
Abstract
Adipose-derived stromal cells (ASCs) are an alternative source of stem cells for cell-based therapies of neurological disorders such as spinal cord injury (SCI). In the present study, we predifferentiated ASCs (pASCs) and compared their behavior with naïve ASCs in vitro and after transplantation into rats with a balloon-induced compression lesion. ASCs were predifferentiated into spheres before transplantation, then pASCs or ASCs were injected intraspinally 1 week after SCI. The cells’ fate and the rats’ functional outcome were assessed using behavioral, histological, and electrophysiological methods. Immunohistological analysis of pASCs in vitro revealed the expression of NCAM, NG2, S100, and p75. Quantitative RT-PCR at different intervals after neural induction showed the up-regulated expression of the glial markers NG2 and p75 and the neural precursor markers NCAM and Nestin. Patch clamp analysis of pASCs revealed three different types of membrane currents; however, none were fast activating Na+ currents indicating a mature neuronal phenotype. Significant improvement in both the pASC and ASC transplanted groups was observed in the BBB motor test. In vivo, pASCs survived better than ASCs did and interacted closely with the host tissue, wrapping host axons and oligodendrocytes. Some transplanted cells were NG2- or CD31-positive, but no neuronal markers were detected. The predifferentiation of ASCs plays a beneficial role in SCI repair by promoting the protection of denuded axons; however, functional improvements were comparable in both the groups, indicating that repair was induced mainly through paracrine mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spinal cord injury (SCI) is one of the unresolved issues in neurosurgery today and in medicine in general. The current therapeutic algorithm includes early surgery, consisting of decompression of the spinal cord and stabilization of the spine in indicated cases. As soon as the patients’ injuries (often multiple) become stable, the patients are transferred to specialized rehabilitation centers. Nonetheless, no treatment that would lead to the repair of the damaged spinal cord tissue is available today.
Nevertheless, stem cell researchers are continuing their attempts to find alternative and accessible cell sources capable of neural differentiation and/or the repair of damaged tissue. Stem cells from fetal and embryonic tissues can undergo expansion and neural differentiation in vitro and in vivo (Hu and Zhang 2010; Neri et al. 2010). However, ethical considerations, a high risk of tumorigenesis, and restricted access limit their clinical utility (Lo et al. 2010; Hess 2009; Abramson 2010).
Mesenchymal stromal cells (MSCs), isolated from bone marrow, represent a promising tool in SCI. Current experimental studies have proven that their application in the acute stage of SCI can be associated with improved function, preservation of tissue, and neuronal regeneration, mainly due to secretion of different growth factors (Urdzikova et al. 2006; Syková et al. 2006; Gu et al. 2010). MSCs have been also used in combination with OEGs (Amemori et al. 2010) or hydrogels (Hejcl et al. 2010) in acute or chronic SCI.
Adipose tissue is an abundant tissue in the body and contains a stromal fraction rich in stem-progenitor cells capable of undergoing differentiation into osteogenic, chondrogenic, and adipogenic lineages (Yamada et al. 2010; Sofroniew et al. 2001). The in vitro as well as in vivo properties of adipose-derived stromal cells (ASCs) resemble those of MSCs obtained from bone marrow, and the liposuction procedure employed to harvest ASCs is minimally invasive for the patient. The pluripotent stem cell populations of MSCs and ASCs are capable of undergoing neural and glial differentiation under appropriate in vitro conditions (Gordon and Scolding 2009; Radtke et al. 2009; Franco Lambert et al. 2009; Khoo et al. 2008). Within in vitro cultures of ASCs, a specific p75+ population has been purified, the behavior of which resembles that of Schwann cells (Yamada et al. 2010); these cells can form myelin-like structures in vitro with PC12 cell neurites (Xu et al. 2008).
There are an increasing number of reports describing the trophic effects of ASCs on the protection, survival, and differentiation of endogenous cells and tissues. In addition, recent studies indicate that ASC-induced repair may not only act through cell differentiation, but also by sharing as a primary reparative function the paracrine mechanisms provided by trophic factors with pro-survival and repair inducing faculties (Wei et al. 2009). Molecules such as hepatocyte growth factor (HGF), granulocyte and macrophage colony stimulating factors, interleukins (ILs) 6, 7, 8, and 11, tumor necrosis factor-alpha (TNF-α), vascular endothelial growth factor (VEGF), brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), adipokines, and others have been identified within the ASCs’ secretome (Salgado et al. 2010). In vivo studies confirmed that ASCs transplantation slowed degeneration in different models of neuronal neurodegenerative diseases (Bae et al. 2010; Lee et al. 2009).
In our comparative study, we obtained stromal cells from the white inguinal fat adipose tissue of the rat. Differentiation was induced by means of a neurosphere-like stage and continued with neurodifferentiation in vitro for 12 days. Concomitantly, we injected undifferentiated ASCs and predifferentiated ASCs (pASCs) (from dissociated spheres) into rats with a spinal cord compression lesion and compared the behavioral outcome of the transplanted animals. Cell fate and survival were evaluated by immunohistochemistry, while transmission electron microscopy (TEM) was used to analyze the level of integration of the cells into the host tissue.
Materials and Methods
Isolation of ASCs
Wild type or transgenic Sprague–Dawley rats (SD-Tg(CAG-EGFP)CZ-004Osb) expressing green fluorescent protein (GFP+) were anesthetized, and adipose tissue from the inguinal pads was dissected, mechanically minced, and treated with 0.2% (w/v) collagenase type I (Worthington Biochemicals, Lakewood, NJ) for 1 h at 37°C. The isolated cellular fraction was resuspended in proliferation medium consisting of DMEM/F12 + Glutamax (Gibco) supplemented with 10% FBS and 0.2% antibiotics (primocin) and plated in culture flasks. Cells were harvested once they reached 90% confluence and re-plated up to the second passage.
Cells from the second passage were then either induced to form spheres or were used as undifferentiated cells for transplantation into a spinal cord lesion.
To assess the multipotency of ASCs, the cells were differentiated into adipogenic, osteogenic, and chondrogenic phenotypes using protocols described elsewhere (Turnovcova et al. 2009).
ASC Predifferentiation and Neural Induction
After reaching 80% confluence, cultured ASCs (passage 2) were plated in 10 cm2 Petri dishes and induced to form spheres by replacing the proliferation medium with sphere-induction medium consisting of DMEM/F12 + Glutamax (3:1 ratio, v/v) with B27 supplement (Gibco, Grand Island, NY), 20 ng/ml EGF (R&D Systems Inc., Minneapolis, MN), 40 ng/ml bFGF (R&D Systems Inc.), and 1% Primocin. Four days later, the formed spheres were collected in a 15 ml Falcon tube. They were dissociated by accutase, plated on laminin (Sigma-Aldrich)-coated dishes, placed in differentiation medium consisting of Neurobasal media (Gibco) with B27 supplement (Gibco, Grand Island, NY), 10 ng/ml NGF (R&D Systems Inc., Minneapolis, MN), 20 ng/ml bFGF (R&D Systems Inc.), and 1% primocin and kept in culture for 6 days. Growth factors were added every second day.
RT-PCR Analysis
At each stage of differentiation, mRNA was isolated from lysed cells using TRI reagent (Sigma-Aldrich) according to the manufacturer’s directions. The expression of target and reference genes was determined by one-step real time RT-PCR using a 7500 Real Time-PCR System (Applied Biosystems) and a QuantiTect® One-step qRT-PCR kit (Qiagen). The 20 μl reaction volume contained 5 μl of extracted RNA. The following thermal profile was used: a single cycle of reverse transcription for 30 min at 50°C, 15 min at 95°C for reverse transcriptase inactivation and DNA polymerase activation, followed by 45 amplification cycles of 15 s at 94°C, and 1 min at 60°C each (combined annealing-extension step). Samples were run in triplicate. The Gene Expression Assay Mix (Applied Biosystems) employed for the study is shown in Table 1. As a housekeeping gene to normalize the data, we used beta actin (ACTB). The results were analyzed using the integrated 7500 System SDS Software version 1.3.1. The relative quantities of mRNA were therefore calculated using a ΔΔCt method with efficiency correction.
Electrophysiological Measurements
Cells in culture, 6 days after neural induction (NI), were evaluated using the patch clamp technique as described in detail in Pivonkova et al. (2010).
Spinal Cord Compression Lesion and Cell Transplantation
All animal experiments were approved by the Animal Committee of the Czech Republic and the Animal Care and Use of Animals Committee of the Institute of Experimental Medicine AS CR. Adult male Wistar rats weighing 280–300 g were anesthetized by isofluorane vapor inhalation (3–5%), and a balloon-induced spinal cord compression lesion was performed according to protocols previously described (Urdzikova et al. 2006). The animals were assisted with manual urination twice a day until the reflex returned, while ampicillin and gentamicin were administered by intramuscular injection twice a day for 3 days. Cell transplantation was carried out 7 days after the SCI according to a previously published procedure (Amemori et al. 2010). SCI animals were randomly divided into two transplanted groups and a control group. Harvested ASCs (passage 2) or pASCs obtained from dissociated spheres were grafted using a stereotaxic injection instrument (Stoelting Co., Wood Dale, IL) and a nanoinjector pump (KD Scientific Inc, Hillstone, MA). A total of 5 × 105 cells suspended in 5 μl PBS were injected into the rostral central caudal part of the lesion site, at a depth of 2 mm, using a capillary glass needle specially adapted to a 10 μl Hamilton syringe (Hamilton Co., Reno, NV). All rats were immunosuppressed by cyclosporine injection (Chong Keun Dang Pharma 1 mg/100 g body weight) 24 h before transplantation and daily until the end of the experiment. The control group was composed of animals with SCI and injected only with 5 μl PBS.
BBB Hind Limb Motor Test
Two observers, blinded to the experimental group, assessed the animals’ hind limb motor function in an open field using the BBB locomotor rating scale (Basso et al. 1995) before SCI and once a week thereafter. Both the left and right hind limb scores were averaged. All data are reported as mean ± SEM. Differences in mean BBB scores between the controls and the two transplanted groups at each post-transplantation interval were assessed for variability using the F-test, and according to the results of the F-test, we analyzed the results using either a parametric or nonparametric unpaired Student’s t test. Values of P < 0.05 were considered statistically significant.
Histology and Immunohistochemistry
The animals were sacrificed and perfused 8 weeks after cell transplantation for histological examination. The rats were deeply anesthetized, and 200 ml of PBS was perfused intracardially into the left ventricle, followed by 300 ml of ice-cold 4% (v/v) paraformaldehyde (PFA) in 0.1 M PBS. The spinal cords were dissected, immersed in 4% (v/v) PFA at 4°C for 24 h, and then placed in 30% (w/v) sucrose for 3 days. After freezing, spinal cords were cryosectioned longitudinally in 20-μm-thick slices. Cells from cell cultures were fixed with a solution of 4% (v/v) formaldehyde (Sigma-Aldrich) in 0.1 M PBS, pH 7.4, for 30 min, and permeabilized with 0.2% (v/v) Triton X-100 (USB Corporation, Cleveland, OH) in PBS. For immunofluorescence studies, the antibodies listed in Table 2 were used. For double-labeling immunocytochemical studies, ASCs isolated from wild type Sprague–Dawley rats were used.
Transmission Electron Microscopy
For electron microscopy, immunolabeled tissue sections were rinsed in 0.1 M PB and then post-fixed for 1 h in 2% osmium tetroxide in 0.1 M PB. They were subsequently dehydrated through a graded series of ethanols followed by propylene oxide, propylene oxide:epon (50:50), and 100% epon (Agar Scientific Ltd., Stansted, UK), before embedding in fresh Epon between sheets of Aclar plastic (Agar Scientific Ltd., Stansted, UK) (Leranth and Pickel 1989). Epon polymerization was carried out by incubation at 60°C over 48 h. Following polymerization, regions of interest were cut from the flat embedded tissue and mounted on the tips of Epon blocks. Ultrathin sections of these regions were cut with a diamond knife (Diatome; TAAB, Gillingham, UK) at a thickness of approximately 70 nm and examined by a transmission electron microscope (Morgagni, Philips, Eindhoven, The Netherlands).
For detection of GFP+ cells in TEM images, sections were incubated in a 1:300 dilution of polyclonal rabbit anti-GFP (Sigma-Aldrich, UK) for 72 h at 4°C in 0.1% BSA/0.1 M TBS/0.25% TRITON X-100. The specificity of the antibody has been previously demonstrated using immunohistochemistry and western blotting (Halliday et al. 1996; Rodríguez et al. 2008). Subsequently, the primary antibody was detected using an immunoperoxidase procedure (Chan et al. 1990). For immunoperoxidase labeling, sections were washed in 0.1 M TBS and placed in a 1:400 dilution of biotinylated goat anti-rabbit IgG (Jackson Immunoresearch, Stratech Scientific Ltd., Soham, UK) for 4 h, followed by two washes in 0.1 M TBS and incubation in a 1:200 dilution of biotin–avidin complex (Vector Laboratories Ltd., Peterborough, UK) for 30 min, followed by washes in 0.1 M TBS. All antisera dilutions were prepared in 0.1 M TBS/0.1% BSA, and the incubations were carried out at room temperature. The peroxidase reaction product was visualized (on TEM images visible as a black color) by incubation in a solution containing 0.022% of 3,30-diaminobenzidine (Sigma-Aldrich, Gillingham, UK) and 0.003% peroxide in 0.1 M TBS for 3 min, followed by washes in 0.1 M TBS and finally in 0.1 M PB. To check for non-specific background labeling or cross reactivity between antibodies derived from different host species, a series of control experiments were performed. Omission of the primary and/or secondary antibodies from the incubation solutions resulted in a total absence of target labeling.
Results
In Vitro Differentiation of ASCs
To test the multipotent capabilities of the ASCs, cells from passage 2 were induced to osteogenic, adipogenic, and chondrogenic differentiation by providing the appropriate conditions. The cells differentiated into all three phenotypes, as proven by staining for calcium deposits (Alizarin red), oil droplets (Oil red O), and acid mucopolysaccharides represented by Alcian blue staining (data not shown).
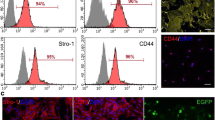
Cells start to form spherical clusters already within 24 h in the induction medium. Five days later, 90% of the cells in monolayer culture formed spheres of different sizes (Fig. 1a). Sphere formation is the first step in testing the ASCs’ neural differentiation capabilities and to induce differentiation toward neural precursors and more mature cellular stages. Immunocytological analysis revealed the strong expression of NCAM on the surface of the spheres. Cells from dissociated spheres were also positive for NG2, S100, and p75, markers of early glial progenitor cells or Schwann cells (Fig. 1b, c). We accentuated neural differentiation by replacing EGF with NGF and decreasing the concentration of bFGF in the medium; 2 days later, the cells remained positive for NCAM (Fig. 1d), while some of them showed positivity for GFAP. We also observed some positivity for βIII-tubulin, a marker of neurons; however, these cells did not show typical neuronal morphology (Fig. 1d). The results obtained from immunocytochemical staining were confirmed by quantitative RT-PCR analysis performed on spheres and on dissociated cells maintained in differentiation medium for 2 or 6 days. We observed robust upregulation of p75 (40-fold) and NG2 (2.5-fold) in the spheres, which slowly decreased during the process of neural differentiation. Nestin and NCAM, early neural progenitor markers, showed the highest expression at the beginning of NI (a 5.7-fold increase for Nestin) or at the end (a 29-fold increase for NCAM), respectively (Fig. 2a). More mature neural markers, such as GFAP, βIII-tubulin, or MAP2, showed only little upregulation, which did not exceed a 0.5-fold increase (Fig. 2b). Regarding receptors, a low expression of the insulin and TGFβ receptors was observed during the whole culturing period, while the expression of the NGF receptor TrkA was upregulated at the end of NI. The expression of the NT3 receptor TrkC decreased with time spent in culture (Fig. 2c).

a Spheres were all positive for NCAM (red). Dissociated spheres plated on laminin-coated coverslips (b, c) and fixed after 6 days in neural medium (d). b S100 (green) NG2 (red); c staining for p75 (red), GFP (green); d NCAM (red), βIII-tubulin (green). e GFP+ ASCs (green) 8 weeks after transplantation into a SCI did not interact with host axons (staining for NF160; red); inset is at higher magnification and shows the lack of interaction between an ASC and the host tissue. f–h In contrast, GFP+ pASCs (green) formed hollow fibers and closely wrapped (white arrows) host axons (f, g; staining for NF160, red) as well as oligodendrocytes (h; staining for O1, red). Inset at higher magnification shows the close interaction of GFP+ cells with neurofilaments. i Some pASCs were positive for NG2 (i, white arrows) and formed guiding strands along the residual host tissue, attracting host NG2-positive progenitors (red) to the lesion site. j TEM image showing a GFP+ pASC (arrow) in close contact with a Schwann cell (S) and a myelinated axon (a). k GFP+ process from a pACS (arrow) in the vicinity of a Schwann cell (S) and an unmyelinated axon (a). l, m Both ASCs (L) as well as pASCs (M) were positive (arrows) for the endothelial marker CD31 (red). Cell nuclei are stained in blue (DAPI). Scale barsa–e, i—20 μm, f, g—50 μm, j—1 μm, k—2.5 μm, h, l, m—25 μm

a–c Quantitative RT-PCR analysis of spheres and dissociated cells from the spheres maintained in neural medium, 48 h and 6 days after NI. The enhanced expression of glial markers and immature neural progenitor markers was found after exposure to differentiation medium. d BBB open-field locomotor test. Hind limb gait was first assessed 1 week after SCI, i.e., before transplantation (Tx), and evaluated weekly thereafter for 7 weeks. Both the ASC- (BBB = 7.0 ± 0.74; n = 11) and the pASC- (BBB = 7.2 ± 0.46; n = 12) treated groups displayed motor improvement compared with controls (BBB = 4.7 ± 0.34; n = 12). There was no significant difference between the transplanted groups
To assess the electrophysiological properties of pASCs, membrane currents were evoked by clamping the cell membrane from a holding potential of −70 mV to values ranging from −160 to +20 mV for 50 ms at 10 mV intervals. We observed three types of membrane currents in the pASC cultures 8 days after NI. However, none of them could be classified as activating Na+ currents supporting cell differentiation into a neuronal lineage.
Behavioral Testing
All the animals had normal motor function of their hind limbs before SCI (BBB = 21 ± 0). One week after SCI, the animals were paraplegic (BBB = 1.41 ± 0.32). At this point, the transplantation of ASCs or pASCs was performed. Over the course of 5 weeks, the control animals achieved BBB scores of 4.7 ± 0.34 (n = 12) with a slight improvement of their hind limb mobility but without any weight support. An improvement of the BBB scores in the transplanted animals was evident 4 weeks after transplantation. The transplanted animals showed a quick recovery supported by their higher dynamism, vigilance, and frequent episodes of weight support. Also, clinical examination after testing gave evidence of better muscle tone. The ASC group achieved BBB scores of 7.0 ± 0.74 (n = 11) 7 weeks after transplantation; the pASC-treated group displayed motor improvement with BBB scores of 7.2 ± 0.46 (n = 12) at the same time point. The transplanted animals improved their gait and the ability to support their body weight. There were no significant differences in functional improvement between the ASC- and pASC-treated animals (Fig. 2d).
Integration of ASCs and pASCs into the Site of SCI
Both types of transplanted cells were detected in host spinal cord tissue 8 weeks after transplantation by GFP fluorescence. The morphology adopted by the transplanted cells differed between the two groups. The ASCs were located within the injected area and showed a similar morphology as observed in culture, appearing as large cell bodies floating in the tissue. Staining for βIII-tubulin clearly documented that the transplanted cells did not attempt to form close contact with the host tissue (Fig. 1e). On the other hand, pASC grafts were more robust than those of the ASCs. In addition, pASCs showed an extensive migration from the injection site and seemed to configure hollow ducts along the neurofilaments, which is more typical of Schwann cells or OEGs (Fig. 1f). Cross-sected axons (staining for NF160) were closely enveloped by GFP+ pASC ringlet-like structures (Fig. 1g). Transplanted cells were not positive for O1, an oligodendrocytic marker (Fig. 1h); however, they were positive for NG2, which is an early marker of developing oligodendrocyte precursor cells (Fig. 1i). Although the pASCs did not differentiate predominantly into NG2+ cells, they might stimulate the inward migration of endogenous NG2+ precursors to the lesion site. This cell-to-cell contact can then promote the differentiation of pASCs into NG2+ cells. This was confirmed by TEM images showing that pASCs were interacting closely with Schwann cells, which migrated to the lesion from the spinal roots, and with myelinated as well as unmyelinated axons (Fig. 1j, k). We did not observe any shift toward neuronal differentiation that was induced by the in vivo environment (i.e., we did not observe any positivity for βIII-tubulin, NCAM, Nestin, or MAP2). The only marker that was present in vitro as well as in vivo was NG2, while a few cells expressed GFAP. Interestingly, we found some transplanted ASCs as well as pASCs positive for the endothelial marker CD31 (Fig. 1l, m).
Discussion
Adipose tissue has been identified as an alternative source of pluripotent stromal cells (Xu et al. 2008; Fang et al. 2010; Chi et al. 2010). Adipogenic, osteogenic, and chondrogenic differentiation assays were performed to evaluate the multipotent differentiation capabilities of our ASCs, with results that are in agreement with other findings (Mizuno 2009). Although cells from passage 2 were used for transplantation, the ASCs had the ability to form spheres, undergo neural differentiation, and differentiate into adipocytes and osteocytes up to five cell passages with a very consistent efficiency, comparable to that seen in the early passages, indicating that these cells had the inherent properties of multipotent stem cells.
After 5 days in NI medium, 80–90% of the adherent cells isolated from white subcutaneous adipose tissue could differentiate into spheres in vitro. This enabled us to prepare enough cells for in vivo experiments. In all likelihood, direct induction will not be as efficient in delivering a large population of precursors for further transplantation. The spheres contained a rather heterogenous population of cells, in which PCR confirmed the robust upregulation of NG2 and p75, typical markers of a glial or Schwann cell lineage. These results are in agreement with immunocytochemical staining of dissociated spheres. Six days after plating the dissociated spheres, we detected GFAP, NCAM, βIII-tubulin, and MAP2 using immunohistochemical staining; however, none of the genes, except for immature markers such as nestin or NCAM, showed upregulated RNA expression. Moreover, further cultivation in neuronal media did not lead to a fully differentiated neuronal phenotype with the expected electrophysiological properties. Therefore, immunocytochemical positivity can be related to changes in the cytoskeleton due to changes in culture conditions and does not have to correspond to full neural differentiation. TrkC expression in the sphere-induction step supports lineage commitment into peripheral nervous system cellular components, whereas exposure to neuronal growth factors (NGF and bFGF) induced a switch toward immature neural precursors resulting in the upregulation of the TrkA receptor.
The main goal of this study was to examine the effect of in vitro predifferentiation on the in vivo survival and fate of the transplanted cells in SCI and to compare the behavior of predifferentiated cells with that of naïve ASCs in SCI. Both groups of cells were transplanted to the lesion center of a balloon contusion SCI, and they successfully engrafted, migrated, and survived during 2 months. Similar work was done by Zhang et al. (2009), who compared ASCs and pACSs using two differentiation protocols in vitro and their effect in rats with a contusion SCI. Their findings from behavioral tests are in agreement with our results. However, their study was more focused on neuronal differentiation detected only by immunohistochemical staining, and therefore, they did not describe differentiation into early glial precursors (positivity for NG2, S100, p75), supported by RT-PCR results. In contrast to their findings, we have described the robust survival of pACSs in a spinal cord lesion. In addition, both the ASC- and pASC-transplanted cells expressed CD31 as an endothelial marker (Harris et al. 2010), which is an important factor in promoting angiogenesis in the lesion. Primary cultures of adipose tissue are a heterogeneous population containing small numbers of hematopoietic cells, pericytes, endothelial cells, and smooth muscle cells (Zuk et al. 2001). Recent findings emphasize (Crisan et al. 2008) that ASCs might originate in pericytes (CD146+) and that long-term culturing leads to the expression of typical MSC markers. We cannot confirm this finding since we do not have anti-rat CD146 available. We used the 2nd passage in our study, and our differentiation assays confirmed the multipotent character of ASCs. Regarding the endothelial cells, we did not find CD31+ cells in cell cultures. Therefore, the CD31+ cells most probably acquired this marker in the ischemic environment of the lesion. Angiogenesis can be also supported by the release of angiogenic factors that are produced by undifferentiated ASCs. Similar to our findings, it has been shown that ASCs can differentiate into endothelial cells in vitro and improve neovascularization in vivo in an ischemic limb model (Hong et al. 2010). The secretion of angiogenic factors was confirmed by RT-PCT analysis (Nakagami et al. 2005).
Functional improvement in animals with SCI has been reported after the transplantation of different types of stem cells, such as MSCs, OEGs, or NSCs (Amemori et al. 2010; Urdzikova et al. 2006; Karimi-Abdolrezaee et al. 2010). Most often this is attributed to the release of trophic factors, such as HGF, VEGF, NGF, BDNF, or GDNF (Park et al. 2010; Chiu et al. 2009). Since the behavioral effect observed in our study appeared rather quickly and was comparable in both the transplanted groups, it is most likely that the cells’ effect on behavioral outcome was also due to paracrine function. However, histological examination revealed an important difference in the in vivo behavior of pASCs and ASCs.
While the ASCs survived in fewer numbers and the cells appeared scattered, the pASCs showed an enhanced expression of neurotrophic factors and neural markers as well as better survival in the host tissue. The major difference was found in the integration of the pASCs into the host tissue. They formed hollow structures that followed host oligodendrocytes, adjacent to the periphery of the lesion. In addition, the new integrated cell layer mimicked in some respects a myelin sheath and was intimately associated with NF160+ axons when myelin was absent. This behavior demonstrated the establishment of a reparative mechanism at the periphery of the lesion site. Such a mechanism seems to work in two ways, protecting the oligodendrocytes from Wallerian degeneration (Weishaupt et al. 2010) and being engaged in the regeneration of the myelin sheath. In addition, the expression of the oligodendrocyte precursor marker NG2 (Knerlich-Lukoschus et al. 2010) by the pASCs suggests that the in vivo environment maintained the glial cell precursor phenotype. This finding is supported by similar findings from other groups who have shown that ASCs can differentiate into Schwann cells and that two months after transplantation these Schwann cell-like cells are able to remyelinate portions of denuded axons (Franco Lambert et al. 2009; Chi et al. 2010; Mizuno 2009), supporting functional recovery (Mantovani et al. 2010; Ohta et al. 2008). We did not find among the transplanted pASCs positive immunofluorescent staining for nestin or for βIII-tubulin. It is therefore evident that the SCI environment is not inducing neuronal differentiation but rather supporting angiogenic or glial differentiation.
The significance of our study lies in highlighting the differences between transplanting predifferentiated or ASCs cells in terms of their behavior in the host tissue and their influence on the fate of host cells at the site of SCI. We demonstrate that in vitro NI and differentiation of ASCs into functionally mature neurons in vivo is not necessary for ASCs to exert neuroprotective effects in our model of SCI, and thus, this abundant source of tissue that is readily accessible and easy to harvest can be considered as a viable tissue source for the supportive treatment of neurological disorders without any further modifications. On the other hand, the differentiation potential of non-engineered ASCs will never match the ability of neural stem cells, embryonic cells, or induced pluripotent cells to generate neural progeny for cell replacement therapies to treat CNS diseases. Lying between those statements is our finding that the predifferentiation of ASCs plays a beneficial role in SCI repair by protecting denuded axons and by recruiting endogenous CNS progenitor cells. Adipose tissue-derived stromal cells can therefore be another important player (besides bone marrow MSCs or OEGs) in multiple strategies using cell therapies together with bioengineered scaffolds for the treatment of SCI.
References
Abramson S (2010) New views of modern medicine regarding treatment with stem cells; its practical and ethical consequences. Klin Onkol 23(1):10–13
Amemori T, Jendelová P, Růzicková K, Arboleda D, Syková E (2010) Co-transplantation of olfactory ensheathing glia and mesenchymal stromal cells does not have synergistic effects after spinal cord injury in the rat. Cytotherapy 12(2):212–225
Bae JS, Carter JE, Jin HK (2010) Adipose tissue-derived stem cells rescue Purkinje neurons and alleviate inflammatory responses in Niemann-Pick disease type C mice. Cell Tissue Res 340(2):357–369
Basso DM, Beattie MS, Bresnahan JC (1995) A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 12(1):1–21
Chan J, Aoki C, Pickel VM (1990) Optimization of differential immunogold-silver and peroxidase labeling with maintenance of ultrastructure in brain sections before plastic embedding. J Neurosci Methods 33(2-3):113–127
Chi GF, Kim MR, Kim DW, Jiang MH, Son Y (2010) Schwann cells differentiated from spheroid-forming cells of rat subcutaneous fat tissue myelinate axons in the spinal cord injury. Exp Neurol 222(2):304–317
Chiu SC, Hung HS, Lin SZ, Chiang E, Liu DD (2009) Therapeutic potential of olfactory ensheathing cells in neurodegenerative diseases. J Mol Med 87(12):1179–1189
Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, Norotte C, Teng PN, Traas J, Schugar R, Deasy BM, Badylak S, Buhring HJ, Giacobino JP, Lazzari L, Huard J, Peault B (2008) A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3:301–313
Fang Z, Yang Q, Xiong W, Li G, Xiao J, Guo F, Li F, Chen A (2010) Neurogenic differentiation of murine adipose derived stem cells transfected with EGFP in vitro. J Huazhong Univ Sci Technol Med Sci 30(1):75–80
Franco Lambert AP, Fraga Zandonai A, Bonatto D, Cantarelli Machado D, Pêgas Henriques JA (2009) Differentiation of human adipose-derived adult stem cells into neuronal tissue: does it work? Differentiation 77(3):221–228
Gordon D, Scolding NJ (2009) Human mesenchymal stem cell culture for neural transplantation. Methods Mol Biol 549:103–118
Gu W, Zhang F, Xue Q, Ma Z, Lu P, Yu B (2010) Transplantation of bone marrow mesenchymal stem cells reduces lesion volume and induces axonal regrowth of injured spinal cord. Neuropathology 30(3):205–217
Halliday GM, Cullen KM, Kril JJ, Harding AJ, Harasty J (1996) Glial fibrillary acidic protein (GFAP) immunohistochemistry in human cortex: a quantitative study using different antisera. Neurosci Lett 209(1):29–32
Harris LJ, Zhang P, Abdollahi H, Tarola NA, DiMatteo C, McIlhenny SE, Tulenko TN, DiMuzio PJ (2010) Availability of adipose-derived stem cells in patients undergoing vascular surgical procedures. J Surg Res 163(2):e105–e112
Hejcl A, Sedý J, Kapcalová M, Toro DA, Amemori T, Lesný P, Likavcanová-Mašínová K, Krumbholcová E, Prádný M, Michálek J, Burian M, Hájek M, Jendelová P, Syková E (2010) HPMA-RGD hydrogels seeded with mesenchymal stem cells improve functional outcome in chronic spinal cord injury. Stem Cells Dev 19(10):1535–1546
Hess PG (2009) Risk of tumorigenesis in first-in-human trials of embryonic stem cell neural derivatives: ethics in the face of long-term uncertainty. Account Res 16(4):175–198
Hong SJ, Traktuev DO, March KL (2010) Therapeutic potential of adipose-derived stem cells in vascular growth and tissue repair. Curr Opin Organ Transplant 15(1):86–91
Hu BY, Zhang SC (2010) Directed differentiation of neural-stem cells and subtype-specific neurons from hESCs. Methods Mol Biol 636:123–137
Karimi-Abdolrezaee S, Eftekharpour E, Wang J, Schut D, Fehlings MG (2010) Synergistic effects of transplanted adult neural stem/progenitor cells, chondroitinase, and growth factors promote functional repair and plasticity of the chronically injured spinal cord. J Neurosci 30(5):1657–1676
Khoo ML, Shen B, Tao H, Ma DD (2008) Long-term serial passage and neuronal differentiation capability of human bone marrow mesenchymal stem cells. Stem Cells Dev 17(5):883–896
Knerlich-Lukoschus F, von der Ropp-Brenner B, Lucius R, Mehdorn HM, Held-Feindt J (2010) Chemokine, expression in the white matter spinal cord precursor niche after force-defined spinal cord contusion injuries in adult rats. Glia 58(8):916–931
Lee ST, Chu K, Jung KH, Im WS, Park JE, Lim HC, Won CH, Shin SH, Lee SK, Kim M, Roh JK (2009) Slowed progression in models of Huntington disease by adipose stem cell transplantation. Ann Neurol 66(5):671–681
Leranth C, Pickel VM (1989) Electron microscopic pre-embedding double immunostaining methods. In: Heimer L, Zaborsky L (eds) Tract-tracing, vol II. Plenum Publishing, New York, pp 129–172
Lo B, Parham L, Cedars M, Fisher S, Gates E, Giudice L, Halme DG, Hershon W, Kriegstein A, Rao R, Roberts C, Wagner R (2010) RESEARCH ETHICS: NIH guidelines for stem cell research and gamete donors. Science 327(5968):962–963
Mantovani C, Mahay D, Kingham M, Terenghi G, Shawcross SG, Wiberg M (2010) Bone marrow- and adipose-derived stem cells show expression of myelin mRNAs and proteins. Regen Med 5(3):403–410
Mizuno H (2009) Adipose-derived stem cells for tissue repair and regeneration: ten years of research and a literature review. J Nippon Med Sch 76(2):56–66
Nakagami H, Maeda K, Morishita R, Iguchi S, Nishikawa T, Takami Y, Kikuchi Y, Saito Y, Tamai K, Ogihara T, Kaneda Y (2005) Novel autologous cell therapy in ischemic limb disease through growth factor secretion by cultured adipose tissue-derived stromal cells. Arterioscler Thromb Vasc Biol 25(12):2542–2547
Neri M, Maderna C, Ferrari D, Cavazzin C, Vescovi AL, Gritti A (2010) Robust generation of oligodendrocyte progenitors from human neural stem cells and engraftment in experimental demyelination models in mice. PLoS One 5(4):e10145
Ohta Y, Takenaga M, Tokura Y, Hamaguchi A, Matsumoto T, Kano K, Mugishima H, Okano H, Igarashi (2008) Mature adipocyte-derived cells, dedifferentiated fat cells (DFAT), promoted functional recovery from spinal cord injury-induced motor dysfunction in rats. Cell Transplant 17(8):877–886
Park HW, Lim MJ, Jung H, Lee SP, Paik KS, Chang MS (2010) Human mesenchymal stem cell-derived Schwann cell-like cells exhibit neurotrophic effects, via distinct growth factor production, in a model of spinal cord injury. Glia 58(9):1118–1132
Pivonkova H, Benesova J, Butenko O, Chvatal A, Anderova M (2010) Impact of global cerebral ischemia on K+ channel expression and membrane properties of glial cells in the rat hippocampus. Neurochem Int 57(7):783–794
Radtke C, Schmitz B, Spies M, Kocsis JD, Vogt PM (2009) Peripheral glial cell differentiation from neurospheres derived from adipose mesenchymal stem cells. Int J Dev Neurosci 27(8):817–823
Rodríguez JJ, Dallérac GM, Tabuchi M, Davies HA, Colyer FM, Stewart MG, Doyère V (2008) N-methyl-d-aspartate receptor independent changes in expression of polysialic acid-neural cell adhesion molecule despite blockade of homosynaptic long-term potentiation and heterosynaptic long-term depression in the awake freely behaving rat dentate gyrus. Neuron Glia Biol 4(3):169–178
Salgado AJ, Reis RL, Sousa NJ, Gimble JM (2010) Adipose tissue derived stem cells secretome: soluble factors and their roles in regenerative medicine. Curr Stem Cell Res Ther 5(2):103–110
Sofroniew MV, Howe CL, Mobley WC (2001) Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci 24:1217–1281
Syková E, Jendelová P, Urdzíková L, Lesný P, Hejcl A (2006) Bone marrow stem cells and polymer hydrogels—two strategies for spinal cord injury repair. Cell Mol Neurobiol 26(7–8):1113–1129
Turnovcova K, Ruzickova K, Vanecek V, Sykova E, Jendelova P (2009) Properties and growth of human bone marrow mesenchymal stromal cells cultivated in different media. Cytotherapy 11(7):874–885
Urdzikova L, Jendelova P, Glogarova K, Burian M, Hajek M, Sykova E (2006) Transplantation of bone marrow stem cells as well as mobilization by granulocyte-colony stimulating factor promotes recovery after spinal cord injury in rats. J Neurotrauma 23:1379–1391
Wei X, Zhao L, Zhong J, Gu H, Feng D, Johnstone BH, March KL, Farlow MR, Du Y (2009) Adipose stromal cells-secreted neuroprotective media against neuronal apoptosis. Neurosci Lett 462(1):76–79
Weishaupt N, Silasi G, Colbourne F, Fouad K (2010) Secondary damage in the spinal cord following motor cortex injury in rats. J Neurotrauma 27(8):1387–1397
Xu Y, Liu Z, Liu L, Zhao C, Xiong F, Zhou C, Li Y, Shan Y, Peng F, Zhang C (2008) Neurospheres from rat adipose-derived stem cells could be induced into functional Schwann cell-like cells in vitro. BMC Neurosci 9:21
Yamada T, Akamatsu H, Hasegawa S, Yamamoto N, Yoshimura T, Hasebe Y, Inoue Y, Mizutani H, Uzawa T, Matsunaga K, Nakata S (2010) Age-related changes of p75 neurotrophin receptor-positive adipose-derived stem cells. J Dermatol Sci 58(1):36–42
Zhang HT, Luo J, Sui LS, Ma X, Yan ZJ, Lin JH, Wang YS, Chen YZ, Jiang XD, Xu RX (2009) Effect of differentiated versus undifferentiated adipose tissue-derived stromal cell grafts on functional recovery after spinal cord contusion. Cell Mol Neurobiol 29:1283–1292
Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, Benhaim P, Lorenz HP, Hedrick MH (2001) Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng 7(2):211–228
Acknowledgments
This study was supported by the grants AV0Z50390703, AVOZ50390512, 1M0538, GA CR 309/08/H079, GA CR 305/09/0717, GA CR 304/10/0320, and IAA50390902. The authors thank James Dutt for the critical reading of the manuscript.
Conflict of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Arboleda, D., Forostyak, S., Jendelova, P. et al. Transplantation of Predifferentiated Adipose-Derived Stromal Cells for the Treatment of Spinal Cord Injury. Cell Mol Neurobiol 31, 1113–1122 (2011). https://doi.org/10.1007/s10571-011-9712-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-011-9712-3