
Abstract
Beta-amyloid peptide (Aβ), a major protein component of senile plaques, has been considered as a critical cause in the pathogenesis of Alzheimer’s disease (AD). Modulation of the Aβ-induced neurotoxicity has emerged as a possible therapeutic approach to ameliorate the onset and progression of AD. The present study aimed to evaluate the protective effect of isorhynchophylline, an oxindole alkaloid isolated from a Chinese herb Uncaria rhynchophylla, on Aβ-induced neurotoxicity in cultured rat pheochromocytoma (PC12) cells. The results showed that pretreatment with isorhynchophylline significantly elevated cell viability, decreased the levels of intracellular reactive oxygen species and malondialdehyde, increased the level of glutathione, and stabilized mitochondrial membrane potential in Aβ25-35-treated PC12 cells. In addition, isorhynchophylline significantly suppressed the formation of DNA fragmentation and the activity of caspase-3 and moderated the ratio of Bcl-2/Bax. These results indicate that isorhynchophylline exerts a neuroprotective effect against Aβ25-35-induced neurotoxicity in PC12 cells, at least in part, via inhibiting oxidative stress and suppressing the mitochondrial pathway of cellular apoptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD), the most common form of dementia in the elderly, is characterized by the progressive deterioration of learning, memory, and other cognitive functions. AD is now known as an irreversible and progressive neurodegenerative disease. Senile plaques, neurofibrillary tangles, and extensive neuronal loss are the main histological hallmarks observed in AD brains (Katzman and Saitoh 1991). Beta-amyloid peptide (Aβ), the major component of senile plaques, has been considered to play an important role in the development and progression of AD (Hardy 1997; Selkoe 2000). Although the precise mechanism of Aβ-induced neurotoxicity is not completely elucidated, several lines of evidence suggest that oxidative stress is closely involved in its pathogenesis, and excessive reactive oxygen species (ROS) production can cause neuronal apoptosis in AD patients (Li et al. 2008; Zhang et al. 2008; Hu et al. 2010). Antioxidants and free radical scavengers have been shown to elicit a beneficial effect both in vitro and in vivo against Aβ-induced neurotoxicity (Heo et al. 2004). Therefore, therapeutic intervention with antioxidants may help to prevent Aβ-induced neurotoxicity and improve neurological outcome in AD.
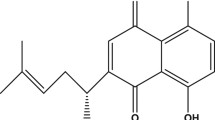
Currently, the pharmacological treatment used to maintain cognitive functions of AD patients primarily consists of two types of drugs in clinical practice, that is, the acetylcholinesterase inhibitors (AChEIs) and the glutamate modulators (Knopman 2006). In addition, several alternative approaches including anti-inflammatory agents, antioxidants, estrogens, and anti-Aβ-peptides agents can also be used to ameliorate the symptoms of AD (Yamada and Nabeshima 2000). However, effective approaches for delaying the progression of AD are yet to be found to date. Thus, searching for safer, better-tolerated, and effective drugs for the treatment of AD remains an important area of drug discovery. During the last decade, many herbal medicines have been tested and demonstrated to be beneficial in different AD-related experimental models as well as in clinical trials (Baum et al. 2008; Huang et al. 2008; Xian et al. 2011). Uncaria rhynchophylla (Miq.) Miq. ex Havil. (Rubiaceae) is one of the best-known herbs in China, Korea, and Japan. It is a component herb of many popular herbal formulae, such as Chotosan (Gouteng-San in Chinese) and Yokukansan (Yigan-San in Chinese), prescribed for the treatment of AD (Watanabe et al. 2003; Tabuchi et al. 2009). Previous study in our laboratory showed that 70% aqueous ethanol extract of Uncaria rhynchophylla was able to ameliorate cognitive deficits induced by D-galactose in mice (Xian et al. 2011). Isorhynchophylline (the chemical structure shown in Fig. 1), an oxindole alkaloid isolated from Uncaria rhynchophylla, has been identified as the main active ingredient responsible for the biological activities of Uncaria rhynchophylla (Kang et al. 2004; Yuan et al. 2009). Recent studies have demonstrated that isorhynchophylline protects against the ischemia- and glutamate-induced neuronal damage or death (Shimada et al. 1999; Kang et al. 2004) and suppresses 5-HT receptor function (Kanatani et al. 1985; Matsumoto et al. 2005). Although the protective effects of isorhynchophylline have been described in different models of neurotoxicity, there is no direct evidence regarding the protective property of isorhynchophylline in the case of Aβ insult. Thus, the aim of the present study was to investigate whether isorhynchophylline has protective effect against Aβ-induced neurotoxicity in PC12 cells and to explore the underlying molecular mechanisms of its neuroprotective action.

Chemical structure of isorhynchophylline
Materials and Methods
Drugs and Reagents
Isorhynchophylline (purity ≥ 98%) was purchased from Chengdu Mansite Pharmaceutical Co. Ltd. (Chengdu, Sichuan, China). Its identity was confirmed by comparing its H1 NMR spectra with that published in the literature (Haginiwa et al., 1973). β-Amyloid peptide (Aβ25-35), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and rhodamine 123 were purchased from Sigma-Aldrich (St. Louis, MO, USA). 2’,7’-Dichlorofluorescin diacetate (DCFH-DA) was obtained from Invitrogen (Carlsbad, CA, USA). Dulbecco’s modified Eagle medium (DMEM), fetal bovine serum (FBS), penicillin, and streptomycin were purchased from Gibco (Grand Island, NY, USA). All other reagents and chemicals used in the study were of analytical grade.
Preparation of Aggregated Aβ25-35
Aβ25-35 was dissolved in deionized distilled water at a concentration of 1 mM and incubated at 37°C for 4 days to induce aggregation (Li et al. 2008). After aggregation, the solution was stored at −20°C until use.
Cell Culture and Treatment
The PC12 cells were obtained from the American Type Culture Collection (Rockville, MD, USA). They were maintained in DMEM supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), 6% FBS, and 6% horse serum at 37°C in a humidified atmosphere of 95% air and 5% CO2. The cells were seeded onto 96-well culture plate at a density of 2 × 104 cells/well, unless otherwise specified. The cells were at first stabilized at 37°C for 48 h and subsequently cultured in free serum medium and incubated with different concentrations of isorhynchophylline (final concentrations: 1, 10, and 50 μM) for 2 h. Aβ25-35 at a final concentration of 20 μM was then added to the culture for an additional 24 h.
Cell Viability Assay
Cell viability in the presence of isorhynchophylline was measured by quantitative colorimetric assay with MTT method as described previously (Mao et al. 2011). Briefly, after drug treatment, 20 μl/well of MTT solution (final concentration, 1 mg/ml) was added, and cells were incubated at 37°C for 4 h. The supernatants were then aspirated off, and formazan crystals were dissolved with 150 μl of DMSO. The optical density of each well was determined at 570 nm using a FLUOstar OPTIMA microplate reader (BMG Labtech, Offenbury, Germany). Cell viability was expressed as percentage of the non-treated control.
Measurement of Intracellular ROS Production
Intracellular ROS level was measured using the 2′,7′-dichlorofluorescein diacetate (DCFH-DA) method (Mao et al. 2011). DCFH-DA is a nonfluorescent compound that can be enzymatically converted to dichlorofluorescein (DCF), a highly fluorescent compound, in the presence of ROS. Briefly, at the end of drug treatment, the cells were washed with D-Hanks solution and incubated with DCFH-DA at a final concentration of 10 μM for 30 min at 37°C in dark. After the cells were washed twice with D-Hanks solution to remove the extracellular DCFH-DA, the fluorescence intensity of DCF was measured in a microplate reader (BMG Labtech) at an excitation wavelength of 485 nm and an emission wavelength of 538 nm. The level of intracellular ROS was expressed as percentage of the non-treated control.
Malondialdehyde (MDA) and Glutathione (GSH) Assay
The PC12 cells were seeded onto 100-mm2 dish at 5 × 106 cells/dish. At the end of drug treatment, the cells were washed with D-Hanks solution, then scraped from the plates into 1 ml ice-cold PBS (0.1 M, containing 0.05 mM EDTA), and homogenized. The homogenate was centrifuged at 4,000×g for 30 min at 4°C. The resulting supernatants were stored at −80°C until the following analyses. Protein content was measured by Bradford method with bovine serum albumin as a standard (Bradford 1976). MDA content was measured as previously described (Xian et al. 2011). Briefly, an aliquot (100 μl) of supernatant was mixed with 1,500 μl acetic acid (20% v/v, pH 3.5), 1,500 μl thiobarbituric acid (0.8%, w/v), and 200 μl sodium dodecyl sulfate (8%, w/v). Each reaction mixture was heated at 95°C for 60 min and then cooled to room temperature. Next, 5,000 μl of n-butanol was added. After mixing and centrifugation at 3,000×g for 10 min, the organic layer was collected and the absorbance measured at 532 nm. MDA level was normalized to the protein concentration of each sample and expressed as percentage of non-treated control. GSH content was measured using a method previously described (Xian et al. 2011). Briefly, an aliquot (100 μl) of supernatant was mixed with 200 μl trichloroacetic acid (25%, v/v) and 200 μl saline. The mixture was centrifuged (3,000×g) for 10 min at 4°C. Then, 200 μl of supernatant was mixed with 1,000 μl phosphate buffer (100 mM, pH 8.0) and 50 μl 5,5-dithiobis-2-nitrobenzoic acid (3 mM). The solution was kept at room temperature for 5 min and the absorbance measured at 412 nm. The GSH level was normalized to the protein concentration of each sample and expressed as percentage of the non-treated control.
Measurement of Mitochondrial Membrane Potential
The mitochondrial membrane potential was measured by using rhodamine 123 fluorescent dye. Rhodamine 123 can enter the mitochondrial matrix and cause photoluminescent quenching dependent on mitochondrial transmembrane potential. The PC12 cells were incubated with 5 mg/l rhodamine 123 for 30 min at 37°C in the dark. After incubation, cells were washed with PBS three times, and the fluorescence intensity was measured at an excitation wavelength of 488 nm and an emission wavelength of 510 nm using a fluorescence microplate reader. Mitochondrial membrane potential was expressed as percentage of the non-treated control.
Quantification of DNA Fragmentation
Quantification of DNA fragmentation was determined by Cell Death Detection ELISAPlus kit (Roche Applied Sciences, Basel, Switzerland) according to the manufacturer’s protocol. In brief, the cells were washed with D-Hanks solution after drug treatment. Then, the cells were incubated with 200 μl of lysis buffer for 30 min at room temperature. The plate was centrifuged at 200×g for 10 min at 4°C. An aliquot (20 μl) of the supernatant from each well was transferred to a streptavidin-coated microplate and incubated with a mixture of anti-histone-biotin and anti-DNA-peroxidase. The apoptotic nucleosomes were captured via their histone component by the anti-histone-biotin antibody that was bound to the streptavidin-coated microplate. Simultaneously, anti-DNA-peroxidase was bound to the DNA part of the nucleosomes. After removing the unbound antibodies, the amount of peroxidase retained in the immunocomplex was quantified by adding 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) as the substrate, and the absorbance of the reaction mixture was measured at 405 nm using a microplate reader. The absorbance is directly proportional to the number of apoptotic nucleosomes. The extent of DNA fragmentation was expressed as percentage of the non-treated control.
Measurement of Caspase-3 Activity
The activity of caspase-3 was measured using a colorimetric assay kit (Sigma-Aldrich, St Louis, MO, USA) according to the manufacturer’s protocol. In brief, the PC12 cells were seeded onto 6-well culture plate at a 2 × 106 cells/well. The cells were washed with D-Hanks solution after drug treatment. Then, the cells were incubated with 500 μl of lysis buffer on ice for 30 min. The cells were transferred to a centrifuge tube and centrifuged at 16,000×g for 10 min at 4°C. An aliquot of the supernatant was incubated with the substrate (acetyl-Asp-Glu-Val-Asp-p-nitroanilide) at 37°C for 90 min. The activity of caspase-3 was measured spectrophotometrically at 405 nm. Data were expressed as percentage of the non-treated control.
Western Blotting Analysis
The PC12 cells were seeded onto 100-mm2 dish at 5 × 106 cells/dish. The cells were washed twice with D-Hanks solution after drug treatment. The cells were harvested and lysed with protein lysis buffer. Protein samples were electrophoresed by SDS-PAGE for 2 h at 80 V. The separated proteins were transferred to PVD membranes using a transblotting apparatus (Bio-Rad Laboratories, USA) for 30 min at 15 V. The membranes were blocked with 5% (w/v) nonfat milk in TBS-T (Tris-buffer saline containing 0.1% Tween-20) at room temperature for 2 h and subsequently incubated at 4°C overnight with appropriate amount of primary antibody against Bcl-2, Bax, and β-actin (Santa Cruz Biotechnology Inc., USA). Next, the membrane was washed with TBS-T three times and probed with horseradish peroxidase–conjugated secondary antibody at room temperature for 1 h. To verify equal loading of samples, the membranes were incubated with monoclonal antibody β-actin, followed by a horseradish peroxidase–conjugated goat anti-mouse IgG. The membrane again was washed with TBS-T for three times, and finally, the protein bands were visualized by the ECL western blotting detection reagents (Amersham Biosciences, Buckinghamshire, UK). The intensity of each band was analyzed using Image J software (NIH Image, Bethesda, MD, USA).
Statistical Analysis
Data were expressed as mean ± SEM. Multiple group comparisons were performed using one-way analysis of variance (ANOVA) followed by Dunnett’s test in order to detect inter-group differences. GraphPad Prism software was used to perform the statistical analysis (Version 4.0; GraphPad Software, Inc., San Diego, CA). A difference was considered statistically significant if the p value was less than 0.05.
Results
Effect of Isorhynchophylline on Aβ25–35-Induced Cytotoxicity in PC12 cells
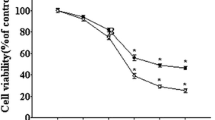
As shown in Fig. 2, treatment of PC12 cells with 20 μM of Aβ25–35 for 24 h induced cytotoxicity as the cell viability was reduced to 67% of the control value (100%). When the cells were pretreated with isorhynchophylline at the concentrations of 1, 10, and 50 μM for 2 h, followed by exposure to 20 μM of Aβ25–35 for 24 h, the cell viability was significantly increased (77, 84, and 94% of the control value, respectively) as compared with the Aβ25–35 group, indicating that isorhynchophylline conferred protection against Aβ25–35-induced cytotoxicity to PC12 cells.

Effect of isorhynchophylline on Aβ25-35-induced cytotoxicity in PC12 cells. Values given are the mean ± SEM (n = 6). # P < 0.001 compared with the control group; *P < 0.05 and **P < 0.01 compared with the Aβ25-35-treated group
Effect of Isorhynchophylline on Aβ25–35-Induced Oxidative Stress in PC12 Cells
As shown in Fig. 3, oxidative stress was assessed by measuring the levels of intracellular ROS (Fig. 3a), MDA (Fig. 3b) and GSH (Fig. 3c). After exposure of PC12 cells to 20 μM Aβ25–35 for 24 h, intracellular ROS and MDA levels were significantly elevated to 234 and 177%, respectively, of the control value, while GSH level was substantially attenuated to 57% of the control value, suggesting that Aβ25–35 induced marked oxidative stress. When PC12 cells were pretreated with isorhynchophylline at the concentrations of 1, 10 and 50 μM for 2 h, followed by exposure to 20 μM of Aβ25–35 for 24 h, intracellular ROS production was significantly reduced (186, 158 and 132% of the control value, respectively) as compared with the Aβ25–35 group. Pretreatment with isorhynchophylline at the concentrations of 10 and 50 μM also significantly decreased MDA level (139 and 124% of the control value, respectively) as compared with the Aβ25–35 group. On the other hand, pretreatment with isorhynchophylline at the concentrations of 10 and 50 μM markedly increased the GSH level (72 and 85% of the control value, respectively) as compared with the Aβ25–35 group. These experimental findings are indicative that isorhynchophylline treatment could significantly reduce the Aβ25–35-induced oxidative stress in PC12 cells.

Effect of isorhynchophylline on Aβ25-35-induced oxidative stress in PC12 cells. Oxidative stress was assessed by measuring intracellular ROS level (a), MDA level (b), and GSH level (c). Values given are the mean ± SEM (n = 6). # P < 0.001 compared with the control group; *P < 0.05 and **P < 0.01 compared with the Aβ25-35-treated group
Effect of Isorhynchophylline on Mitochondrial Membrane Potential in Aβ25–35-Treated PC12 Cells
As shown in Fig. 4, treating PC12 cells with 20 μM of Aβ25–35 for 24 h caused a significant decrease in the mitochondrial membrane potential (67% of the control value). When the cells were pretreated with isorhynchophylline at the concentrations of 10 and 50 μM for 2 h, followed by exposure to 20 μM of Aβ25–35 for 24 h, the mitochondrial membrane potential of the PC12 cells was significantly increased (84 and 89% of the control value, respectively) as compared with the Aβ25–35 group.

Effect of isorhynchophylline on mitochondrial membrane potential in Aβ25-35-treated PC12 cells. Values given are the mean ± SEM (n = 6). # P < 0.001 compared with the control group; *P < 0.05 and **P < 0.01 compared with the Aβ25-35-treated group
Effect of Isorhynchophylline on Aβ25–35-Induced Apoptosis in PC12 Cells
Cellular apoptosis was assessed using a number of apoptotic assays including measuring the extent of DNA fragmentation (Fig. 5a), the activity of caspase-3 (Fig. 5b), and the Bcl-2/Bax expression ratio (Fig. 5c, d). Treating PC12 cells with 20 μM of Aβ25–35 for 24 h caused a significant augmentation in the amount of DNA fragmentation (208% of the control value) and the activity of caspase-3 (229% of the control value), while it caused a decrease in the ratio of Bcl-2/Bax (44% of the control value), suggesting that Aβ25–35 treatment was capable of inducing apoptosis of PC12 cells. When PC12 cells were pretreated with isorhynchophylline at the concentrations of 10 and 50 μM for 2 h, followed by exposure to 20 μM of Aβ25–35 for 24 h, the extent of DNA fragmentation (157 and 135% of the control value, respectively) was significantly decreased as compared with the Aβ25–35 group. Pretreated with isorhynchophylline at the concentrations of 1, 10, and 50 μM also significantly decreased the activity of caspase-3 (195, 164, and 149% of the control value, respectively) and accentuated the Bcl-2/Bax expression ratio (85, 70, and 82% of the control value, respectively) as compared with the Aβ25–35 group. These experimental results unambiguously indicate the ability isorhynchophylline to protect PC12 cells from Aβ25–35-mediated cellular apoptosis.

Effect of isorhynchophylline on Aβ25-35-induced apoptosis in PC12 cells. Apoptosis was assessed by measuring the extent of DNA fragmentation (a), the activity of caspase-3 (b), quantitative analysis of expression of Bcl-2 and Bax (c), and the ratio of values of Bcl-2/Bax (d). Values given are the mean ± SEM (n = 3-6). # P < 0.001 compared with the control group; *P < 0.05, **P < 0.01 and **P < 0.001 compared with the Aβ25-35-treated group
Discussion
Uncaria rhynchophylla has long been used in Chinese medicine and Japanese Kampo medicine to treat vascular dementia, stroke, and other neurodegenerative diseases (Yuan et al. 2009). Isorhynchophylline, an oxindole alkaloid isolated from the stem with hooks of this plant, is easily absorbed into the body and readily penetrates the blood–brain barrier (Huang et al. 2001), rendering it a promising therapeutic agent for the treatment of various neurodegenerative diseases. It is worth noting that Uncaria rhynchophylla contains about 0.020% of isorhynchophylline (Xian et al. 2011). Although Uncaria rhynchophylla has been a subject of many pharmacological investigations both in vitro and in vivo experimental systems (Fujiwara et al. 2006; Xian et al. 2011), for neuroprotective effect, our present study for the first time demonstrated the neuroprotective effect of isorhynchophylline, one of the active principles of Uncaria rhynchophylla, against Aβ-mediated cytotoxicity in cultured PC12 cells. Treatment with isorhynchophylline significantly increased the cell viability and mitochondrial membrane potential, while decreased oxidative stress. In addition, we also found that alteration of Bcl-2 protein family was involved in neuroprotective action of isorhynchophylline against Aβ-induced cellular toxicity.
Oxidative stress, defined as a disturbance in the balance between the production of ROS and antioxidant defense systems, has been implicated in the neuronal injury induced by Aβ (Li et al. 2008; Zhang et al. 2008). Growing evidence from experimental models and human brain studies suggests that oxidative stress plays an important role in neuronal degeneration in AD (Chauhan and Chauhan 2006). Previous in vitro and in vivo studies showed that Aβ treatment caused a significant increase in the level of ROS (Li et al. 2008; Peng et al. 2009). Excessive ROS production is known to cause oxidative damage to major macromolecules in cells, including DNA, lipids, and proteins, thereby disrupting cellular functions and integrity (Gardner et al. 1997; Fiers et al. 1999). In this study, Aβ25-35 was found to cause a marked elevation of oxidative stress characterized by excessive ROS and MDA production, and a reduction in GSH level. Pretreatment with isorhynchophylline effectively mitigated these changes, indicating that the neuroprotective effect of isorhynchophylline may be attributed to its antioxidant ability.
Mitochondrial membrane potential is crucial in determining cell survival and death, particularly under the influence of oxidative stress (Perez and Cederbaum 2003). Mitochondrial dysfunction has been found in cells treated with Aβ (Zhang et al. 2008), AD transgenic mice (Eckert et al. 2008), platelets from AD patients (Parker et al. 1990), as well as postmortem brains of AD patients (Devi et al. 2006). Mitochondrial dysfunction, a prominent feature of Aβ-induced neuronal toxicity in AD patients (Chen and Yan 2007), is central to the development of oxidative stress because the mitochondrion is a primary source of cellular oxidants (Beal 2005). Mitochondrial disruption would increase ROS production and lead to abnormalities in mitochondrial functions in cells. Our finding indicated that treating PC12 cells with Aβ25-35 reduced the mitochondrial membrane potential, and pretreatment with isorhynchophylline was able to restore the mitochondrial membrane potential, suggesting that mitochondria are the subcellular organelle sites associated with the protective effect of isorhynchophylline against Aβ25-35-induced neurotoxicity.
It is well known that neuronal apoptosis is a leading pathway for Aβ-induced neurotoxicity and prevention of Aβ-triggered apoptosis is viewed as a reasonable therapeutic strategy for AD (Hardy and Selkoe 2002; Bachurin 2003). Apoptosis is actively regulated by several members of the caspase family, including caspase-3, which is a major executioner of apoptotic signals that catalyzes the cleavage of many cellular regulatory proteins (May and Madge 2007; Cheung et al. 2008). The mitochondrial pathway of apoptosis is regulated by the Bcl-2 family proteins consisting of several homologous proteins including anti-apoptotic proteins such as Bcl-2 and pro-apoptotic proteins including Bax (Kosten et al. 2008). Anti-apoptotic Bcl-2 appeared to inhibit the mitochondria depolarization and ROS production, while pro-apoptotic Bax induced mitochondrial depolarization and ROS production. Upon the occurrence of the mitochondrial depolarization induced by excessive ROS, the permeability transition pore opened, and intermembrane proteins are released out of the mitochondria, and the event subsequently activates the downstream executive caspase-3 that causes cell death (Tamatani et al. 1998; Gross et al. 1999). Thus, the balance of pro- and anti-apoptotic proteins is an important determinant for cell survival or death (Li et al. 2008; Wang et al. 2008). The findings of parallel increases in caspase-3 activity and Bcl-2/Bax expression ratio in Aβ25-35-treated PC12 cells indicate the involvement of mitochondrial pathway in triggering the Aβ25-35-induced apoptosis. Our above experimental findings are consistent with the observations in previous studies (Li et al. 2008; Zhang et al. 2008). The increase in Bcl-2/Bax expression ratio and the inhibition of caspase-3 activity in Aβ25-35-treated cells by isorhynchophylline suggest the ability of isorhynchophylline to suppress the mitochondrial apoptotic pathway.
In conclusion, our results show that isorhynchophylline exerts a protective effect against Aβ25-35-induced neurotoxicity in PC12 cells. The protective effect of isorhynchophylline was mediated through inhibiting oxidative stress, stabilizing mitochondrial function, and reducing neuronal apoptosis. These observations not only place the use of Uncaria rhynchophylla in traditional medicine for the treatment of various neurological diseases on a scientific foundation but also render isorhynchophylline a promising naturally occurring chemical constituent worthy of further development into pharmaceutical therapy for AD.
References
Bachurin SO (2003) Medicinal chemistry approaches for the treatment and prevention of Alzheimer’s disease. Med Res Rev 23:48–88
Baum L, Lam CW, Cheung SK, Kwok T, Liu V, Tsoh J, Lam L, Leung v, Hui E, Ng C, Woo J, Chiu HF, Goggins WB, Zee BC, Cheng KF, Fong CY, Wong A, Mok H, Chow MS, Ho PC, Ip SP, Ho CS, Yu XW, Lai CY, Chan MH, Szeto S, Chan IH, Mok V (2008) Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer’s disease. J Clin Psychopharmacol 28:110–113
Beal MF (2005) Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58:495–505
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chauhan V, Chauhan A (2006) Oxidative stress in Alzheimer’s disease. Pathophysiology 13:195–208
Chen JX, Yan SD (2007) Pathogenic role of mitochondrial [correction of mitochondrial] amyloid-beta peptide. Expert Rev Neurother 7:1517–1525
Cheung ZH, Leung MC, Yip HK, Wu W, Siu FK, So KF (2008) A neuroprotective herbal mixture inhibits caspase-3-independent apoptosis in retinal ganglion cells. Cell Mol Neurobiol 28:137–155
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26:9057–9068
Eckert A, Hauptmann S, Scherping I, Rhein V, Muller-Spahn F, Götz J, Muller WE (2008) Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener Dis 5:157–159
Fiers W, Beyaert R, Declercq W, Vandenabeele P (1999) More than one way to die: apoptosis and necrosis and reactive oxygen damage. Oncogene 18:7719–7730
Fujiwara H, Iwasaki K, Furukawa K, Seki S, He M, Maruyama M, Tomita N, Kudo Y, Higuchi M, Saido TC, Maeda S, Takashima A, Hara M, Ohizumi Y, Arai H (2006) Uncaria rhynchophylla, a Chinese medicinal herb, has potent antiaggregation effects on Alzheimer’s beta-amyloid proteins. J Neurosci Res 84:427–433
Gardner AM, Xu FH, Fady C, Jacoby FJ, Duffey DC, Tu Y, Lichtenstein A (1997) Apoptotic versus nonapoptotic cytotoxicity induced by hydrogen peroxide. Free Radic Biol Med 22:73–83
Gross A, McDonnell JM, Korsmeyer SJ (1999) Bcl-2 family members and the mitochondria in apoptosis. Genes Dev 13:1899–1911
Haginiwa J, Sakai S, Aimi N, Yamanaka E, Shinma N (1973) Studies of plants containing indole alkaloids. 2. On the alkaloids of Uncaria rhynchophylla Miq. Yakugaku Zasshi 93:448–452
Hardy H (1997) Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci 20:154–159
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356
Heo H, Kim DO, Choi SJ, Shin DH, Lee CY (2004) Potent inhibitory effect of flavonoids in Scutellaria baicalensis on amyloid β protein-induced neurotoxicity. J Agric Food Chem 52:4128–4132
Hu JF, Chu SF, Ning N, Yuan YH, Xue W, Chen NH, Zhang JT (2010) Protective effect of (-) clausenamide against Aβ-induced neurotoxicity in differentiated PC12 cells. Neurosci Lett 483:78–82
Huang B, Wu Q, Wen G, Lu Y, Shi J (2001) The distribution of isorhynchophylline in the tissues of the rats and the determination of its plasma half-time. Acta Academiae Medicinae Zunyi 24:119–120
Huang SH, Lin CM, Chiang BH (2008) Protective effects of Angelica sinensis extract on amyloid β-peptide-induced neurotoxicity. Phytomedicine 15:710–721
Kanatani H, Kohda H, Yamasaki K, Hotta I, Nakata Y, Segawa T, Yamanaka E, Aimi N, Sakai S (1985) The active principle of the branchlets and hook of Uncaria sinensis Oliv. examined with a 5-hydroxytryptamine receptor-binding assay. J Pharm Pharmacol 37:401–404
Kang TH, Murakami Y, Takayama H, Kitajima M, Aimi N, Watanabe H, Matsumoto K (2004) Protective effect of rhynchophylline and isorhynchophylline on in vitro ischemia-induced neuronal damage in the hippocampus: putative neurotransmitter receptors involved in their action. Life Sci 76:331–343
Katzman R, Saitoh T (1991) Advances in Alzheimer’s disease. FASEB J 5:278–286
Knopman DS (2006) Current treatment of mild cognitive impairment and Alzheimer’s disease. Curr Neurol Neurosci Rep 6:365–371
Kosten TA, Galloway MP, Duman RS, Russell DS, D’Sa C (2008) Repeated unpredictable stress and antidepressants differentially regulate expression of the bcl-2 family of apoptotic genes in rat cortical, hippocampal, and limbic brain structures. Neuropsychopharmacology 33:1545–1558
Li G, Ma R, Huang C, Tang Q, Fu Q, Liu H, Hu B, Xiang J (2008) Protective effect of erythropoietin on β-amyloid-induced PC12 cell death through antioxidant mechanisms. Neurosci Lett 442:143–147
Mao QQ, Xian YF, Ip SP, Tsai SH, Che CT (2011) Protective effects of peony glycosides against corticosterone-induced cell death in PC12 cells through antioxidant action. J Ethnopharmacol 133:1121–1125
Matsumoto K, Morishige R, Murakami Y, Tohda M, Takayama H, Sakakibara I, Watanabe H (2005) Suppressive effects of isorhynchophylline on 5-HT2A receptor function in the brain: behavioural and electrophysiological studies. Eur J Pharmacol 517:191–199
May MJ, Madge LA (2007) Caspase inhibition sensitizes inhibitor of NF-kappaB kinase beta-deficient fibroblasts to caspase-independent cell death via the generation of reactive oxygen species. J Biol Chem 282:16105–16116
Parker WD Jr, Fillery CM, Parks JK (1990) Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 40:1302–1303
Peng Y, Xing C, Xu S, Lemere CA, Chen G, Liu B, Wang L, Feng Y, Wang X (2009) L-3-n-butylphthalide improves cognitive impairment induced by intracerebroventricular infusion of amyloid-β peptide in rats. Eur J Pharmacol 621:38–45
Perez MJ, Cederbaum AI (2003) Adenovirus-mediated expression of Cu/Zn- or Mn- superoxide dismutase protects against CYP2E1-dependent toxicity. Hepatology 38:1146–1158
Selkoe DJ (2000) Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci 924:17–25
Shimada Y, Goto H, Itoh T, Sakakibara I, Kubo M, Sasaki H, Terasawa K (1999) Evaluation of the protective effects of alkaloids isolated from the hooks and stems of Uncaria sinensis on glutamate-induced neuronal death in cultured cerebellar granule cells from rats. J Pharm Pharmacol 51:715–722
Tabuchi M, Yamaguchi T, Lizuka S, Imamura S, Ikarashi Y, Kase Y (2009) Ameliorative effects of yokukansan, a traditional Japanese medicine, on learning and non-cognitive disturbances in the Tg2576 mouse model of Alzheimer’s disease. J Ethnopharmacol 122:157–162
Tamatani M, Ogawa S, Nunez G, Tohyama M (1998) Growth factors prevent changes in Bcl-2 and Bax expression and neuronal apoptosis induced by nitric oxide. Cell Death Differ 5:911–919
Wang W, Huang W, Li L, Ai H, Sun F, Liu C, An Y (2008) Morroniside prevents peroxide-induced apoptosis by induction of endogenous glutathione in human neuroblastoma cells. Cell Mol Neurobiol 28:293–305
Watanabe H, Zhao Q, Matsumoto K, Tohda M, Murakami Y, Zhang SH, Kang TH, Mahakunakorn P, Maruyama Y, Sakakibara I, Aimi N, Takayama H (2003) Pharmacological evidence for antidementia effect of Choto-san (Gouteng-san), a traditional Kampo medicine. Pharmacol Biochem Behav 75:635–643
Xian YF, Lin ZX, Zhao M, Mao QQ, Ip SP, Che CT (2011) Uncaria rhynchophylla ameliorates cognitive deficits induced by D-galactose in mice. Planta Med 77:1–7
Yamada K, Nabeshima T (2000) Animal models of Alzheimer’s disease and evaluation of anti-dementia drugs. Pharmacol Ther 88:93–113
Yuan D, Ma B, Yang JY, Xie YY, Wang L, Zhang LJ (2009) Anti-inflammatory effects of rhynchophylline and isorhynchophylline in mouse N9 microglial cells and the molecular mechanism. Inter Immunopharmacol 9:1549–1554
Zhang HY, Liu YH, Wang HQ, Xu JH, Hu HT (2008) Puerarin protects PC12 cells against β-amyloid-induced cell injury. Cell Biol Int 32:1230–1237
Acknowledgment
This study was supported by a Direct Grant of The Chinese University of Hong Kong (Project no. 2030409).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Xian, YF., Lin, ZX., Mao, QQ. et al. Protective Effect of Isorhynchophylline Against β-Amyloid-Induced Neurotoxicity in PC12 Cells. Cell Mol Neurobiol 32, 353–360 (2012). https://doi.org/10.1007/s10571-011-9763-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-011-9763-5