
Abstract
The pathological hallmarks of Alzheimer’s disease (AD) include formation of extracellular amyloid-β peptide (Aβ) and inflammatory responses. Numerous studies have reported that cerebral microvascular Aβ deposition promotes neuroinflammation in AD. Matrix metalloproteinases (MMPs) are involved in the cleavage of extracellular matrix proteins and regulation of growth factors, receptors, and adhesion molecules. Relatively little is known about the involvement of MMPs as inflammatory mediators in the pathological processes of AD. In this study, we explored the signaling pathway of MMP-2 up-regulation by Aβ in brain endothelial cells (BECs) of mice. Using Western blots, we found that inhibitors of extracellular-signal-regulated kinases (ERK) and c-Jun N-terminal kinase (JNK) significantly decreased Aβ-induced MMP-2 expression in BECs. Furthermore, antibody neutralization of the receptor for advanced glycation endproducts effectively blocked Aβ-induced activation of ERK and JNK and their contribution to elevated MMP-2 expression in BECs. Our results suggest that increased MMP-2 expression induced by the interaction of Aβ with RAGE in BECs may contribute to enhanced vascular inflammatory stress in Aβ-related vascular disorders, such as cerebral amyloid angiopathy and AD. This study offers new insights into neuroinflammation in the progression of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive neuronal degenerative disorder of the central nervous system (CNS) that ultimately results in the loss of cognitive function (Yankner 1996). This disease is characterized by the accumulation of toxic amyloid-β peptide (Aβ) proteins in the extracellular space of the brain and on the wall of blood vessels in the brain (De la Torre 2002; Deane and Zlokovic 2007). Hardy and Cullen concluded that vascular deposition of Aβ is critical to the development of AD (Hardy and Cullen 2006). Indeed, in AD cases with a clinical history of cerebral bleeding, the muscular wall of blood vessels is sometimes completely replaced by Aβ deposits, suggesting that the vascular system may be an initiator of the disease (Kawai et al. 1990; Tagliavini et al. 1990).
Aβ 39- to 43-amino acid peptide, the major constituent of senile plaques and cerebrovascular deposits, is thought to play a significant role in the pathophysiology of AD due to its cytotoxic properties. The predominant forms of Aβ are Aβ40 and Aβ42, formed by the normal proteolytic processing of amyloid precursor protein (APP). Aβ42, a minor product of APP, has been the subject of extensive focus in rare familial AD. In contrast, Aβ40, which is overproduced predominantly around vessels and accumulates as a major component of vascular amyloid deposits (Golde et al. 2000), may be involved in neurovascular dysfunction in AD, but the nature of Aβ40’ s involvement in the pathogenesis of AD remains unclear.
Matrix metalloproteinases (MMPs) are a family of zinc-dependent enzymes that regulate both the integrity and composition of extracellular matrix (ECM). In the CNS, MMPs can be released by astrocytes, neurons, oligodendrocytes, microglia, endothelial cells, and leukocytes (Lorenzl et al. 2003). Membrane bound MMPs act at the cell surface and have several functions, including activation of other proteases and growth factors. Secreted MMPs cleave protein components of the ECM and regulate growth factors, receptors, and adhesion molecules. Particularly, Mun-Bryce et al. (2002) reported that increased expression and activation of matrix metalloproteinase-2 (MMP-2) is associated with neuroinflammation. Recently, it was shown that MMP-2 is directly linked to Aβ in the brain and dysfunction in this enzyme may influence the processing of Aβ (Mlekusch and Humpel 2009). It was concluded that MMP-2 is both effector and regulator of the inflammatory response. In addition, we recently discovered that both activity and expression of MMP-2 are increased in a mouse model of AD (Huan Du, unpublished observation). The aim of this study is to explore the mechanism of MMP-2 up-regulation by Aβ40 in vitro. Our results show that Aβ40 up-regulates MMP-2 expression via the receptor for advanced glycation endproducts (RAGEs) of brain endothelial cells (BECs).
Methods
Reagents and Antibodies
DMEM, MCDB131, and Trypsin–EDTA were purchased from Gibco (Gaithersburg, MD). Fetal bovine serum (FBS) was purchased from Jianghai Biotech Co. (Haerbing, Heilongjiang, China). Endothelial cell growth supplement (ECGS), Collagenase, l-glutamine, Dextran, Heparin, Insulin, Biotin–avidin peroxide kit, and Oligomeric Aβ40 were from Sigma-Aldrich (St. Louis, MO). Penicillin and Streptomycin were from the North China Pharmaceutical Group Corporation (Shijiazhuang, Hebei, China). All the other chemicals used were analytical grade reagents. Platelet/endothelial cell adhesion molecule-1 (PECAM-1/CD31) polyclonal antibody (sc-1506) and rabbit anti-MMP-2 polyclonal antibody (sc-10736) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phospho and anti-total ERK1/2 and JNK MAP kinase IgG were purchased from Cell Signaling Technology Inc (Shanghai, China). Affinity-purified anti-RAGE IgG was from R&D Systems (Shanghai, China).
Isolation of Primary Microvascular Endothelial Cells from Mouse Brains
All studies were performed with the approval of the experimental animal committee at Beijing University of Chinese Medicine. Male C57 mice (Beijing Vital River Laboratory Animals Co., Ltd, Beijing, China) were killed by decapitation at 6–8 weeks of age. Primary cultures of microvascular endothelial cells were obtained from cerebral gray matter of the mice, after we removed cerebellum, striatum, optic nerve, and brain white matter tissue. Outer vessels and meninges were then removed using dry cotton swabs.
We used a scalpel to cut the isolated cerebral gray matter into small pieces, and homogenized about every 0.5 g tissue in a 2 ml of washing medium (DMEM supplemented with 2% FBS) using a Dounce homogenizer with a loose fitting. Using 4-5 up-and-down strokes and the resulting homogenate was mixed with 30% dextran (v/v, molecular weight 100,000–200,000) in washing medium. This suspension was centrifuged at 10,000g for 10 min at 4°C. The neural component and the dextran layer were discarded. The pellet containing the vascular component was resuspended in washing medium. The resulting suspension was filtered through 40 μm nylon mesh. The homogenate remaining on the nylon mesh was washed in PBS and then centrifuged at 2,000g for 5 min at room temperature (RT). The pellet was digested by type II collagenase (0.1%) at 37°C for 20 min, and centrifuged at 1,000g for 5 min at RT. Fragments of microvessels were then collected and suspended in complete culture medium (MCDB131, 10% FBS, 75 μg/ml ECGS, 40 U/ml heparin sodium, 0.2 U/ml insulin, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, pH 7.2–7.4), and the suspension was seeded in gelatin-coated Petri dishes. Cultures were incubated at 37°C in a 5% CO2/95% air humidified atmosphere.
Culturing Endothelial Cells
At 72 h after the initial plating, the medium was changed. Subsequently, the medium was changed every other day. When the cells reached 80% confluence, they were passaged with 0.1% trypsin–EDTA. We characterized the endothelial cells via PECAM-1 staining, and the cultures eventually achieved 95% purity of BECs. Cells were used for experiments after the third passage. To control for possible differences in cell growth, all measurements were normalized to total cell protein and to a housekeeping protein as indicated. For all experiments, cells were grown to between 70 and 80% confluence, serum-starved overnight, and then incubated with the indicated concentration of Aβ or anti-RAGE for the appropriate time periods. Cells were pretreated with ERK kinase inhibitor, PD98059 (Promega), or JNK inhibitor, SP600125 (Sigma-Aldrich), at a final concentration of 10 μM.
Immunostaining
Immunostaining was performed for PECAM-1 expression. In brief, endothelial cells grown on glass coverslips in 6-well dishes were fixed in 4% paraformaldehyde for 30 min at 4°C and washed with PBS. Primary antibodies (anti-PECAM-1, 1:100) were applied for 12 h at 4°C, followed by incubation with anti-rabbit biotin solution (1:50) and ExtraAvidin Peroxide solution (1:50) at RT for 30 min, respectively. Immunostaining images were recorded with a Nikon E800 microscope (Nikon, Japan).
Western Blot Analysis
Western blot analysis was performed as described previously (Du et al. 2010). Total lysate from cultured cells was used for detection of MMP-2 and cell signaling expression. Protein concentration was determined by a BCA protein assay. Protein (75 mg) was boiled at 100°C in SDS sample buffer for 5 min, electrophoresed on 7–15% SDS–PAGE gels for 180 min at 120 volts, and transferred to polyvinyldifluoridine (PVDF) membranes. These were incubated overnight at 4°C with the following IgGs: MMP-2 (1:1000), anti-phospho ERK1/2 (1:1000), and anti-phospho JNK MAP kinase IgG (1:250). Membranes were washed with PBS/0.1% Tween 20, incubated with Goat anti-rabbit or goat anti-mouse peroxidase conjugated IgG (both 1:4,000) at RT for 60 min, and washed three times for 15 min with PBS/Tween. Peroxidase activity was visualized with an enhanced chemiluminescence substrate. Membranes which were incubated with anti-phospho ERK1/2 and anti-phospho JNK MAP kinase IgG were washed with stripping buffer for 20 min, and then were incubated overnight at 4°C with the following IgGs: anti-total ERK1/2 and anti-total JNK MAP kinase IgG (both 1:2,000). Goat anti-rabbit or goat anti-mouse peroxidase conjugated IgG (both 1:5,000) were used to identify binding sites of the primary antibody. In all Western blot studies, at least three cell lysates per group were used. Results of representative experiments are shown.
Statistical Analysis
Data were obtained from at least three independent experiments and expressed as mean ± SD. Statistical significance was evaluated by one-way ANOVA by SPSS11.0 for repeated measures. P-values less than 0.05 were considered significant.
Results
Aβ Induces MMP-2 Expression in BECs
Having established mouse BECs cultures in our laboratory, we used immunostaining of the cultured cells to examine expression of Platelet/endothelial cell adhesion molecule-1 (PECAM-1/CD31). We found that PECAM-1 was significantly expressed in the cytoplasm of BECs (Fig. 1a). Because Aβ at micromolar concentration has been shown to cause necrosis or cell death in endothelial cells (Basta 2008), we first examined the effect of various concentrations of Aβ40 on the expression of MMP-2 in BECs (Fig. 1b). After a 6 h exposure of BECs to 0.1 μM Aβ40, expression of MMP-2 was not significantly different than in control BECs (P = 0.15). However, a higher concentration (0.5, 1, and 2.5 μM) of Aβ40 caused significantly elevated MMP-2 expression at the 6-h time period (P < 0.001). As there was no discernible change of cell death in the morphology of BECs treated with 0.5 μM Aβ40 for 6 h, we used this concentration of Aβ40 and this time period for most of the experiments described here.

Identification of BECs (a) and characterization of MMP-2 expression in BECs (b). a Immunostaining of BECs using PECAM-1 antibody. Immunoreactivity indicates significant expression of PECAM-1 in the cytoplasm of BECs (arrows, BECs positive for PECAM-1). Scale bar = 20 μm. b Aβ induced MMP-2 expression. BECs were incubated for 6 h with various concentrations of Aβ40 (0, 0.1, 0.5, 1, and 2.5 μM). Significant MMP-2 expression was detected by Western blot, ##P < 0.001, compared with control samples. β-actin demonstrates equal protein loading. t test (n = 3 independent experiments)
MMP-2 Expression is Involved in Signal Transduction in BECs
To determine the signal pathway of Aβ-dependent MMP-2 expression, we first examined the effect of pharmacological inhibitors of ERK and JNK on MMP-2 expression (Harja et al. 2008; Li et al. 2009). As shown in Fig. 2a, pretreatment of BECs with the pERK MAP kinase inhibitor PD98059 (PD), or with the JNK MAP kinase inhibitor SP600125 (SP) for 1 h before Aβ40 for 6 h, reduced MMP-2 expression in BECs (P = 0.036 and P = 0.017, respectively). Subsequently, we examined whether Aβ40 treatment could activate ERK and JNK in BECs (Fig. 2b). Our results showed that Aβ40 activated ERK and JNK (P = 0.022 and P = 0.035, respectively). These results suggested that ERK and JNK activation were involved in Aβ40-induced MMP-2 expression in BECs.

MMP-2 expression involved signal transduction in BECs. a Effects of inhibition of ERK and JNK on Aβ-induced MMP-2 expression. Confluent cultures of BECs were pretreated with ERK inhibitor PD98059 (PD), and JNK inhibitor SP600125 (SP) for 1 h before Aβ40 was added to the culture medium and incubated with the cells for 6 h. Significantly greater MMP-2 expression by BECs was detected with Western blot, #P < 0.05, ##P < 0.001 compared to controls, and significantly depressed MMP expression, *P < 0.05, was found for ERK and JNK-inhibited samples compared to Aβ-exposed cells without ERK and JNK inhibitor (Aβ). β-actin demonstrates equal protein loading. b Aβ induced signal transduction in BECs. Confluent BECs were pretreated with Aβ40 for 6 h (Aβ). Western blots with phosphorylation-specific antibodies show significant increases in phosphorylated forms of ERK and JNK. #P < 0.05 compared to controls. The blots were reprobed for total ERK and JNK. t test (n = 3 independent experiments)
RAGE-Mediated Aβ Induces MMP-2 Expression in BECs
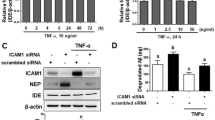
RAGE, a multiligand receptor in the immunoglobulin superfamily, binds a broad repertoire of ligands. Several investigators have shown that Aβ can interact with RAGE (Basta 2008; Koyama et al. 2007). In the CNS, RAGE is expressed on microglia and neurons, as well as endothelial cells and smooth muscle cells of the vasculature. In light of these findings, we explored the involvement of RAGE in Aβ-dependent MMP-2 expression in BECs. We pretreated BECs with RAGE antibody for 2 h before the Aβ40 incubation and Western blotting to detect MMP-2 protein expression in the cells (Fig. 3a). This experiment showed that neutralizing RAGE with antibodies abrogated Aβ-induced MMP-2 expression (P < 0.001).

RAGE-mediated Aβ induces MMP-2 expression in BECs. a RAGE-neutralizing antibody abrogates Aβ-induced MMP-2 expression in BECs. BECs were pretreated with RAGE-neutralizing antibody (RAGE, 10 μg/ml) for 2 h, before Aβ40 was added to the culture medium (Aβ + RAGE). Western blotting shows MMP-2 expression in BECs was significantly boosted by Aβ compared to controls, ##P < 0.001, but RAGE neutralization significantly abrogated Aβ induction of MMP, **P < 0.001, compared to BECs exposed to the Aβ group without RAGE neutralization. b RAGE-neutralizing antibody inhibits activation of ERK and JNK. BECs were pretreated with RAGE-neutralizing antibody (RAGE, 10 μg/ml) for 2 h, and then incubated with Aβ40 (Aβ + RAGE). The expression of ERK and JNK was analyzed by Western blot. In the absence of RAGE neutralization, Aβ40-incubated cells (Aβ) show significant increases in ERK and JNK compared to controls without Aβ40, ##P < 0.001, but with RAGE neutralization, ERK and JNK induction is significantly depressed, *P < 0.05, compared to the Aβ-exposed cells without RAGE neutralization. t test (n = 3 independent experiments)
RAGE consists of an extracellular domain (V-type followed by two C-type regions) and a single transmembrane domain followed by a short cytosolic tail, which mediates signal transduction. In vitro, interaction of Aβ with RAGE-bearing neuronal-like cells leads to activation of NF-κB and expression of macrophage-colony stimulating factor and interleukin-6, suggesting a possible role for neuronal RAGE in inflammation (Yan et al. 1997). Additional studies have suggested that JNK and ERK are downstream signal molecules coupled to RAGE (Basta 2008). Knowing that RAGE was involved in Aβ-induced MMP-2 expression in BECs, we next tested whether RAGE is associated with activation of ERK and JNK in contributing to MMP-2 expression in the cells (Fig. 3b). We found that neutralization of RAGE with antibodies significantly inhibited the activation of ERK and JNK induced by Aβ (P = 0.014 and P = 0.0058, respectively). Together, these data indicated that RAGE in BECs is a receptor for Aβ, and also can activate ERK and JNK signal transduction to induce MMP-2 expression.
Discussion
The pathological hallmarks of AD include accumulation of neurofibrillary tangles, accumulation of extracellular Aβ, and chronic inflammation. Signs of inflammatory response are evident around the Aβ deposits. A number of studies have implicated cerebral microvascular Aβ deposition in promoting neuroinflammation and dementia in AD and related familial cerebral amyloid angiopathy (CAA) disorders (Vinters 2001; Atterns and Jellinger 2004; Bailey et al. 2004; Greenberg et al. 2004). In addition, the presence of inflammatory mediators and high levels of expression of the full complement cascade in the vicinity of Aβ deposits in the brains of AD patients strongly indicates that inflammation contributes to the pathogenesis of AD (McGeer et al. 1989). A number of cells and cytokines are involved in the neuroinflammatory cascade. BECs can produce some of these cytokines (such as VEGF and BDNF) and participate in the regulation of neuronal activity (Wang et al. 2006). In previously published studies, we found paracrine signaling of brain microvascular endothelial cells plays important roles in the survival of neurons under normal conditions and following injury (Du et al. 2010; Li et al. 2009). In this study, we isolated and cultured mouse BECs (Fig. 1a) and investigated the consequences of exposing them to Aβ.
MMPs contribute to remodeling of the pericellular environment, primarily by cleaving ECM proteins and cell surface components. It has also been reported that MMP-2 is capable of cleaving collagen IV and V, laminin, and chondroitin sulfate proteoglycan, which are associated with cell adhesion (Yong et al. 2001). MMP-2 is initially expressed as an inactive proenzyme. It is cleaved into active forms after cellular release (Van den Steen et al. 2002). MMP-2-mediated degradation of ECM components leads to loss of specific ECM–integrin interactions, resulting in apoptosis of vascular cells (Frisch and Ruoslahti 1997). Numerous studies have demonstrated activation of inflammatory processes in pathologically vulnerable regions of the AD brain and have documented the presence of a large number of inflammatory molecules (Tarkowski 2002; McGeer and McGeer 2003). Plasma levels of inflammatory proteins are increased before clinical onset of dementia, AD, and vascular dementia (Engelhart et al. 2004). Particularly, Mun-Bryce et al. (2002) have reported that increased expression and activation of MMP-2 is associated with neuroinflammation. Since Aβ clearance at the blood–brain barrier (BBB) may be dysregulated in AD, and MMP-2 may be involved in breakdown of the BBB, an interaction of MMP-2 with Aβ seems likely. Recent studies have shown that there was an increase of MMP-2 expression in astrocytes surrounding senile plaques in transgenic mice brain (Li et al. 2011). We also discovered that both activity and expression of MMP-2 are increased in a mouse model of AD, and we found that there was a positive correlation between expression levels of MMP-2 and inflammatory injury, which may be an important pathway of inflammatory reaction in AD. In this study, we found similar results in vitro: MMP-2 expression was increased in Aβ-induced endothelial cells (Fig. 1). On the other hand, it was also reported that Aβ protein can induce the expression of MMPs, which could be involved in the degradation of Aβ (Mizoguchi et al. 2009; Li et al. 2011). With accumulation evidence implicating that there could be a complex regulation of MMP-2 expression by oligomeric Aβ, so the relationship between the expression of MMP-2 and the pathological process of AD need further research. In our study, to confirm the mechanisms of this MMP-2 expression in BECs, we first used ERK and JNK inhibitors to detect MMP-2 expression (Fig. 2a), then directly examined ERK and JNK signaling transduction (Fig. 2b). The results of our experiments revealed that ERK and JNK activation are involved in MMP-2 expression in Aβ40-induced BECs.
Advanced Glycation Endproducts (AGEs), the products of nonenzymatic glycation and oxidation of proteins and lipids, accumulate in diverse biological settings, such as inflammation, diabetes, and aging. AGEs have multiple potential effects on the vessels and tissues. It was reported that AGEs are further increased in the brain in the presence of vascular or Alzheimer’s dementia, and AGEs may be central to the exacerbation of dementia and enhanced predilection of stroke (Girones et al. 2004; Ramasamy et al. 2005). RAGE, a multiligand receptor in the immunoglobulin superfamily, binds a broad repertoire of ligands, including AGEs, Aβ, the S100/calgranulin family of proinflammatory cytokine-like mediators, and high mobility group box 1 nonhistone DNA binding protein (HMGB1 or amphoterin) (Yan et al. 1996; Chen et al. 2007; Hofmann et al. 1999). A 60% increase in RAGE protein has been found in the vessels of CAA patients, compared to age-matched non-dementia controls (Lue et al. 2005). A significant role for RAGE-related inflammation is supported by previous studies linking RAGE to the progression of chronic immunopathies, ranging from colitis to atherosclerosis (Schmidt et al. 2001). We sought to determine whether RAGE is involved in Aβ40-induced MMP-2 expression in BECs. Our experiments highlighted the role of RAGE as a signal transduction receptor mediating the effects of an Aβ-rich environment on these cells. Ligand–receptor interactions of Aβ and RAGE initiate cellular signaling, leading to increased levels of MMP-2 protein. Activation of JNK MAP kinase and ERK1/2 are involved in these interactions (Fig. 3), supporting the concept that RAGE functions as a signaling receptor, rather than just tethering ligands to the cell surface.
In summary, this study provides clear evidence that Aβ, interacting with RAGE on BECs, triggers intracellular ERK and JNK activation and thereby promotes endothelial MMP-2 expression. Our results suggest that increased MMP-2 expression, brought about by the interaction of Aβ with RAGE in BECs, may contribute to enhanced vascular inflammatory stress in Aβ-related vascular disorders, such as CAA and AD. RAGE and MMP-2 expression by BECs may thus be potential research and therapeutic targets for AD.
References
Atterns J, Jellinger KA (2004) Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—a pilot study. Acta Neuropathol 107:83–90
Bailey TL, Rivara CB, Rocher AB, Hof PR (2004) The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res 26:573–578
Basta G (2008) Receptor for advanced glycation end products and atherosclerosis: from basic mechanisms to clinical implications. Atherosclerosis 196:9–21
Chen X, Walker DG, Schmidt AM, Arancio O, Lue LF, Yan SD (2007) RAGE: a potential target for Abeta-mediated cellular perturbation in Alzheimer’s disease. Curr Mol Med 7:735–742
De la Torre JC (2002) Alzheimer disease as a vascular disorder: nosological evidence. Stroke 33:1152–1162
Deane R, Zlokovic BV (2007) Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 4:191–197
Du H, Li P, Pan Y, Li W, Hou J, Chen H, Wang J, Tang H (2010) Vascular endothelial growth factor signaling implicated in neuroprotective effects of placental growth factor in an in vitro ischemic model. Brain Res 1357:1–8
Engelhart MJ, Geerlings MI, Meijie J, Kiliaan A, Ruitenberg A, van Swieten JC, Stijnen T, Hofman A, Witteman JC, Breteler MM (2004) Inflammatory proteins in plasma and the risk of dementia: the Rotterdam study. Arch Neurol 61:668–672
Frisch SM, Ruoslahti E (1997) Integrins and anoikis. Curr Opin Cell Biol 9:701–706
Girones X, Guuimera A, Cruz-Sanchez CZ, Ortega A, Sasaki N, Makita Z, Lafuente JV, Kalaria R, Cruz-Sanchez FF (2004) N epsilon carboxymethyllysine in brain aging, diabetes mellitus, and Alzheimer’s disease. Free Radic Biol Med 36:1241–1247
Golde TE, Eckman CB, Younkin SG (2000) Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Biochem Biophys Acta 1502:172–187
Greenberg SM, Gurol ME, Rosand J, Smith EE (2004) Amyloid angiopathy related vascular cognitive impairment. Stroke 35:2616–2619
Hardy J, Cullen K (2006) Amyloid at the blood vessel wall. Nat Med 12:756–757
Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, Nikolla Z, Belov D, Lalla E, Ramasamy R, Yan SF, Schmidt AM (2008) Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J Clin Invest 118:183–194
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901
Kawai M, Kalaria RN, Harik SI, Perry G (1990) The relationship of amyloid plaques to cerebral capillaries in Alzheimer’s disease. Am J Pathol 137:1435–1446
Koyama H, Yamamoto H, Nishizawa Y (2007) RAGE and soluble RAGE: potential therapeutic targets for cardiovascular disease. Mol Med 13:625–635
Li M, Shang DS, Zhao WD, Tian L, Li B, Fang WG, Zhu L, Man SM, Chen YH (2009a) Amyloid βinteraction with receptor for advanced glycation end production upregulates brain endothelial CCR5 expression and promotes T cells crossing the blood-brain barrier. J Immunol 182:5778–5788
Li W, Li P, Hua Q, Hou J, Wang J, Du H, Tang H, Xu Y (2009b) The impact of paracrine signaling in brain microvascular endothelial cells on the survival of neurons. Brain Res 1287:28–38
Li W, Poteet E, Xie L, Liu R, Wen Y, Yang SH (2011) Regulation of matrix metalloproteinase 2 by oligomeric amyloid βprotein. Brain Res 1387:141–148
Lorenzl S, Albers DS, LeWitt PA, Chirichigno JW, Hilgenberg SL, Cudkowicz ME, Beal MF (2003) Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative disease. J Neurol Sci 207:71–76
Lue LF, Yan SD, Stern DM, Walker DG (2005) Preventing activation of receptor for advanced glycation endproducts in Alzheimer’s disease. Curr Drug Targets CNS Neurol Disord 4:249–266
McGeer EG, McGeer PL (2003) Inflammatory processes in Alzheimer’s disease. Prog Neuropsycholpharmacol Biol Psychiatry 27:741–749
McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Immune system response in Alzheimer’s disease. Can J Neurol Sci 16:516–527
Mizoguchi H, Takuma K, Fukuzaki E, Ibi D, Someya E, Akazawa K, Alkam T, Tsunekawa H, Mouri A, Noda Y, Nabeshima T, Yamada K (2009) Matrix metalloprotease-9 inhibition improves amyloid β-mediated cognitive impairment and neurotoxicity in mice. J Pharmacol Exp Ther 331:14–22
Mlekusch R, Humpel C (2009) Matrix metalloproteinases-2 and -3 are reduced in cerebrospinal fluid with low beta-amyloid1–42 levels. Neurosci Lett 466:135–138
Mun-Bryce S, Lukes A, Wallace J, Lukes-Marx M, Rosenberg GA (2002) Stromelysin-1 and gelatinase A are upregulated before TNF-α in LPS-stimulated neuroinflammation. Brain Res 933:42–49
Ramasamy R, Vannucci SJ, Yan SD, Herold K, Yan SF, Schmidt AM (2005) Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration and inflammation. Glycobiology 15:16R–28R
Schmidt AM, Yan SD, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108:949–955
Tagliavini F, Ghiso J, Timmers WF, Giaccone G, Bugiani O, Frangione B (1990) Coexistence of Alzheimer’s amyloid precursor protein and amyloid protein in cerebral vessel walls. Lab Invest 62:761–767
Tarkowski E (2002) Cytokines in dementia. Curr Drug Targets Inflamm Allergy 1:193–200
Van den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G (2002) Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9. Crit Rev Biochem Mol Biol 37:375–536
Vinters HV (2001) Cerebral amyloid angiopathy: a microvascular link between parenchymal and vascular dementia? Ann Neurol 49:691–692
Wang H, Ward N, Boswell M, Katz DM (2006) Secretion of Brain-derived neurotrophic factor from brain microvascular endothelial cells. Eur J Neurosci 23:1665–1670
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382:685–691
Yan SD, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, Rajavashisth T, Chen X, Goldman GC, Stern D, Schmidt AM (1997) RAGE–Aβ interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci USA 94:5296–5301
Yankner BA (1996) Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron 16:921–932
Yong VW, Power C, Forsyth P, Edwards DR (2001) Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2:502–511
Acknowledgments
The Grant sponsors are from Ministry of Education and Key laboratory of Chinese Internal Medicine; The National Basic Research Program (Grant No. 2005CB523311); The National significant science and technology special projects (Grant No. 2009ZX0950214).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Du, H., Li, P., Wang, J. et al. The Interaction of Amyloid β and the Receptor for Advanced Glycation Endproducts Induces Matrix Metalloproteinase-2 Expression in Brain Endothelial Cells. Cell Mol Neurobiol 32, 141–147 (2012). https://doi.org/10.1007/s10571-011-9744-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-011-9744-8