
Abstract
Resveratrol is a naturally occurring phytoalexin found in red grapes, and believed to have neuroprotective, anti-oxidant, and anti-inflammatory effects. But little is known about its effect on the neural impairments induced by microglial over-activation, which leads to neuroinflammation and multiple pathophysiological damages. In this study, we aimed to investigate the protective effects of resveratrol on the impairments of neural development by microglial over-activation insult. The results indicated that resveratrol inhibited the lipopolysaccharide (LPS)-dependent release of cytokines from activated microglia and LPS-dependent changes in NF-κB signaling pathway. Conditioned medium (CM) from activated microglia treated by resveratrol directly protected primary cultured hippocampal neurons against LPS-CM-induced neuronal death, and restored the inhibitory effects of LPS-CM on dendrite sprouting and outgrowth. Finally, neurons cultured in CM from LPS-stimulated microglia treated by resveratrol exhibited increased spine density compared to those without resveratrol treatment. Our findings support that resveratrol inhibits microglial over-activation and alleviates neuronal injuries induced by microglial activation. Our study suggests the use of resveratrol as an alternative intervention approach that could prevent further neuronal insults.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Microglia comprise approximately 12 % of cells in the brain and function as the dominant immune cell of the brain, which is analogous to the macrophages of the systemic immune system. Microglial cells are of the monocytic lineage and typically exist in a quiescent state in the normal brain, characteristic of ramified morphology and short branched processes (Fetler and Amigorena 2005; Nimmerjahn et al. 2005). Following injury, microglia exhibit various behaviors, including activation, division, and migration to the injury site. Therefore, shortly after injury, there is an influx of microglial cells, which behave more macrophage-like with time and are often neuroprotective. Vascular damages, as with all spinal injuries, cause extensive inflammation and invasion of a large number of microglia from the blood stream (Fawcett and Asher 1999). The state of microglial activation, e.g., the number of microglia and their phagocytic activity, is an important indicator of neuropathology hence an ideal target for pharmacological manipulation. Over-activated microglia, in response to brain insults, can produce synergistic effects on neurotoxicity via release of inflammatory cytokines and chemokines including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), inducible nitric oxide synthase (iNOS), nitric oxide (NO), reactive oxygen species (ROS), and cyclooxygenase-2 (COX-2), which contribute to neuronal damage, particularly in neurodegenerative diseases (Cao et al. 2010; Choi et al. 2009; Nam et al. 2008). Recent evidences have indicated that activated microglia inhibit axonal growth and regeneration (Horn et al. 2008; Kitayama et al. 2011). Therefore, modulation of microglia-mediated inflammatory responses is important in designing new therapeutic approach against neuronal inflammatory diseases.
Resveratrol (3,5,4′-trihydroxy-trans-stilbene, RV) is a naturally occurring phytoalexin found in red grapes, and has attracted considerable attention due to its wide variety of pharmacological properties. RV exhibits anti-oxidative effect, anti-platelet activity, cardioprotective ability, neuroprotective effects, and an anti-inflammatory activity via modulating NO production, inhibiting cytochrome P450, cyclooxygenase (COX), and the nuclear factor-kappa B (NF-κB) pathways (Saiko et al. 2008b). Although a large number of studies have shown that resveratrol is neuroprotective in both acute central nervous system (CNS) injuries and chronic neurodegenerative diseases such as Huntington’s, Alzheimer’s, and Parkinson’s diseases (Anekonda and Reddy 2006; Ates et al. 2007a, b; Constant et al. 2012; Karlsson et al. 2000; Parker et al. 2005), the exact mechanisms for its beneficial effects, particularly how resveratrol suppresses the inflammatory response in microglia and the potential roles of resveratrol plays in neuronal regeneration, are still not fully understood.
In this study, we aimed to determine the capacity of resveratrol in protecting microglia over-activation, using lipopolysaccharide (LPS) as the stimuli to induce microglia over-activation, which mimics neurotoxicity caused by accumulation of microglia followed by CNS injuries. We further identified the role of resveratrol in microglia-mediated neuronal impairments, using conditioned medium (CM) from over-activated microglia to culture the hippocampal neurons, and evaluated neuronal growth parameters. We found that resveratrol inhibits LPS-dependent release of cytokines in over-activated microglia and LPS-dependent changes in NF-kB signaling pathway. Moreover, resveratrol rescues the impairments of neurons stimulated with LPS-CM in vitro. Our results support the use of resveratrol as potential therapeutic agent against neuronal inflammatory diseases in the future.
Materials and Methods
Chemicals
Resveratrol (trans-3,5,4′-trihydroxystilbene; purity >99 %) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were obtained from Sigma-Aldrich (St. Louis, MO). Resveratrol was dissolved with dimethyl sulfoxide (DMSO) at a stock concentration of 20 mM. For the cell viability assay, an equivalent amount of DMSO (0.1 % final concentration) was added to control cultures. For other experiments, DMSO was excluded in the control cultures.
Cell Culture Methods
Primary Microglial Cultures
Primary microglia were isolated according to previously published method (Griffin et al. 2007). Briefly, cerebral cortices dissected from postnatal day 1 to 2 mice were trypsinized, mechanically dissociated, and seeded on the flasks. The cultures were maintained in DMEM (Gibco, Carlsbad, CA) supplemented with 10 % fetal bovine serum (FBS; Gibco, Carlsbad, CA) and 5 ng/ml macrophage colony stimulating factor (Peprotech). Then, the suspended microglia were retrieved from the culture medium or subsequent experiments. For MTT and ELISA experiments, cell cultured on plates for overnight and then treated with resveratrol or LPS (100 ng/ml) for 24 h.
Hippocampal Neuron Culture, Transfection, and Immunostaining
Low-density of primary hippocampal neurons were prepared from both of the hemispheres hippocampus of postnatal day 1 mouse according to the literatures (Amin et al. 2013; Song et al. 2014). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of The Second Hospital of Hebei Medical University. The IACUC committee members at The Second Hospital of Hebei Medical University approved this study. All surgeries were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize animal suffering, to reduce the number of animals used, and to utilize alternatives to in vivo techniques, if available. Briefly, Mice were sacrificed by decapitation after being anesthetized by CO2, hippocampi were dissection, cut into slices, and washed twice with the dissection medium (Ruaro et al. 2005). Cells were gently dissociated by approximately 50 passages through a Gilson P1000 tip after digested in TrypIE (Life Technologies, Grand Island, NY) for 10 min at 37 °C. The cell suspension was then centrifuged at 300×g for 5 min, and the pellet was resuspended in culture medium (DMEM medium containing 10 % FBS, 2 % B-27 supplement, penicillin–streptomycin, Ham’s F-12 Nutrient Mixture). Cultures were maintained at 37 °C in humidified atmosphere with 5 % CO2, and medium was changed 24 h after plating and every 3 days thereafter. To get highly purified hippocampal neurons and arrest glial cell proliferation, on the second day, we treated the culture with 5 μM cytosine-β-D-arabinofuranoside (Ara-C), Sigma-Aldrich, St. Louis, MO). Neurons cultured for 7–21 days were used in the experiments. For transfection, the neurons were plated on glass coverslips coated with poly-l-lysine (Sigma-Aldrich, St. Louis, MO) at an approximate density of 70 cells/mm2 and were transfected (performed at DIV17) using modified calcium phosphate precipitation method according to descriptions by Zhang et al. (2003). Immunostaining was performed at DIV 21. The transfected cells were fixed with 2 % paraformaldehyde and blocked with 2 % normal goat serum (Sigma-Aldrich), and permeabilized with 0.1 % Triton X-100 in PBS. Then, the cells were incubated with primary antibodies diluted in PBS-BSA solution for 1 h, following by Alexa Fluor 488- or 647-labeled secondary antibodies (1:1000, Life Technologies, Grand Island, NY) were used.
Conditioned Medium (CM) for Hippocampal Neurons Cultures
CMs were collected according to the literatures (Song et al. 2014; Wu et al. 2009). Briefly, primary microglial cells were first cultured overnight, and then the cell culture supernatants were replaced by DMEM-F12 FBS-free medium (neuron culture medium) with or without LPS stimulation in the absence or presence of resveratrol continue to culture 24 h. CM was harvested, centrifuged for 5 min at 1000×g, filtered through 0.22-μm-pore-diameter Millipore filters, and then dialyzed overnight using the Slide-A-Lyzer Dialysis Cassette (10 K MWCO, Pierce Biotechnology Inc., Rockford, IL) to exclude the possible influence of LPS. CMs were stored at −80 °C until use. For the cultures of primary hippocampal neurons, the fresh media with CMs at 1:1 were supplemented to the cultures every 3 days.
MTT Assay
Cell viability was measured using an MTT assay according to previous report (Park et al. 2012). Briefly, primary microglial cells (1 × 104 cells/well) were seeded in 96-well plates, and after 24 h, treated with different concentrations of resveratrol (0.1–20 μM). After treatment, media were gently aspirated, cells were washed twice with PBS, and 200 μl of a 0.5 mg/ml MTT solution in PBS was added to each well. Plates were incubated at 37 °C for 4 h, MTT solution was removed, and the solubilization solution was added into each well to dissolve the formazan by shaking at room temperature for 10 min. The dye product was quantified using a microplate reader (SpectraMax M Series, multi-mode, Molecular Devices, CA) at a wavelength of 560 nm (A560). For hippocampal neuronal cells viability assay, the neurons (1 × 104 cells/well) were seeded on laminin-coated 96-well plates and cultured in proliferative medium containing DMEM-F12 with 2 % B27 supplement (Life Technologies, Grand Island, NY), EGF (20 ng/ml, Sigma-Aldrich, St. Louis, MO), and FGF-2 (20 ng/ml, Sigma-Aldrich, St. Louis, MO). After seeding overnight, hippocampal neuron culture supernatants were changed with various CMs supplemented with 2 % B-27 supplement, 20 ng/ml EGF, and 20 ng/ml FGF-2 cultured for 24 h. Neuronal viability was measured by MTT assay as described in primary microglial cells.
LDH Assay
The membrane integrity of neurons was evaluated by using lactate dehydrogenase (LDH) assay, according to the protocol of The Thermo Scientific Pierce LDH Cytotoxicity Assay Kit (Thermo Scientific, Rockford, IL). The assays were performed after 7-day culture to evaluate the membrane integrity of neurons, and data were recorded using a microplate reader (SpectraMax M Series, multi-mode, Molecular Devices, CA) at a wavelength of 490 and 680 nm (A490-A680).
Tumor Necrosis Factor-Alpha and IL-1β Assays
The production of tumor necrosis factor-alpha (TNF-α) and IL-1β was measured with a commercial ELISA kit from R&D Systems (Minneapolis, MN) according to the manufacturer’s instructions. 100 μl of the supernatant was sampled for cytokine analysis under different stimulatory conditions before cells were harvested for RNA extraction. The absorbance at 450 nm was determined using a microplate reader (SpectraMax M series, Multi-mode, Molecular Devices, Sunnyvale, CA). The concentrations of TNF-α and IL-1β in culture supernatants were calculated using TNF-α and IL-1β standards.
RNA Extraction and SYBR Green Quantitative Real-Time PCR
After incubation, media were removed and primary microglial cells were washed with phosphate-buffered saline (PBS) twice. Total RNA was extracted using Trizol reagent as recommended by the manufacturer (Invitrogen, Carlsbad, CA). RNA quality and concentration were evaluated spectroscopically using a NanoDrop 2000c instrument (Thermo Scientific, Rockford, IL). For mRNA analysis, real-time PCR was performed using Superscript TM-ш kit (Invitrogen, Carlsbad, CA) on an ABI 7900HT PCR machine Applied Biosystems, and data were normalized to β-actin and further normalized to the control without LPS stimulation unless otherwise indicated. The sequences of PCR primers were as follows:
(1) iNOS sense 5′-GGACGAGACGGATAGGCAGAGATT-3′, anti-sense 5′-AAGCCACTGACACTTCGCACAA-3′;
(2) COX-2 sense 5′-TCTCCAACCTCTCCTACTAC-3′, anti-sense 5′-GCACGTAGTCTTCGATCACT-3′;
(3) β-actin sense 5′-GATGGTGGGAATGGGTCAGA-3′, anti-sense 5′-TCCATGTCGTCCCAGTTGGT-3′.
Data analysis was performed using the 2−ΔΔCt method.
Preparation of Cytoplasmic and Nuclear Extracts and Immunoblotting Analysis
Cells were washed with ice-cold PBS twice, scraped, collected, and transferred into clean centrifuge tubes. Cell pellets were collected to separate the cytoplasmic and nuclear fractions using the CHEMICON’s nuclear extraction kit (EMD Millipore Corporation, Temecula, CA) according to the manufacturer’s instructions. Protein concentration in the supernatant was quantified by Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Rockford, IL). 10–30 µg reduced protein in Laemmli sample buffer was resolved using 6–12 % sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) and transferred to nitrocellulose membrane (Bio-Rad, Hercules, CA) for immunoblotting analysis. Blots were stripped using stripping buffer (Thermo Scientific, Rockford, IL) before reprobing. β-actin was used as an endogenous protein for normalization. Images were analyzed by Quantity One software (Bio-Rad, Hercules, CA).
Image Acquisition and Morphometry Analysis
Fluorescence images were acquired either by confocal laser microscopy (Zeiss LSM710) or Box-Type Fluorescence Imaging Device (Olympus-FSX100). The lengths of the dendrites and filopodia were determined using NIH software Image J, with Neuron J plug-in (Copyright from Erik Meijering). Three independent experiments were performed, in each of which 100 neurons were counted per group. To visualize growth cones, neurons were fixed for 10 min with 2 % PFA and stained with Alexa Fluor® 488-conjugated phalloidin probe (1:500; Invitrogen, Carlsbad, CA) and Neuron-specific β-tubulin (anti-TuJ1) (1:1,000; R&D systems). Data were collected from 3 independent experiments. Results are shown as relative area of GC compared to control neurons without LPS stimulation, considered as 1 (Pool et al. 2008).
Statistical Analysis
The statistical significances of differences between the vehicle and resveratrol-treated groups were determined by analysis of variance (ANOVA) followed by a Tukey’s post hoc test procedure. Values are shown as mean ± SEM unless otherwise specified. A value of p < 0.05 is considered statistically significant and is reported. All protein biochemical experiments were carried out a minimum of three times.
Results
Toxicity of RV to Primary Microglial Cells
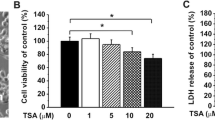
We first investigated the effect of RV on the proliferation of primary microglial cells. Treatment of primary microglial cells with both 10 and 20 µM resveratrol clearly decreased the cell number and arrested cell growth (Fig. 1a), indicating that resveratrol can induce cell death in primary microglial cells at higher concentrations. It is worth mentioning that the DMSO concentration was 0.1 % in the culture medium of 20 µM resveratrol group, while it was 0.005 % in 1 µM RV group. The DMSO at the highest concentration of 0.1 % had neglectable effects on cell viability (Fig. 1b). RV concentrations of up to 1 µM had no significant effect on proliferation of primary microglial cells. However, viability of primary microglial cells was significantly inhibited by higher doses (10 and 20 µM, Fig. 1b). To exclude the toxic effect of RV on cell growth, we selected 1 μM as optimal RV dose for the following experiments.

Effects of resveratrol on the cell viability of primary microglial cells. a Microglial cells were exposed to resveratrol (0.1, 1, 10, and 20 µM) for 24 h and labeled for α-tubulin (Green). Scale bar 100 µM. b Cells proliferation was determined using an MTT assay. Cells were seeded into 96-well plates (1 × 104 cells/well) and cultured for 24 h. Cells were treated with either vehicle or the indicated concentrations of resveratrol for 24 h. The lower concentrations of resveratrol slightly inhibited microglial proliferation. Values are the mean ± SEM (n = 4). *p < 0.05 versus vehicle and #p < 0 .01 versus vehicle. (One-way ANOVA with Tukey’s post hoc test procedure) (Color figure online)
RV Reduces LPS-Induced Pro-inflammatory Mediator Release from Microglia
Activated microglia produce an array of pro-inflammatory factors that mediate LPS-induced neurotoxicity (Block et al. 2007). To provide more evidence of the anti-inflammatory effect of RV in neurophysiology, LPS-induced inflammatory model was used in the primary microglial cells. LPS, an endotoxin from bacteria as a pro-inflammatory stimulus to induce neurotoxicity, could be a perfect positive control for us to evaluate the function of RV on microglia activation. LPS induces the synthesis of inflammatory mediators, including various chemokines and cytokines (Montine et al. 2002; Quan et al. 1994), which have been shown to affect neural cell behavior and function. We first examined the microglial morphology in the experimental groups (Fig. 2a). The microglia cells presented the typical morphology of unipolar and/or ameboid shapes. We then determined the levels of several major inflammation-related factors released from microglia after LPS treatment. Before LPS activation, primary microglial cultures in either vehicle control or RV treatment groups expressed low levels of TNF-α and IL-1β, iNOS and COX-2 (Fig. 2b–e, lane 1 vs. lane 2, p < 0.05). Exposure of primary microglial cells to LPS (100 ng/ml) for 24 h resulted in significantly increased expressions of pro-inflammatory cytokines TNF-α and IL-1β released into the culture medium, as shown in Fig. 2b, c (lane 1 compared to lane 3, p < 0.01). Furthermore, we examined the mRNA levels of iNOS and COX-2, both of which are involved in the microglia-mediated inflammation. As expected, when primary microglial cells were activated by LPS, the expressions of iNOS and COX-2 were strongly enhanced (Fig. 2d, e, lane 1 vs. lane 3, p < 0.01). These hallmark shifts reflected a successful inflammatory response in primary microglial cells, indicative of microglia over-activation upon LPS stimulation. Next, we evaluated the effect of RV (1 μM) on LPS-induced over-activation in primary microglial cells. As shown in Fig. 2b, c (lane 3 vs. lane 4), primary microglial cells stimulated with LPS responded to RV treatment with >70 % reduction of TNF-α, and >60 % reduction of IL-1β expressions, respectively. mRNA expressions of iNOS and COX-2 were also significantly decreased (both >70 % reduction) in RV group compared with those in vehicle control under LPS-stimulated condition (Fig. 2d, e, lane 3 compared to lane 4, p < 0.01). Besides, the cell densities were comparable (p < 0.05) in these groups (control without LPS 1973 ± 124 per mm2; RV without LPS 1945 ± 136 per mm2; control with LPS 2025 ± 143 per mm2; RV with LPS 2008 ± 164 per mm2), which excluded the influence of cell density on the cytokine release. Overall, these results indicated that RV is capable of inhibiting inflammatory response in primary microglial cells by decreasing levels of pro-inflammatory cytokines.

Resveratrol attenuates the LPS-induced inflammatory responses. a Primary microglial cells were seeded into 6-well plates (6 × 105) with glass coverslips in it and cultured for 24 h. Cells were undergone resveratrol (1 µM) challenge either with or without 100 ng/ml lipopolysaccharide (LPS) stimulation 24 h. Representative microglia morphological changes were photographed by Olympus-FSX 100 Fluorescence microscopy. Images of microglia labeled for α-tubulin (green) to visualize microtubules and DAPI (blue) to label nuclei. Scale bar 100 µM. Expression of molecules that are hallmarks of microglial activation TNF-α (b) and IL-1β (c) was quantified by ELISA kits. Gene expression of iNOS (d) and COX-2 (e) was assessed by quantitative polymerase chain reaction (qPCR) and normalized to the housekeeping gene, β-actin. Values are expressed as mean ± SEM for 3–6 replicates using different cell cultures. One-way ANOVA with Tukey’s post hoc test revealed differences from control microglia without LPS stimulation. #p < 0.01 (Color figure online)
RV Inhibits the LPS-Induced Changes in NF-kB Signaling Pathway in Primary Microglial Cells
It was reported that inhibition of inflammation by RV was mediated through the NF-κB pathway (Holmes-McNary and Baldwin 2000; Kundu and Surh 2004). To confirm the involvement of the NF-κB pathway in the RV-mediated suppression of the LPS-induced neuroinflammation, primary microglial cells were treated with RV (1 µM) in the presence of LPS, and the activity of the NF-κB pathway was evaluated by measuring the protein expression levels of p65, IκBα, and p-IκBα in the cytosolic and nuclear extracts. Primary microglial cells responded to LPS stimulation with >50 % reduction of p65 (Fig. 3a, 1st panel, b, lane 1 compared to lane 3) and IκBα (Fig. 3a, 2nd panel, c, lane 1 vs. lane 3), and >40 % increase of p-IκBα expression (Fig. 3a, 3rd panel, d, compared lane 1 to lane 3) in cytosolic extract compared to the control. Moreover, LPS enhanced the nuclear translocation of NF-κB p65 subunit (Fig. 3e, upper panel, f, lane 1 vs. lane 3), and this enhancement was prevented by RV treatment (Fig. 3e, upper panel, f, compared lane 3 to lane 4). These target gene expression changes at the protein level indicated the activation of NF-κB signaling pathway in primary microglial cells was stimulated by LPS, and the stimulation was blocked by RV treatment, demonstrating that RV can effectively inhibit the LPS-dependent changes in NF-kB signaling in activated microglia.

Resveratrol inhibits the LPS-induced activation of NF-κB in primary microglial cells. a Cells were seeded into 100-mm dishes (1.5 × 106/dish) and cultured for 24 h. Cells were then undergone resveratrol (1 µM) challenge either with or without 100 ng/ml lipopolysaccharide (LPS) stimulation 24 h. Levels of target proteins p65, IκBα, and p- IκBα in the cytosolic extract were evaluated with immunoblotting. b–d The Western blotting results in (a) were quantified and shown in a graph format. The expression of those target proteins (p65, IκBα, and p-IκBα) was normalized to β-actin, and the expression for different groups was determined as a relative change from vehicle control in the absence of LPS treatment and shown as mean ± SEM. e Cells were treated with resveratrol (1 µM) either with or without 100 ng/ml lipopolysaccharide (LPS) stimulation 24 h. The level of p65 in the nuclear extract was determined by immunoblotting. f The results from the Western analysis in (e) were quantified and subjected to densitometry and shown as a graph. HDAC1 was used as the nuclear protein marker and β-actin as the cytosolic protein marker. The relative expression of p65 was normalized to HDAC1 expression, and the expression for various groups was determined as a relative change from vehicle control and shown as mean ± SEM of 3 independent experiments. #p < 0.01 compared to the control
Conditioned Medium from RV-Treated Microglia Promotes Survival of Hippocampal Neurons
We next investigated the role of RV in promoting survival of cultured hippocampal neurons upon LPS-induced microglia over-activation. First, we collected CM before performing the following experiments (see “Cell Culture Methods” section). Conditioned media from microglia-enriched cultures in the absence (control-CM) or presence of 1 μM RV (RV-CM) and with (LPS-CM) or without LPS stimulation were prepared. Microglia cells were incubated first with or without RV for 24 h, and CM was collected and dialyzed to remove RV. This RV-free CM was then added to neuron-enriched cultures and incubated for 7 days before assays. We examined the proliferation of hippocampal neurons. ANOVA analysis showed no significant differences in cell viability between control-CM and RV-CM (1 µM) in the absence of LPS stimulation group (Fig. 4a, lane 1 vs. lane 2, p > 0.05). However, after stimulation with LPS-CM, the cell viability was decreased by about 25 % (Fig. 4a, compared lane 1 to lane 3, p < 0.05). This decrease was restored by addition of RV-CM (Fig. 4a, lane 3 vs. lane 4, p < 0.05). Extracellular lactate levels, which reflect the anaerobic metabolism of neurons, did not change in the presence of 1 µM RV-CM (Fig. 4b, compared lane 1 to lane 2, p > 0.05). However, LPS-CM induced an approximately 50 % increase in lactate levels, indicating LPS had an obvious impairment on the integrity of the cultured neuronal cell membrane (Fig. 4b, lane 1 vs. lane 3, p < 0.05), as LDH release in culture media indicates membrane damage and is a biomarker for cellular cytotoxicity and cytolysis. As expected, we found that the presence of RV with LPS stimulation CM was able to prevent this alteration (Fig. 4b, lane 3 vs. lane 4, p < 0.05).

Effect of resveratrol on metabolic changes induced by LPS in Hippocampal neurons. After overnight culture on 96-well laminin-coated plates, as described in “Materials and Methods” section, hippocampal neurons culture medium was replaced with four different CM (control-CM, RV-CM, LPS-CM, and LPS + RV-CM) supplemented with 2 % B27 supplement, 20 ng/ml EGF, and 20 ng/ml FGF-2 cultured for 24 h. After treatment, a MTT-measured viability of hippocampal neurons cultured in different stimulatory conditions. b Lactate was determined in the extracellular medium. The data (expressed as a percentage of the control) represent mean ± SEM of 3 independent experiments performed in triplicate. *p < 0.05, significant difference from control
RV Rescues the LPS-Induced Impairments of Growth Cones (GC) in the Cultured Hippocampal Neurons
Microglia are one of the key modulators of axonal growth and regeneration following injury (Fawcett and Asher 1999). Reactive microglia increases the production of a variety of cytokines that have been shown to inhibit axonal growth (Aldskogius and Kozlova 1998; Hung and Colicos 2008; Streit et al. 1988). We next investigated whether RV affects these secreted factors that would interfere with morphological properties of neurons and dendrite outgrowth, using an in vitro model. When the hippocampal neurons were cultured with CM from LPS-activated microglia, the average of size of GC and lamellipodia (Fig. 5b, compared lane 1 to lane 3, p < 0.05), ratio between the number of filopodia and GC area (Fig. 5e, lane 1 compared to lane 3, p < 0.05), and the number of filopodia and their length (Fig. 5c, d, lane 1 vs. lane 3, p < 0.05) were all significantly decreased. As expected, we found that RV has the capacity to reverse these decreased parameters (Fig. 5b–d, lanes 3 and 4).

Morphological properties and differences of hippocampal GCs responded to LPS and resveratrol. The hippocampal neurons were cultured for 7 days in the different stimulatory conditions. a Confocal fluorescence images of a hippocampal GC stained for F-actin (phalloidin), Tuj1 (β-tubulin-ш antibody) and merge of the two stainings. The right image shows the quantification of growth cone area (white area), filopodia numbers (white arrows), and length (yellow line). b Normalized area of hippocampal GCs. c Average number of filopodia emerging from hippocampal GCs. d Average filopodium lengths from the tip of each filopodia to the edge of the hippocampal GCs. e Ratio of number of filopodia and area of GC in hippocampal GCs (Color figure online)
RV Treatment Restores Dendrite Outgrowth in LPS-Conditioned Medium-Induced Neuronal Cells
Figure 6a shows a representative image of a typical hippocampal neuron with extending dendrites in the culture. The dendrite sprouting and outgrowth were characterized by the number of dendrites per cell, number of dendritic end tips, and the average length of dendrite. Dendrite length was also used as a parameter to evaluate the neuron ability to maintain the dynamics of the tubulin and actin cytoskeletons which are essential for the establishment of synapses. As shown in Fig. 6b, c, LPS-CM-treated neurons showed a dramatic impairment of dendrite outgrowth, evident by decreased numbers of primary dendrites per cell and dendritic end tips, with reduction of about 20 and 40 %, respectively (p < 0.05). Additionally, LPS-CM significantly reduced the dendrite length (Fig. 6d, with reduction of about 20 %; p < 0.05), exhibiting deleterious effect of neurotoxicity. When co-cultured with RV-CM, RV reverted the impairment of dendrite outgrowth parameters, as well as inhibited further impairment of dendrite outgrowth induced by LPS-CM (increase of about >20 % when compared with control-CM).

Average number and length of dendrites under different conditioned cultures. The hippocampal neurons were cultured for 7 days in the different conditioned media of the experimental group. a A typical hippocampal neuron with extending dendrites in the culture, stained for MAP-2. Scale bar 20 µM. The neurite sprouting and outgrowth were characterized by the number of primary dendrites per cell. b Average number of primary dendrites per neuron. c Average number of dendritic end tips. d Average dendrite lengths from the tip of each dendrite to the edge of the hippocampal soma. e Western blot analysis of GAP-43 expression in the cultures of different conditioned media. The figure is representative of three experiments with similar results. f The Western blot results in (e) were quantified to determine whether a statistically significant difference exists between the groups and shown in a graph format (p < 0.05 compared to the control). The intensities of the bands corresponding to GAP-43 were compared to those corresponding to β-actin. The expression of GAP-43 was determined as a relative change from control without LPS stimulation and shown as mean ± SEM GAP-43, growth-associated protein-43. #p < 0.01, *p < 0.05
Growth-associated protein (GAP)-43 is a neuronal protein associated with neurite outgrowth and has been shown to be an efficient marker for the presence of neuronal growth cones (Meiri et al. 1986). To examine whether dendrite outgrowth enhancement by RV-CM correlated with GAP-43 upregulation, we performed Western blot analysis on DIV14 mature neurons. GAP-43 expression was significantly higher in RV-CM group than control-CM with LPS stimulation (Fig. 6e, lane 3 and 4). This result suggested that RV promote dendrite outgrowth, which was evident by correlated GAP-43 upregulation.
RV Increases Spine Density in Hippocampal Neurons
We next investigated the effect of RV on spinogenesis. To obtain a higher yield of fluorescently labeled neurons, we transfected hippocampal neurons with mCherry-actin vectors at DIV17. Figure 7a, b (higher magnification) shows the image of a representative mCherry-actin-transfected hippocampal neuron in culture, where the features of a normal neuron with several primary dendrites can be clearly visualized. In addition, the spine density and the relative frequency of three different spine types (stubby, mushroom, and thin shaped) were quantified in different CM cultured hippocampal neurons. We cultured hippocampal neurons with LPS-CM in the absence or presence of RV-CM, and found significantly reduced spine density in LPS-CM cultures alone (Fig. 7d, lanes 1 and 3, p < 0.05). As expected, we also found that RV-CM markedly increased spine density in cultured hippocampal neurons (Fig. 7d, lanes 3 and 4, p < 0.05). Furthermore, LPS-CM resulted in a considerable deficit of 15 % in stubby-shaped spine. Meanwhile, we did not observe any significant difference in the number of the other two types of spine (mushroom and thin shaped) (Fig. 7c). In the presence of RV-CM, we found that RV induced the formation of dendritic protrusions in stubby-shaped spines (Fig. 7c, left). Taken together, these results clearly demonstrated that RV remarkably improved spine morphology and density in the in vitro hippocampal neuronal culture.

Effects of resveratrol treatment on spine morphology and density in the hippocampal neurons at DIV14. a The cultured hippocampal neurons were transfected with mCherry-actin at DIV17 by the calcium phosphate method and imaged at DIV21. Scale bar 10 µm. b Higher magnification views of representative segments of spine. Scale bar 2 µm. c Proportion of different spine types expressed as percentage of total spines in the neurons in the experimental groups. d Quantification of spine density, expressed per 10 µM of dendrites of the neurons in the experimental groups. n = 10–15. Data were presented by mean ± SEM *p < 0.05 versus control group without LPS stimulation
Discussion
RV exerts a wide variety of anti-oxidation and anti-inflammation activities (Kundu and Surh 2004; Saiko et al. 2008a, b). Recent studies both in vitro and in vivo have demonstrated its neuroprotective roles in spinal cord injuries, traumatic brain injury in rats, H2O2-induced oxidative injury in acute hippocampal slice preparation from Wistar rats and early brain injury after subarachnoid hemorrhage (Ates et al. 2007a; de Almeida et al. 2008; Liu et al. 2011; Shao et al. 2014).
However, the signaling cascades that mediate the effects RV on pro-inflammatory factors production have not been fully characterized. Possible underlying mechanism was demonstrated to be, at least partially, via inhibiting the NF-κB pathway (Kundu and Surh 2004). We therefore evaluated the protein expression targeting NF-κB pathway. Under normal physiological conditions, the heterodimer NF-κB, which consists of three subunits (p65, p50 and IκBα), exists in the cytoplasm in an inactive state. Inhibitory kappa B (IκBα) plays an important role in maintaining the inactivation of NF-κB. Various stimuli including TNF-α can induce the activation of inhibitory kappa kinases (IKK), resulting in phosphorylation of IκBα and proteasomal degradation of NF-κB. Then, the p65 subunits of NF-κB are released from the heterodimer and translocate into the nucleus, where they promote transcription of cytokines (e.g., TNF-α, IL-1β, and IL-6). After LPS stimulation, the expression of IκBα in primary microglial cells was down-regulated, and phosphorylated IκBα was up-regulated in comparison with their corresponding non-LPS-treated groups, as evidenced by the Western blot analysis (Fig. 3a, c, d). Meanwhile, p65 successfully entered the nucleus fraction in the primary microglial cells after LPS stimulation as shown in Fig. 3e, f. Consistent with previous study (Henn et al. 2009), our results also showed that RV inhibited the nuclear translocation of p65, suggesting an inhibitory effect of RV on NF-κB activity. These findings suggest that the inflammatory responses of primary microglial cells in RV culturing may partially inhibit the degradation of IκBα in over-activated primary microglial cells, which may result in the down-regulation of the cytokines observed in Fig. 2b, c, d, e, lanes 4.
The second part of our investigation involves the isolation of individual cell types to measure their specific cellular parameters responding to different stimuli, therefore aiding in an understanding of the role microglia play in the function of larger structures in the brain. We then used CM culture system to evaluate the effects of microglia over-activation on isolated hippocampal neurons in vitro, as well as to identify the role of RV plays during this process. Here, hippocampus was chosen to be the model of investigation, as it is an important brain area owing to its vital role in the consolidation of several forms of learning and memory (Lynch 2004). Conditioned culturing was employed instead of co-culturing the microglia with the hippocampal neurons to avoid the interference of RV itself on the cell viability and growth (Song et al. 2014). These culture systems provide the in vitro model to investigate changes that were initiated in vivo. We first investigated whether microglia could affect primary cultured neuron viability using MTT assay. Cell survival was evaluated after treatment with control-CM, RV-CM with or without LPS stimulation for 24 h, as detailed in the Methods section. Compared to cells in control-CM, the viability of LPS-CM cultured neurons was significantly decreased, whereas no obvious change in the cells in control-CM and RV-CM without LPS stimulation can be observed. However, neurons in LPS-treated CM produced with RV presence exhibited much higher viability than those in LPS-treated CM alone, suggesting RV could alleviate the LPS-induced cytotoxicity in primary cultured hippocampal neurons. In addition, LDH assay provided consistent result that RV restored the impairment on the integrity of neuron cell membrane caused by LPS-treated CM (Fig. 4b, compared lane 3 and 4), as LDH release indicated membrane damage and is a biomarker of necrosis. This result indicates RV behaved very similarly to the effects observed in primary microglial cells. Therefore, we could use this culture system to perform functional assays in primary neuronal cultures.
The glial reaction to CNS damage recruits microglia, oligodendrocyte precursors, meningeal cells, astrocytes, and stem cells, many of which produce molecules that inhibit axon regeneration (Fawcett and Asher 1999). Neuronal motility is the basis of major functions including neuronal development, memory, repair, and cell migration (Ghashghaei et al. 2007). In performing these functions, neurites protrude from neurons, searching for the appropriate chemical or mechanical cues guiding the formation of correct connections (Solecki et al. 2006). Neurite exploration is guided by growth cones (GCs) located at their end tip (Bray et al. 1978; Goodman 1996; Song and Poo 2001), formed by extended lamellipodia from which thin filopodia emerge (Mongiu et al. 2007). In our study, LPS-CM cultured neurons showed decreased area of GCs compared with CM without LPS stimulation. Moreover, the number of filopodia per GC and the length of filopodia were also decreased in these cultures. However, when co-cultured with LPS-CM prepared in the presence of RV, all the decreased parameters were restored to the level of untreated control. Furthermore, we found that RV was capable of promoting dendrite sprouting and outgrowth of mouse hippocampal neurons, as evidenced by the increased average dendrite numbers and length after stimulation with LPS-CM (Fig. 6b–d, compared lane 3 and 4). GAP-43 participates in neurite formation and regeneration (Benowitz and Routtenberg 1997). As expected, GAP-43 expression was also significantly increased in LPS + RV-CM group compared in LPS-CM group (Fig. 6e, f), which may explain the enhanced dendrite sprouting and outgrowth of neurons when treated with LPS + RV-CM.
Finally, we tested the effect of our culture system on spinogenesis by observing the changes of spine morphology and density in different stimulatory conditions in vitro. Noticeably, we found LPS + RV-CM could increase spine density compared with the LPS-CM group. Meanwhile, it also induced the formation of dendritic protrusions in stubby-shaped spine. This finding could reflect the potential therapeutic application of RV in CNS insults and recovery. Activated microglia produce free radicals, nitric oxide, and arachidonic acid derivatives, many of which must participate in rendering the damaged CNS inhibitory for axon regeneration. RV could indirectly influence the molecules released during microglial over-activation to exhibit the neuroprotective roles during this process.
Conclusions
We hereby demonstrate that RV exhibits neuroprotective roles for primary culture of mouse hippocampal neurons by promoting dendrite sprouting and outgrowth. Many neurodegenerative diseases or acute injuries to CNS result in loss of neurons and damage of neurite. Based on our results, RV may be potentially used as an alternative therapeutic approach to this insult.
References
Aldskogius H, Kozlova EN (1998) Central neuron-glial and glial-glial interactions following axon injury. Prog Neurobiol 55:1–26
Amin L, Ercolini E, Ban J, Torre V (2013) Comparison of the force exerted by hippocampal and DRG growth cones. PLoS ONE 8:e73025
Anekonda TS, Reddy PH (2006) Neuronal protection by sirtuins in Alzheimer’s disease. J Neurochem 96:305–313
Ates O, Cayli S, Altinoz E, Gurses I, Yucel N, Sener M, Kocak A, Yologlu S (2007a) Neuroprotection by resveratrol against traumatic brain injury in rats. Mol Cell Biochem 294:137–144
Ates O, Cayli SR, Yucel N, Altinoz E, Kocak A, Durak MA, Turkoz Y, Yologlu S (2007b) Central nervous system protection by resveratrol in streptozotocin-induced diabetic rats. J Clin Neurosci 14:256–260
Benowitz LI, Routtenberg A (1997) GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci 20:84–91
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Bray D, Thomas C, Shaw G (1978) Growth cone formation in cultures of sensory neurons. Proc Natl Acad Sci USA 75:5226–5229
Cao Q, Li P, Lu J, Dheen ST, Kaur C, Ling EA (2010) Nuclear factor-kappaB/p65 responds to changes in the Notch signaling pathway in murine BV-2 cells and in amoeboid microglia in postnatal rats treated with the gamma-secretase complex blocker DAPT. J Neurosci Res 88:2701–2714
Choi Y, Lee MK, Lim SY, Sung SH, Kim YC (2009) Inhibition of inducible NO synthase, cyclooxygenase-2 and interleukin-1beta by torilin is mediated by mitogen-activated protein kinases in microglial BV2 cells. Br J Pharmacol 156:933–940
Constant JP, Fraley GS, Forbes E, Hallas BH, Leheste JR, Torres G (2012) Resveratrol protects neurons from cannulae implantation injury: implications for deep brain stimulation. Neuroscience 222:333–342
de Almeida LM, Leite MC, Thomazi AP, Battu C, Nardin P, Tortorelli LS, Zanotto C, Posser T, Wofchuk ST, Leal RB, Goncalves CA, Gottfried C (2008) Resveratrol protects against oxidative injury induced by H2O2 in acute hippocampal slice preparations from Wistar rats. Arch Biochem Biophys 480:27–32
Fawcett JW, Asher RA (1999) The glial scar and central nervous system repair. Brain Res Bull 49:377–391
Fetler L, Amigorena S (2005) Neuroscience. Brain under surveillance: the microglia patrol. Science 309:392–393
Ghashghaei HT, Lai C, Anton ES (2007) Neuronal migration in the adult brain: are we there yet? Nat Rev Neurosci 8:141–151
Goodman CS (1996) Mechanisms and molecules that control growth cone guidance. Annu Rev Neurosci 19:341–377
Griffin RS, Costigan M, Brenner GJ, Ma CH, Scholz J, Moss A, Allchorne AJ, Stahl GL, Woolf CJ (2007) Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci 27:8699–8708
Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M (2009) The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 26:83–94
Holmes-McNary M, Baldwin AS Jr (2000) Chemopreventive properties of trans-resveratrol are associated with inhibition of activation of the IkappaB kinase. Cancer Res 60:3477–3483
Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J (2008) Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci 28:9330–9341
Hung J, Colicos MA (2008) Astrocytic Ca(2+) waves guide CNS growth cones to remote regions of neuronal activity. PLoS ONE 3:e3692
Karlsson J, Emgard M, Brundin P, Burkitt MJ (2000) Trans-resveratrol protects embryonic mesencephalic cells from tert-butyl hydroperoxide: electron paramagnetic resonance spin trapping evidence for a radical scavenging mechanism. J Neurochem 75:141–150
Kitayama M, Ueno M, Itakura T, Yamashita T (2011) Activated microglia inhibit axonal growth through RGMa. PLoS ONE 6:e25234
Kundu JK, Surh YJ (2004) Molecular basis of chemoprevention by resveratrol: NF-kappaB and AP-1 as potential targets. Mutat Res 555:65–80
Liu C, Shi Z, Fan L, Zhang C, Wang K, Wang B (2011) Resveratrol improves neuron protection and functional recovery in rat model of spinal cord injury. Brain Res 1374:100–109
Lynch MA (2004) Long-term potentiation and memory. Physiol Rev 84:87–136
Meiri KF, Pfenninger KH, Willard MB (1986) Growth-associated protein, GAP-43, a polypeptide that is induced when neurons extend axons, is a component of growth cones and corresponds to pp46, a major polypeptide of a subcellular fraction enriched in growth cones. Proc Natl Acad Sci USA 83:3537–3541
Mongiu AK, Weitzke EL, Chaga OY, Borisy GG (2007) Kinetic-structural analysis of neuronal growth cone veil motility. J Cell Sci 120:1113–1125
Montine TJ, Milatovic D, Gupta RC, Valyi-Nagy T, Morrow JD, Breyer RM (2002) Neuronal oxidative damage from activated innate immunity is EP2 receptor-dependent. J Neurochem 83:463–470
Nam KN, Koketsu M, Lee EH (2008) 5-Chloroacetyl-2-amino-1,3-selenazoles attenuate microglial inflammatory responses through NF-kappaB inhibition. Eur J Pharmacol 589:53–57
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318
Park HR, Kong KH, Yu BP, Mattson MP, Lee J (2012) Resveratrol inhibits the proliferation of neural progenitor cells and hippocampal neurogenesis. J Biol Chem 287:42588–42600
Parker JA, Arango M, Abderrahmane S, Lambert E, Tourette C, Catoire H, Neri C (2005) Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet 37:349–350
Pool M, Thiemann J, Bar-Or A, Fournier AE (2008) NeuriteTracer: a novel ImageJ plugin for automated quantification of neurite outgrowth. J Neurosci Methods 168:134–139
Quan N, Sundar SK, Weiss JM (1994) Induction of interleukin-1 in various brain regions after peripheral and central injections of lipopolysaccharide. J Neuroimmunol 49:125–134
Ruaro ME, Bonifazi P, Torre V (2005) Toward the neurocomputer: image processing and pattern recognition with neuronal cultures. IEEE Trans Biomed Eng 52:371–383
Saiko P, Pemberger M, Horvath Z, Savinc I, Grusch M, Handler N, Erker T, Jaeger W, Fritzer-Szekeres M, Szekeres T (2008a) Novel resveratrol analogs induce apoptosis and cause cell cycle arrest in HT29 human colon cancer cells: inhibition of ribonucleotide reductase activity. Oncol Rep 19:1621–1626
Saiko P, Szakmary A, Jaeger W, Szekeres T (2008b) Resveratrol and its analogs: defense against cancer, coronary disease and neurodegenerative maladies or just a fad? Mutat Res 658:68–94
Shao AW, Wu HJ, Chen S, Ammar AB, Zhang JM, Hong Y (2014) Resveratrol attenuates early brain injury after subarachnoid hemorrhage through inhibition of NF-kappaB-dependent inflammatory/MMP-9 pathway. CNS Neurosci Ther 20:182–185
Solecki DJ, Govek EE, Hatten ME (2006) mPar6 alpha controls neuronal migration. J Neurosci 26:10624–10625
Song H, Poo M (2001) The cell biology of neuronal navigation. Nat Cell Biol 3:E81–88
Song Q, Jiang Z, Li N, Liu P, Liu L, Tang M, Cheng G (2014) Anti-inflammatory effects of three-dimensional graphene foams cultured with microglial cells. Biomaterials 35:6930–6940
Streit WJ, Graeber MB, Kreutzberg GW (1988) Functional plasticity of microglia: a review. Glia 1:301–307
Wu HM, Tzeng NS, Qian L, Wei SJ, Hu X, Chen SH, Rawls SM, Flood P, Hong JS, Lu RB (2009) Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology 34:2344–2357
Zhang H, Webb DJ, Asmussen H, Horwitz AF (2003) Synapse formation is regulated by the signaling adaptor GIT1. J Cell Biol 161:131–142
Acknowledgment
This work was supported by Major Program of Medical Scientific Research of Hebei Province, China (Grant No. ZL20140145).
Conflict of interest
The authors (Feng Wang, Na Cui, Lijun Yang, Lin Shi, Qian Li, Gengshen Zhang, Jianliang Wu, Jun Zheng, and Baohua Jiao) declare there is no conflict of interest.
Ethical standard
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Wang, F., Cui, N., Yang, L. et al. Resveratrol Rescues the Impairments of Hippocampal Neurons Stimulated by Microglial Over-Activation In Vitro. Cell Mol Neurobiol 35, 1003–1015 (2015). https://doi.org/10.1007/s10571-015-0195-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-015-0195-5