
Abstract
Rotenone is an inhibitor of mitochondrial complex I-induced neurotoxicity in PC12 cells and has been widely studied to elucidate the pathogenesis of Parkinson’s disease. We investigated the neuroprotective effects of betaine on rotenone-induced neurotoxicity in PC12 cells. Betaine inhibited rotenone-induced apoptosis in a dose-dependent manner, with cell viability increasing from 50 % with rotenone treatment alone to 71 % with rotenone plus 100-μM betaine treatment. Flow cytometric analysis demonstrated cell death in the rotenone-treated cells to be over 50 %; the number of live cells increased with betaine pretreatment. Betaine pretreatment of PC12 cells attenuated rotenone-mediated mitochondrial dysfunction, including nuclear fragmentation, ATP depletion, mitochondrial membrane depolarization, caspase-3/7 activation, and reactive oxygen species production. Western blots demonstrated activation of caspase-3 and caspase-9, and their increased expression levels in rotenone-treated cells; betaine decreased caspase-3 and caspase-9 expression levels and suppressed their activation. Together, these results suggest that betaine may serve as a neuroprotective agent in the treatment of neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There is large variability in the physiological, structural, functional, and cognitive changes that occur during the normal aging of the brain (Riederer et al. 2011). However, structural and functional neuroimaging studies of older adults with intact cognition have consistently shown loss of both volume and structural integrity of the white matter, particularly in the prefrontal cortex (Caserta et al. 2009). The intrinsic mitochondrial apoptotic pathway represents the most common mechanism of neuronal cell death. In particular, Bax and Bak, pro-apoptotic members of the Bcl-2 family, cause the release of cytochrome c from the intermembrane space into the cytosol, where it activates the caspase cascade (Pettmann and Henderson 1998; Ribe et al. 2008).
Mitochondrial dysfunction has long been implicated in both the physiological process of aging and in age-related neurodegenerative disorders such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) (Fukae et al. 2007; Vila et al. 2008; Keane et al. 2011). Neurodegenerative disorders are the result of aberrant neuronal cell death (Mehler and Gokhan 2000). For example, in PD, impaired mitochondrial respiration due to abnormalities in complex I activity in the dopaminergic neurons located in the substantia nigra leads to the generation of reactive oxygen species (ROS), energy depletion, and activation of mitochondrial-dependent apoptotic pathways (Dauer and Przedborski 2003).
Rotenone, an inhibitor of mitochondrial complex I, suppresses ATP production and induces apoptosis by enhancing the production of mitochondrial ROS (Kirkinezos and Moraes 2001). Rotenone-induced neurotoxicity in PC12 cells is widely used to study the pathogenesis of PD (Wei et al. 2008), and is associated with increased processing of pro-caspase-12, pro-caspase-9, and pro-caspase-3, suggesting that both mitochondrial- and endoplasmic reticulum (ER)-dependent caspases are activated. Moreover, rotenone-induced degeneration of spinal cord motor neurons in male Lewis rats is accompanied by upregulation of calpain and caspase-3 levels (Samantaray et al. 2007).
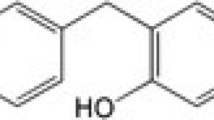
Herbal products with anti-aging properties have drawn considerable interest as candidates for the treatment of neurodegenerative disease (Ho et al. 2010). The natural product betaine is widely distributed in animals, plants, and microorganisms, and is present in many foods such as Lycium chinense (Shin et al. 1999; Craig 2004). Betaine has been shown to be beneficial for a number of conditions and diseases, including heart and liver diseases (Bidulescu et al. 2007; Cave et al. 2007; Du et al. 2009; Ganesan and Anandan 2009). It also has a protective effect against ethanol-induced hepatotoxicity (Kanbak et al. 2001), and against stress-induced oxidative damage in Wistar albino rats (Ganesan et al. 2011).
The purpose of the present study was to determine the neuroprotective properties of betaine in PC12 cells treated with rotenone. We evaluated the effects of betaine on cellular energy metabolism, mitochondrial membrane potential, and changes in the levels of caspase-3 and caspase-9 proteins.
Materials and Methods
Materials
PC12 cells were purchased from the Korean Cell Line Bank (Seoul, Korea). Betaine was purchased from Wako Pure (Tokyo, Japan). Rotenone and 4′,6-diamidino-2-phenylindole (DAPI) solution were purchased from Sigma Chemical (St. Louis, MO, USA). RPMI 1640 and fetal bovine serum (FBS) were purchased from Gibco BRL (Grand Island, NY, USA). CellTiter Aqueous One Solution Cell proliferation assay kit (MTS), DeadEndTM Colorimetric TUNEL System, and Homogeneous Caspase-3/7 Assay kits were purchased from Promega Co. (Madison, WI, USA). Luminescence ATP detection kit (PerkinElmer, Waltham, MA, USA) and JC-1 mitochondrial membrane potential detection kit (Biotium, Hayward, CA, USA) were used. Mitotracker, Image-iT live green ROS detection kit, and MitoSOX, annexin V, and a propidium iodide (PI) double staining kit were purchased from Invitrogen Molecular Probes (San Diego, CA, USA). Ninety-six-well collagen IV-coated plates were purchased from BD Biosciences (Bedford, MA, USA). Bcl-2, Bax, cytochrome c, and the secondary antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Actin and p53 antibodies were obtained from Abcam Ltd (Cambridge, UK). Cleaved caspase-3, caspase-3, cleaved caspase-9, caspase-9, actin, and anti-rabbit antibody were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell Culture and Cell Viability
PC12 cells were cultured in 96-well collagen IV-coated plates in RPMI 1640 with 10 % FBS at 37 °C in a humidified atmosphere of 95 % air and 5 % CO2. The cells were seeded at a density of 1 × 104 cells/well and incubated for 24 h. They were either untreated or pretreated with betaine (10, 50, or 100 μM) for 24 h before the addition of rotenone (5 μM). Cell viability was assessed after 1 h on the basis of reduction of [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt (MTS) to formazan according to the manufacturer’s instruction; samples were assayed at 490 nm using a microplate fluorimeter (Molecular devices, Sunnyvale, CA, USA).
Annexin V Externalization Assay for Apoptosis
Cell apoptosis was determined by flow cytometry using annexin V-FITC and PI double staining (invitrogen) according to the manufacturer’s instructions. Cells were pretreated with betaine (10, 50, or 100 μM) for 1 h before addition of rotenone (5 μM). After 24 h, the cells were trypsinized, washed in PBS, and resuspended in 1× annexin-binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2; pH 7.4). The cell suspension was incubated with 5 μl of FITC-conjugated annexin V and 1 μl of PI for 15 min in the dark. A total of 10,000 cells were counted using a FACSCalibur flow cytometer, and data were analyzed using the CellQuest software (Becton–Dickinson, San Jose, CA, USA).
TUNEL Assay
Rotenone-induced DNA fragmentation was quantified using the TdT-mediated dUTP Nick-End Labeling (TUNEL) assay (Promega) according to the manufacturer’s instructions. Briefly, the cells were fixed with methanol-free 4 % paraformaldehyde at 4 °C and washed with PBS. The slides were immersed in an equilibration buffer for 10 min, which was then replaced by a mixture of 1 μl of TdT enzyme, 5 μl of nucleotide mix, and 45 μl of equilibration buffer. The slides were then kept in the dark for 60 min. SSC (2×) was added for 15 min at room temperature to stop the TdT enzyme reaction. The slides were then immersed in PI for 15 min in the dark. Images were visualized using confocal microscopy with a standard fluorescein filter set (fluorescein: 520 nm; PI: 620 nm).
Caspase-3-like Activity
Cells (5 × 104 cells/well) were seeded in 96-well white plates. They were either untreated or pretreated with betaine (10, 50, or 100 μM) for 1 h before addition of rotenone (5 μM). After 24 h, caspase-3-like activity was measured using Apo-ONETM Homogeneous Caspase 3/7 Assay Kit according to the manufacturer’s instructions. The cells were lysed in 100 μl of homogeneous caspase-3/7 buffer containing the caspase-3 substrate, and the lysates were incubated for 1 h at room temperature. Caspase-3/7 activity was determined using a luminometer.
Western Blotting
Cells were either untreated or pretreated with betaine (10, 50, or 100 μM) for 1 h before addition of rotenone (5 μM). After 24 h, the cells were lysed; 20 μg of protein lysate from each sample was electrophoresed on a 12 % sodium dodecyl sulfate (SDS)-polyacrylamide gel and transferred to PVDF membranes. The membranes were blocked with a 5 % skim milk solution for 1 h at room temperature. Blots were incubated overnight at 4 °C with 1:1,000 dilutions of monoclonal antibodies to cleaved caspase-3, caspase-3, cleaved caspase-9, caspase-9, or actin. The blots were washed thrice for 10 min each with PBS-T. The membranes were then incubated with anti-rabbit secondary antibody for 2 h. Proteins were detected using an enhanced chemiluminescence kit (ECL solution) and quantified using Image J densitometry software (Rasband, W.S., ImageJ, US National Institutes of Health, Bethesda, MD, USA, http://imagej.nih.gov/ij/, 1997–2011).
ATP Measurement
PC12 cells were left either untreated or pretreated with betaine (10, 50, or 100 μM) for 1 h before addition of rotenone (5 μM). After 24 h, the total cellular ATP content was determined using a luminescence ATP detection kit (Petty et al. 1995). The ATP content was determined by running an internal standard and expressed as percentages of untreated cells (control).
Metabolic Flux Analysis
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured with a Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA, USA). The mitochondrial capability to exceed the OCR requires maintaining basic cellular metabolic needs—versus glycolysis (Moran et al. 2010). Cells were seeded at a density of 5 × 104 cells/well in 24-well Seahorse tissue culture microplates and incubated overnight at 37 °C. The cells were either left untreated or pretreated with 100-μM betaine for 24 h. On the day of metabolic flux analysis, the cells were transferred to unbuffered DMEM (DMEM base medium supplemented with 25 mM glucose, 2 mM sodium pyruvate, 31 mM NaCl, and 2 mM GlutaMax; pH 7.4) and incubated at 37 °C in a non-CO2 incubator for 1 h. The effects of 5-μM rotenone were tested on untreated and betaine-treated cells; controls received neither betaine nor rotenone. Baseline measurements reflect oxygen consumption rates prior to rotenone addition.
Mitochondrial Membrane Potential (MMP)
MMP in PC12 cells was measured using the JC-1 mitochondrial membrane potential detection method (Biotium). Cells were seeded in 96-well plates at a density of 1 × 104 cells/well; the cells were either left untreated or treated with betaine (10, 50, or 100 μM) for 1 h before addition of 5-μM rotenone. After 1 h, the media were aspirated from the plates containing untreated or betaine-treated cells, and adherent cells were washed with PBS. The plates were then incubated at 37 °C for 20 min after the addition of 100 μl of 1× JC-1 reagent into the wells. Cells were washed twice more with PBS, and sufficient PBS was added to the wells to cover the cell layer. Red (excitation: 550 nm; emission: 600 nm) and green (excitation: 485 nm; emission: 535 nm) fluorescence were determined using a Softmax Pro 5 fluorescence plate reader (molecular devices). JC-1 fluoresces red when it aggregates in healthy mitochondria with high membrane potentials and fluoresces green when in monomeric form in mitochondria with diminished membrane potential. Thus, compared to the ratio observed in healthy cells, the ratio of red to green fluorescence is lower in dead cells and in cells undergoing apoptosis. For analysis by confocal microscopy, the cells were treated with 1× JC-1 and incubated for 15 min at 37 °C. The cells were imaged using the FV10i-LIV confocal microscope (Olympus, Tokyo, Japan).
Mitochondrial Superoxide
Cells were seeded in 96-well white plates at a density of 1 × 104 cells for detecting quantitative fluorescence; 1 × 103 cells were added to cover slips for confocal analysis. The cells were either untreated or pretreated with betaine (10, 50, or 100 μM) for 1 h before addition of 5-μM rotenone. After 24 h, the media were removed from both the untreated and betaine-treated cells, and cells were washed with PBS. Cells were incubated with 5-μM MitoSOX Red for 20 min at 37 °C. MitoSOX Red fluorescence intensity (excitation: 510 nm; emission: 580 nm) was measured using a Softmax Pro 5 fluorescence plate reader (Molecular Devices).
Confocal Microscopy Analysis
PC12 cells (5 × 104) were plated on a chamber slide. The media were removed and the cells were washed with PBS. The cells were either untreated or pretreated with betaine (10, 50, or 100 μM) for 1 h before adding 5-μM rotenone. Then, the cells were incubated for 45 min with 10-μM Mitotracker Red and 1-μg/ml DAPI to detect mitochondria. Excitation/emission spectra for Mitotracker Red and DAPI were 578/598 and 359/461 nm, respectively. Image-iT™ LIVE Green Reactive Oxygen Species detection kit was used to detect intracellular ROS in live cells. The cells were incubated with 25-μM carboxy-H2DCFDA for 30 min in the dark at 37 °C; 1-μM Hoechst 33342 was added during the last 5 min.
Statistics
All data were expressed as mean ± standard deviation of at least three independent experiments. Data were analyzed by one-way ANOVA, followed by multiple comparisons using the Student’s t test. Differences were considered significant at P < 0.05.
Results
Betaine Shows Neuroprotective Effects in PC12 cells
In pilot studies, we found that a 1-h treatment of PC12 cells with 5-μM rotenone was sufficient to reduce cell viability by 50 % (data not shown). To examine the protective effect of betaine, we performed a cell viability test on the cells treated with vehicle alone, with 5-μM rotenone, or with 5-μM rotenone following pretreatment with betaine. Betaine inhibited rotenone-mediated toxicity in a dose-dependent manner (Fig. 1), with viability increasing from 50 to 71 % of that of the control cells in the rotenone-treated cells and those treated with rotenone and 100-μM betaine. Results from the flow cytometry studies are shown in Fig. 2a. Quadrant statistics demonstrated increasing levels of cell death of greater than 50 % in rotenone-induced cells. Compartmental analysis suggested that rotenone induced apoptosis and necrosis in PC12 cells, whereas pretreatment with betaine protected the cell from apoptosis. The control cells exhibited no TUNEL-positive nuclei, while the rotenone-treated cells showed some TUNEL-positive nuclei (Fig. 2b). Pretreatment with betaine prevented rotenone-induced nuclear fragmentation.

Protective effect of betaine on rotenone-induced neurotoxicity in PC12 cells. Cells were either untreated or pretreated with 10, 50, or 100 μM betaine for 1 h before the addition of 5-μM rotenone. Data are expressed as a percentage of control (mean ± standard deviation). * P < 0.05 versus control, # P < 0.05 versus rotenone only

Representative flow cytometric analysis and TUNEL assay. a Flow cytometric analyses of PC12 cells in which the graphs depict the distinction between apoptosis and necrosis by using a dual-parameter dot plot of FITC-labeled annexin V versus propidium iodide (PI) fluorescence, shown as logarithmic fluorescence intensity. Quadrants refer to live cells (lower left), early apoptotic cells (lower right), necrotic cells (upper left), and late apoptotic cells (upper right). b Apoptosis detection in PC12 cells using the TUNEL assay as viewed by laser scanning confocal microscopy (60 × 1.5). Red fluorescence indicates the uptake of PI; green fluorescence indicates the uptake of fluorescein-12-dUTP, which is reflective of apoptotic cells (Color figure online)
Betaine Prevents Caspase Activation in PC12 Cells
To analyze the apoptotic pathway through which rotenone-induced cell death occurs, caspase-3/7 activity was assessed after exposure to rotenone. Rotenone elevated caspase-3/7 activity to 138.1 % of the activity observed in the control cells (Fig. 3a). Pretreatment with betaine suppressed the increase in caspase-3/7 activity in a dose-dependent manner. Western blotting analysis demonstrated that the relative expression levels of both isoforms (caspase-3 and caspase-9) increased by rotenone treatment and decreased by pretreatment with betaine (Fig. 3b). Increased expression levels of cleaved caspase-3 and caspase-9 suggested that both isoforms were activated up to 155 and 158 %, respectively, by rotenone. However, caspase cleavage was protected by pretreatment with 100 μM of betaine until 73.5 and 85 %, respectively.

Effect of betaine on rotenone-mediated caspase activity. a Cells were either untreated or pretreated with betaine (10, 50, or 100 μM) for 1 h before addition of rotenone (5 μM). After 24 h, the apoptotic activity of caspase-3/7 was assessed using the luminescence-based caspase-3/7 activity kit. b Western blot analysis of the mitochondrion-dependent cell death pathway in rotenone-treated PC12 cells. The phosphorylated and total protein levels were detected by Western blot analysis using specific antibodies. β-actin served as the loading control. Quantitative analysis was performed by measuring the signal intensity relative to the control (n = 3). Data are expressed as a percentage of control (mean ± standard deviation). * P < 0.05 versus control, # P < 0.05 versus rotenone only
Betaine Prevents Rotenone-Mediated Cellular Energy Depletion
Next, we determined whether betaine affects energy production. Rotenone lowered cellular ATP levels to 57.7 % of the control levels (Fig. 4a), while betaine pretreatment attenuated this effect. To assess the impact of rotenone on mitochondrial function in PC12 cells, we measured OCR and ECAR to estimate spare respiratory capacity for oxidative stress. The OCR and ECAR can be assigned to oxidative phosphorylation and glycolysis, respectively. Control cells did not show a significant change in OCR and ECAR, indicating a higher relative level of glycolytic activity to mitochondrial metabolism. Cells have enough substrates for energy production under normal conditions. In contrast, exposure to rotenone resulted in a decrease in OCR and ECAR, indicating a decrease in basal mitochondrial efficiency (Fig. 4b, c). Rotenone treatment also affected oxygen consumption via the involvement of extramitochondrial metabolism. However, the betaine-treated cells did not show alteration of either of these effects.

Effect of betaine on energy metabolism in PC12 cells. a PC12 cells were either untreated or pretreated with betaine (10, 50, or 100 μM) for 1 h before adding rotenone (5 μM). ATP levels were measured using the ATP Lite luminescence-based assay. b The oxygen consumption rate (OCR) or c extracellular acidification rate (ECAR) were monitored after the addition of buffer alone (control), rotenone, or rotenone following pretreatment with 100-μM betaine. Rotenone was applied at the time points indicated by the dashed line. Data are expressed as a percentage of control (mean ± standard deviation). *P < 0.05 versus control, # P < 0.05 versus rotenone only
Betaine Attenuates Rotenone-Induced Mitochondrial Membrane Depolarization
Rotenone significantly depolarized the mitochondrial membrane, as demonstrated by a reduction in the ratio of red to green fluorescence emitted by the probe JC-1 (Fig. 5a, b). Betaine pretreatment suppressed rotenone-induced mitochondrial membrane depolarization; membrane potential was 84.3 % relative to that in the controls upon pretreatment with 100-μM betaine (Fig. 5a).

Mitochondrial membrane potential measured by JC-1 fluorescence. a Cells were either left untreated or treated with betaine (10, 50, or 100 μM) for 1 h before addition of 5 μM rotenone. Red (excitation: 550 nm; emission: 600 nm) and green fluorescence (excitation: 485 nm; emission: 535 nm) was determined using a fluorescence plate reader. b Confocal images of JC-1 fluorescence (60 × 3.5). As a monomer, JC-1 exhibits green fluorescence, reflecting a depolarized mitochondrial membrane. As an aggregate, it exhibits red fluorescence, reflecting a polarized mitochondrial membrane. Data are expressed as a percentage of control (mean ± standard deviation). *P < 0.05 versus control, # P < 0.05 versus rotenone only (Color figure online)
Betaine Suppresses Mitochondrial Superoxide Generation
The generation of mitochondrial superoxide was measured using the MitoSOX Red mitochondrial superoxide indicator. (Fig. 6a, b). Rotenone increased mitochondrial superoxide levels to 187.8 % relative to those in the controls (Fig. 6a). In another study, the addition of the antioxidant ROS-scavenging N-acetyl-l-cysteine (NAC) significantly reduced MitoSOX fluorescence, and thus, mitochondrial ROS levels (Weir et al. 2012). Similarly, betaine pretreatment abolished this response; in the presence of 10-μM betaine, the level of rotenone-stimulated superoxide was 103.2 % of that in the controls.

Determination of mitochondrial superoxide production measured using MitoSOX Red. a Mitochondrial-derived superoxide generation measured by fluorescence (excitation: 510 nm; emission: 580 nm). Cells were either untreated or pretreated with betaine (10, 50, or 100 μM) for 1 h before adding 5-μM rotenone. b Confocal microscopy images of MitoSOX fluorescence (60 × 3.5). Cells were treated as previously described. Data are expressed as a percentage of control (mean ± standard deviation). * P < 0.05 versus control, # P < 0.05 versus rotenone only
Betaine Suppresses Rotenone-Mediated Mitochondrial Dysfunction
Confocal microscopy analysis using Mitotracker probes revealed an even distribution of mitochondrial staining throughout control cells (Fig. 7a). Treatment with rotenone reduced the red fluorescence intensity in mitochondria, reflecting the depolarization of the inner mitochondrial membrane. Pretreatment with betaine for 1 h resulted in a mitochondrial staining pattern similar to that of the control cells. Betaine pretreatment also increased the intensity of red fluorescence and the membrane permeability to DAPI. Finally, we compared ROS production in PC12 cells using the DCF fluorescence method with the LIVE Green Reactive Oxygen Species detection kit. Although rotenone-treated PC12 cells were found to produce more ROS than control cells (Fig. 7b), ROS production was significantly lower in the cells pretreated with betaine.

Confocal microscopy images of mitochondria by using Mitotracker red and DCF dye. a Cells were either untreated or pretreated with betaine (10, 50, or 100 μM) for 1 h before addition of 5-μM rotenone. The distribution of Mitotracker red fluorescence indicates mitochondria; merged images show the colocalization with DAPI blue fluorescence (10 × 3.5). b Images of intracellular reactive oxygen species (ROS) and nuclear chromatin were obtained using confocal microscopy. The Image-iT™ LIVE green reactive oxygen species detection kit was used to detect intracellular ROS in live cells (Color figure online)
Discussion
Herbal treatments have drawn increased interest as alternative therapies to treat neurodegenerative diseases. Therefore, we tested the protective effect of betaine, a widely distributed natural product, on rotenone-mediated toxicity in PC12 cells. Rotenone is a specific inhibitor of the mitochondrial complex and reproduces the neurochemical, behavioral, and pathological features of PD in rats. We demonstrated the anti-apoptotic effect of betaine by multiple approaches, including flow cytometry, DNA fragmentation (TUNEL), and Western detection of caspase activation. Moreover, we showed that betaine protects PC12 cells against rotenone-induced cell death, suggesting that betaine may be a neuroprotective agent. FACS analysis showed an increase in the frequency of apoptosis in rotenone-treated cells; however, compared to treatment with rotenone alone, treatment with betaine increased the number of live cells.
We also found that betaine attenuated the reduction in intracellular ATP, elevation in ROS, mitochondrial membrane depolarization, and caspase-3 activation induced by exposure to rotenone. The maintenance of mitochondrial function is crucial to cell survival, not only in terms of providing energy in the form of ATP, but also for regulating apoptosis. These 2 roles are related in that depletion of ATP by inhibition of the electron transport chain leads to depolarization of the mitochondrial membrane. This in turn results in the release of cytochrome c from the mitochondria into the cytosol. Cytochrome c is a key regulator of caspases, a family of proteins that drive apoptosis. In our study, rotenone decreased the spare respiratory capacity, leading to ATP depletion. Thus, these data provide a link between the inhibition of complex I by rotenone and mitochondrial membrane depolarization. Betaine attenuated both ATP depletion and mitochondrial membrane depolarization. Thus, this represents one mechanism by which betaine prevents the activation of the cascade-dependent apoptotic cascade.
Under conditions of oxidative stress, increased ROS production and oxidative damage to biomolecules are associated with a variety of pathological events including neurodegenerative disorders (Batandier et al. 2002). The production of free radicals by mitochondria has been considered to play an important role in the degradation of cellular function. Numerous studies have shown that the inhibition of complex I, which is the mechanism of action of rotenone, causes an increase in mitochondrial oxidative stress. Similar to mitochondrial membrane depolarization, the subsequent compromise of mitochondrial membrane integrity because of an increase in oxidative stress leads to the release of pro-apoptotic factors into the cytosol. Caspase-dependent pathways represent an important mechanism of the neuronal apoptotic cascade (Velier et al. 1999). In our study, the activities of caspase-3 and caspase-9, and the incidence of apoptotic cell death, increased in PC12 cells concomitant with an elevation in oxidative stress. Betaine suppressed the increase in caspase-3 activity, suggesting that it prevents cell death by down-regulating pro-apoptotic factors.
Neurodegenerative diseases are associated with an impairment of mitochondrial metabolism that leads to increased ROS production and mitochondrial dysfunction. Several studies have shown that neuronal cell death due to aberrant apoptosis is the major reason for cognitive decline in AZ and PD (Martin 2010). Antioxidants, mitochondrial enhancers, anti-apoptotic agents, dopaminergic agents, N-methyl-d-aspartate (NMDA) antagonists, and protein chaperones have all been suggested as putative neuroprotective targets (Tenenbaum et al. 2002). Our results demonstrating that betaine has protective effects in neuronal PC12 cells against rotenone-induced neurotoxicity suggest that betaine may also have a therapeutic neuroprotective effect.
References
Batandier C, Fontaine E, Keriel C, Leverve XM (2002) Determination of mitochondrial reactive oxygen species: methodological aspects. J Cell Mol Med 6:175–187
Bidulescu A, Chambless LE, Siega-Riz AM, Zeisel SH, Heiss G (2007) Usual choline and betaine dietary intake and incident coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) study. BMC Cardiovasc Disord 7:20
Caserta MT, Bannon Y, Fernandez F, Giunta B, Schoenberg MR, Tan J (2009) Chapter 1 Normal brain aging: clinical, immunological, neuropsychological, and neuroimaging features. Int Rev Neurobiol 84:1–19
Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, McClain C (2007) Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem 18:184–195
Craig SA (2004) Betaine in human nutrition. Am J Clin Nutr 80:539–549
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39:889–909
Du YP, Peng JS, Sun A, Tang ZH, Ling WH, Zhu HL (2009) Assessment of the effect of betaine on p16 and c-myc DNA methylation and mRNA expression in a chemical induced rat liver cancer model. BMC Cancer 9:261
Fukae J, Mizuno Y, Hattori N (2007) Mitochondrial dysfunction in Parkinson’s disease. Mitochondrion 7:58–62
Ganesan B, Anandan R (2009) Protective effect of betaine on changes in the levels of lysosomal enzyme activities in heart tissue in isoprenaline-induced myocardial infarction in Wistar rats. Cell Stress Chaperones 14:661–667
Ganesan B, Anandan R, Lakshmanan PT (2011) Studies on the protective effects of betaine against oxidative damage during experimentally induced restraint stress in Wistar albino rats. Cell Stress Chaperones 16:641–652
Ho YS, So KF, Chang RC (2010) Anti-aging herbal medicine–how and why can they be used in aging-associated neurodegenerative diseases? Ageing Res Rev 9:354–362
Kanbak G, Inal M, Baycu C (2001) Ethanol-induced hepatotoxicity and protective effect of betaine. Cell Biochem Funct 19:281–285
Keane PC, Kurzawa M, Blain PG, Morris CM (2011) Mitochondrial dysfunction in Parkinson’s disease. Parkinsons Dis 2011:716871
Kirkinezos IG, Moraes CT (2001) Reactive oxygen species and mitochondrial diseases. Semin Cell Dev Biol 12:449–457
Martin LJ (2010) Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals (Basel) 3:839–915
Mehler MF, Gokhan S (2000) Mechanisms underlying neural cell death in neurodegenerative diseases: alterations of a developmentally-mediated cellular rheostat. Trends Neurosci 23:599–605
Moran M, Rivera H, Sanchez-Arago M, Blazquez A, Merinero B, Ugalde C, Arenas J, Cuezva JM, Martin MA (2010) Mitochondrial bioenergetics and dynamics interplay in complex I-deficient fibroblasts. Biochim Biophys Acta 802:443–453
Pettmann B, Henderson CE (1998) Neuronal cell death. Neuron 20:633–647
Petty RD, Sutherland LA, Hunter EM, Cree IA (1995) Comparison of MTT and ATP based assays for the measurement of viable cell number. J Biolumin Chemilumin 10:29–34
Ribe EM, Serrano-Saiz E, Akpan N, Troy CM (2008) Mechanisms of neuronal death in disease: defining the models and the players. Biochem J 415:165–182
Riederer BM, Leuba G, Vernay A, Riederer IM (2011) The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp Biol Med (Maywood) 236:268–276
Samantaray S, Knaryan VH, Guyton MK, Matzelle DD, Ray SK, Banik NL (2007) The parkinsonian neurotoxin rotenone activates calpain and caspase-3 leading to motoneuron degeneration in spinal cord of Lewis rats. Neuroscience 146:741–755
Shin YG, Cho KH, Kim JM, Park MK, Park JH (1999) Determination of betaine in Lycium chinense fruits by liquid chromatography-electrospray ionization mass spectrometry. J Chromatogr A 857:331–335
Tenenbaum L, Chtarto A, Lehtonen E, Blum D, Baekelandt V, Velu T, Brotchi J, Levivier M (2002) Neuroprotective gene therapy for Parkinson’s disease. Curr Gene Ther 2:451–483
Velier JJ, Ellison JA, Kikly KK, Spera PA, Barone FC, Feuerstein GZ (1999) Caspase-8 and caspase-3 are expressed by different populations of cortical neurons undergoing delayed cell death after focal stroke in the rat. J Neurosci 19:5932–5941
Vila M, Ramonet D, Perier C (2008) Mitochondrial alterations in Parkinson’s disease: new clues. J Neurochem 107:317–328
Wei HL, Jiafeng S, Ming C, Hongyan C, Linsen H (2008) Mechanisms of rotenone-induced neurotoxicity in PC 12 cells. Neural Regen Res 3:1281–1285
Weir HJ, Murray TK, Kehoe PG, Love S, Verdin EM, O’Neill MJ, Lane JD, Balthasar N (2012) CNS SIRT3 expression is altered by reactive oxygen species and in Alzheimer’s disease. PLoS One 7:e48225
Acknowledgments
This research was supported by the “Study of aging-control by energy metabolism based on oriental medicine (K12101)” funded by the “KM-Based Herbal Drug Research Group” of the Korea Institute of Oriental Medicine.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Im, AR., Kim, YH., Uddin, M.R. et al. Betaine Protects Against Rotenone-Induced Neurotoxicity in PC12 Cells. Cell Mol Neurobiol 33, 625–635 (2013). https://doi.org/10.1007/s10571-013-9921-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-013-9921-z