiol.14140680
Xu G, Antuono PG, Jones J et al (2007) Perfusion fMRI detects deficits in regional CBF during memory-encoding tasks in MCI subjects. Neurology 69:1650–1656. doi:10.1212/01.wnl.0000296941.06685.22
Yip AG, McKee AC, Green RC et al (2005) APOE, vascular pathology, and the AD brain. Neurology 65:259–265. doi:10.1212/01.wnl.0000168863.49053.4d
Yoon HJ, Park KW, Jeong YJ, Kang D-Y (2012) Correlation between neuropsychological tests and hypoperfusion in MCI patients: anatomical labeling using xj view and Talairach Daemon software. Ann Nucl Med 26:656–664. doi:10.1007/s12149-012-0625-0
Zavoreo I, Bašić Kes V, Lisak M et al (2013) Cognitive decline and cerebral vasoreactivity in asymptomatic patients with severe internal carotid artery stenosis. Acta Neurol Belg 113:453–458. doi:10.1007/s13760-013-0196-4
Zlatar ZZ, Wierenga CE, Bangen KJ et al (2014) Increased hippocampal blood flow in sedentary older adults at genetic risk for Alzheimer’s disease. J Alzheimers Dis JAD. doi:10.3233/JAD-132252
Acknowledgments
This work was supported by VA CSR&D Merit Award 5I01CX000565 (to C.E.W.), National Institutes of Health T32 MH019934 (to Z.Z.Z.), and the National Science Foundation Graduate Research Fellowship Program 2015207525 (to C.C.H.)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hays, C.C., Zlatar, Z.Z. & Wierenga, C.E. The Utility of Cerebral Blood Flow as a Biomarker of Preclinical Alzheimer’s Disease. Cell Mol Neurobiol 36, 167–179 (2016). https://doi.org/10.1007/s10571-015-0261-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-015-0261-z