
Abstract
Spontaneously hypertensive rats (SHR) are widely used as model to investigate the pathophysiological mechanisms of essential hypertension. Catecholamine plasma levels are elevated in SHR, suggesting alterations of the sympathoadrenal axis. The residual hypertension in sympathectomized SHR is reduced after demedullation, suggesting a dysfunction of the adrenal medulla. Intact adrenal glands exposed to acetylcholine or high K+ release more catecholamine in SHR than in normotensive Wistar Kyoto (WKY) rats, and adrenal chromaffin cells (CCs) from SHR secrete more catecholamines than CCs from WKY rats. Since Ca2+ entry through voltage-gated Ca2+ channels (VGCC) triggers exocytosis, alterations in the functional properties of these channels might underlie the enhanced catecholamine release in SHR. This study compares the electrophysiological properties of VGCC from CCs in acute adrenal slices from WKY rats and SHR at an early stage of hypertension. No significant differences were found in the macroscopic Ca2+ currents (current density, I–V curve, voltage dependence of activation and inactivation, kinetics) between CCs of SHR and WKY rats, suggesting that Ca2+ entry through VGCC is not significantly different between these strains, at least at early stages of hypertension. Ca2+ buffering, sequestration and extrusion mechanisms, as well as Ca2+ release from intracellular stores, must now be evaluated to determine if alterations in their function can explain the enhanced catecholamine secretion reported in CCs from SHR.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
High blood pressure (HBP) is a leading risk factor for heart disease, stroke, and kidney failure. Most patients suffer essential hypertension, whose primary causes are still under debate. An anomalous hyperactivity of the sympathetic nervous system (SNS) has been implicated in the pathophysiology of HBP, because sympathetic nerve activity and plasma levels of epinephrine and norepinephrine are elevated in hypertensive patients (Anderson et al. 1989; Goldstein et al. 1983). Higher catecholamine levels probably result from increased release from sympathetic neurons and adrenal chromaffin cells.
The spontaneously hypertensive rats (SHR), a strain developed in Japan in the 1960s, are widely used as a model to investigate the mechanisms underlying essential hypertension. In SHR, plasma catecholamine levels are higher relative to normotensive Wistar Kyoto (WKY) rats (Pak 1981) and the development of hypertension is partially prevented by neonatal sympathectomy (Lee et al. 1991). The adrenal medulla’s role in the origin and maintenance of hypertension in SHR is unclear: bilateral adrenalectomy of pre-hypertensive and adult SHR produced normotensive animals, even though some BP reduction was also observed in WKY rats (Aoki et al. 1973). Bilateral removal of the adrenal medulla (demedullation) in pre-hypertensive juvenile SHR attenuated the hypertension developed in the adult (Borkowski and Quinn 1983), whereas demedullation in hypertensive adult SHR either had no effect (Borkowski and Quinn 1983, Aoki et al. 1973) or lowered BP (Sakaguchi et al. 1983). When sympathectomized SHR are demedullated, BP returned to normotensive levels (Lee et al. 1991). These findings implicate a possible dysfunction of the adrenal medulla in SHR.
When stimulated with acetylcholine (ACh) or high K+, intact adrenal glands from SHR release more catecholamines than WKY rats (Lim et al. 2002). Accordingly, catecholamine secretion, measured with amperometry is higher in isolated chromaffin cells (CCs) from SHR compared with CC from normotensive rats (Miranda-Ferreira et al. 2008, 2009). The elevation of intracellular calcium concentration [Ca2+]i in the vicinity of the release sites triggers catecholamine release, and the most important source of [Ca2+]i elevation in CCs is Ca2+ entry through voltage-dependent Ca2+ channels (VDCC; García et al. 2006). In rat chromaffin cells, Ca2+ current is carried mainly by L, N, P, and Q-type Ca2+ channels (Gandia et al. 1995, Hernández-Guijo et al. 1999), which open when the cell membrane is depolarized after the activation of cholinergic receptors by acetylcholine released from splanchnic nerve terminals. The enhanced catecholamine release reported in SHR CCs could result from an augmented function of VDCC. This is not implausible: vascular smooth muscle cells from SHR have a higher density of L-type Ca2+ current and an elevated expression of mRNA for α1C (Cav1.2) Ca2+ channel subunits (Cox and Lozinskaya 1995; Pratt et al. 2002).
The aim of this study is to compare the electrophysiological properties of voltage-gated Ca2+ currents in adrenal CCs from SHR and WKY rats at early stages of hypertension. Since the enzymatic dissociation of CCs and the tissue culture conditions can alter the expression and regulation of VDCC subtypes, (García et al. 2006), these experiments were performed in adrenal gland slices (Barbara et al. 1998), where CCs remain in their native environment and maintain close to normal cell-to-cell interactions.
Methods
Animals
All protocols used are in accordance with the guidelines of the National Council on Animal Care. Male spontaneously hypertensive (SHR) and Wistar Kyoto (WKY) normotensive rats were obtained from Charles River Laboratories, Inc., Wilmington and have been bred and housed in our animal facility for >6 months. The facility is temperature-controlled (22°C) and maintained on a 12-h light/dark cycle. Rats received standard laboratory chow and tap water ad libitum.
Blood Pressure Measurements
Male SHR and WKY rats of 7–12-weeks-old were subject to blood pressure measurements via a non-invasive tail-cuff plethysmograph device (LE 5002, Panlab Harvard Apparatus). The animal is introduced into a plexiglass restraint device with the blood pressure measurement cuff placed on the base of the tail. The thermal heating pad underneath was set to 30°C. Blood pressure measurements (systolic and diastolic) were conducted between 10:00 am and 2:00 pm at least three times per day over a 2-day period before sacrifice. Animals were anesthetized by administration of ketamine (75 mg/kg) and xylazine (5 mg/kg, i.p.) and killed by decapitation.
Adrenal Slice Preparation
Adrenal glands were removed and immersed in ice-cold, low Ca2+ bicarbonate-buffered saline (BBS) containing (in mM): 140 NaCl, 2 KCl, 0.1 CaCl2, 5 MgCl2, 26 NaHCO3, and 10 glucose. The solution was continuously gassed with 95% O2 and 5% CO2. Glands were embedded in 3% low gelling point agarose at 35°C. The agarose was gelled on ice and the gland was sectioned into 200 μm thick slices with a vibrating blade microtome (Leica VT-1000S). Slices were kept at room temperature in a holding chamber in low Ca2+ BBS until recording. Experiments were performed 2–6 h after slice preparation.
ICa2+ Recording, Data Acquisition, and Analysis
Whole-cell voltage-clamp recordings of macroscopic calcium currents (ICa2+) were performed with glass pipettes (2–5 MΩ) filled with intracellular solution containing (in mM): 10 NaCl, 14 ethylene glycol tetraacetic acid (EGTA), 100 CsCl, 20 TEA.Cl, 2 MgCl2, 5 Mg.ATP, 0.3 Na.GTP, 20 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)/CsOH (pH 7.3, 270 mOsM). The patch pipette was connected to the head stage of an EPC-10 patch-clamp amplifier (HEKA Electronic, Lambrecht, Germany). Data were acquired at 5–10 kHz and filtered at 1–2 kHz. Recordings where leak currents >25 pA or series resistance >20 MΩ were discarded. Slices were placed in the bottom of a plexiglass chamber mounted on the stage of a Nikon upright microscope (Eclipse FN1, Nikon Corporation, Tokyo, Japan). The patch pipette approached the cell with the aid of a Sutter micromanipulator (MPC-200, Sutter Instrument Company, USA). During seal formation the slice was superfused with BBS solution containing (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3 and 10 glucose, gassed with 95% O2/5% CO2. Once the whole-cell recording was established, the cell was locally superfused with a Krebs-HEPES extracellular solution containing 10 mM CaCl2 and 1 μM tetrodotoxin (TTX). The common outlet of the local perfusion system was placed within 100 μm of the cell and the flow rate was regulated to achieve complete replacement of the cell surroundings within less than 200 ms. Data acquisition and analysis was performed using the Patchmaster program (HEKA Elektronics). All experiments were performed at room temperature (22–25°C).
Voltage-clamped chromaffin cells were held at −90 mV and ICa2+ was elicited by depolarizing square pulses from −60 mV to +40 mV (5 mV steps) of 60 ms in duration. Once a cell was selected for recording, K+ currents were blocked by dialyzing Cs+ and TEA through the pipette and Na+ currents were eliminated by bathing with 1 μM TTX. An external CaCl2 concentration of 10 mM was used to increase ICa2+ and I–V relationships were constructed by measuring ICa2+ amplitude at the peak (first 5 ms of the pulse) and at the steady-state (last 5 ms of the pulse) and plotted as current densities (pA/pF). The ICa2+ activation curve was obtained by transforming current data to conductance (g Ca = I Ca/(V m − E Ca). The apparent reversal potential of I Ca (E Ca) was estimated by extrapolation (~+60 mV). The Boltzmann equation g/g max = {1 + exp[−(V − V a1/2)/k a]}−1 was used to fit the data. (g max = peak conductance; V a1/2 = half-activation voltage; k a = steepness parameter). For the ICa2+ steady-state inactivation curve, a test voltage step to −20 mV was applied to preferentially activate the transient and to +5 mV to activate the sustained ICa2+ components. Pre-pulse conditioning voltage steps of 1.5 s in duration (range from −100 to +10 mV) were applied before the test pulse in 3 mV (transient) and 10 mV increments (sustained). Steady-state inactivation curves were constructed by plotting the current elicited during the test pulse as a function of the pre-pulse voltage. The Boltzmann relationship I/I max = {1 + exp[(V − V i1/2)/k i]}−1 was used to fit the data (I max = current elicited by the test pulse after a −100 mV pre-pulse; V i1/2 = half-inactivation voltage; k i = steepness parameter).
To assess the autocrine/paracrine modulation of ICa2+ by endogenous neurotransmitters, CCs were held at −80 mV and pulses to 0 mV were applied every 10 s to measure ICa2+ peak amplitude. A depolarizing pre-pulse (+80 mV; 30 ms) was applied occasionally to assess the voltage-dependent component of ICa2+ modulation. Then, bath flow was stopped to allow accumulation of neurotransmitters released spontaneously and autocrine modulation of ICa2+. Voltage-dependent and -independent ICa2+ modulation was evaluated with these depolarizing pre-pulses both in the flow- and flow-stop conditions.
Nifedipine was prepared in ethanol and diluted in extracellular solution to the required final concentration (5 μM). At the concentration used (0.1%), ethanol did not affect ICa2+. Nifedipine, agarose, and salts were purchased from Sigma-Aldrich (Mexico DF, Mexico). TTX was purchased from Tocris (Ellsville, MO, USA). Results are expressed as means ± S.E.M. One-way ANOVA followed by Tukey’s multiple comparison test were used. In some cases an unpaired t-test was preferred. A P-value <0.05 was taken as the limit of significance.
Results and Discussion
Blood Pressure Measurements in Normotensive WKY Rats and Hypertensive SHR
Rats 7–12 weeks of age were used. At approximately this age, blood pressure in SHR reaches 150 mmHg (Lee et al. 1991). In the WKY group (n = 15), systolic and diastolic BP were, respectively, 111.2 ± 2.8 and 81.4 ± 1.5 mmHg (mean ± SE). In the SHR group (n = 16), systolic and diastolic BP were, respectively, 147.1 ± 3.2 and 108.7 ± 4.2 mmHg (mean ± SE). Inter-strain differences were significant (P < 0.0001).
ICa2+ in CCs from Normotensive WKY Rats and from Hypertensive SHR
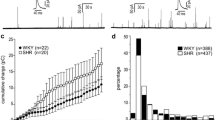
ICa2+ recorded from both WKY and SHR CCs comprised two current components, an early, fast transient component that peaked and inactivated rapidly and a late, non-inactivating, sustained component (see records in Fig. 1a, b). At more depolarized potentials, these components became difficult to distinguish. The sustained component inactivated only slightly during the depolarizing pulse. The kinetics and voltage dependence of ICa2+ in CCs from WKY and SHR rats are very similar, but they differ from those recorded under the same conditions in rat CCs from the Wistar strain (data not shown, see García et al. 2006). A characterization of voltage-gated Ca2+ currents in rat CCs from the WKY strain is underway (Segura-Chama, in preparation). Given the obvious differences in kinetics and voltage-dependence of ICa2+ in CCs from WKY/SHR and Wistar strains, experiments where CCs from SHR are compared to Wistar, Sprague–Dawley or other normotensive strains, rather than WKY should be taken with caution.

Comparison of ICa2+ in WKY and SHR CCs. a, b Representative Ca2+ current traces elicited by depolarizing pulses indicated (holding potential = −90 mV). Notice in both strains the transient inactivating component and the sustained, non-inactivating component. Dotted lines indicate times at which transient and sustained components were measured. c, d Current density–voltage relationship of transient (triangles) and sustained (circles) ICa2+ components in CCs from WKY rats (closed symbols) and SHR (open symbols), respectively (mean ± SEM; n = 19)
Voltage Dependence of Activation and Inactivation of ICa2+
Activation and inactivation of ICa2+ between normotensive and hypertensive rat strains are similar (Fig. 1a, b). The amplitude of early and late current components was measured during the first 5 ms of the pulse and at 5 ms before the end of the pulse, respectively, (dotted vertical lines in Fig. 1a) and current amplitudes were divided by the cell capacitance to obtain an indication of current density (pA/pF). Then, current density–voltage step relationship wAS plotted (I–V curve). CCs capacitances were 8.1 ± 1.1 and 7.8 ± 1.2 pF in WKY in SHR CCs, respectively (mean ± SE; n = 19 SHR; 19 WKY; P = 0.41). Figure 1c, d illustrates mean I–V curves obtained from 19 CCs (WKY and SHR, respectively). The transient component began to activate at ~−40 or −45 mV and peaked at around −5 mV. In contrast, the sustained component activated at ~−30 to 35 mV and peaked at approximately +5 mV. Both Ca2+ current components exhibited an apparent reversal potential at about +50 mV. The mean peak amplitude of the transient component was 61.7 ± 3.2 and 55.5 ± 4.9 pA/pF (mean ± SE). Similarly, the mean amplitude of the sustained component was 40.7 ± 2.1 and 35.7 ± 3.6 pA/pF (mean ± SE; n = 19) in WKY and SHR CCs, respectively. Inter-strain differences are not significant (P = 0.29 and 0.23; transient and sustained components, respectively).
Activation and Steady-State Inactivation Curves of ICa2+
Activation curves were constructed by transforming current data to conductance, and plotting them as a function of the corresponding voltage steps. Figure 2a summarizes data obtained for WKY and SHR CCs. When the Boltzmann relationship (see “Methods” section) was used to fit the data the half-activation potentials (V a1/2) of the transient ICa2+ component were −19.6 and −20.1 mV; with k a values of 9.5 and 9.15 (WKY and SHR, respectively). In contrast, for the sustained ICa2+ component, V a1/2 values were −6.2 and −9.1 mV; with k a values 7.5 and 7.0 (WKY and SHR, respectively). The V a1/2 and k a values for transient and sustained ICa2+ components are significantly different (P < 0.0001 and P < 0.0006) suggesting the activation of two Ca2+ channel subtypes. On the other hand, V a1/2 and k a were not statistically different between the WKY and SHR (P = 0.52 and P = 0.6, respectively) suggesting that voltage dependence of both ICa2+ components is indistinguishable between SHR and WKY rats.

Activation and steady-state inactivation of ICa2+ in WKY and SHR CCs. a Normalized Ca2+ conductance values plotted against the depolarizing step potential. Values of transient (triangles) and sustained (circles) components were fitted to the Boltzmann activation relationship (continuous lines; see “Methods” section). b Steady-state inactivation curves. ICa2+ data were normalized and plotted against the prepulse potentials. Transient (triangles) and sustained (circles) components were fitted with the Boltzmann inactivation relationship (continuous lines). WKY closed symbols, SHR open symbols
Steady-state inactivation curves were constructed by plotting peak current amplitude elicited during the test pulse as a function of the conditioning pre-pulse (see “Methods” section; Fig. 2b). Half-inactivation potentials (V i1/2) of the transient ICa2+ component were −46.9 and −46.8 mV, with k i values of 4.0 and 4.3 (WKY and SHR, respectively). For the sustained ICa2+ component, half-inactivation potentials (V i1/2) were −20.8 and −18.8 mV with k i values of 9.8 and 9.8 (WKY and SHR, respectively). V i1/2 and k i values of transient and sustained components were significantly different (P < 0.0001 in both cases), suggesting two ICa2+ components with different voltage-dependent inactivation. These parameters were not statistically different when WKY and the SHR CCs were compared (P = 0.8 and P = 0.23). Thus, the voltage dependence of activation and inactivation of both ICa2+ components is not appreciably different in CCs from hypertensive and normotensive rats.
Blockade of ICa2+ by Ni2+
T-type Ca2+-channels are present in CCs from immature animals (Marcantoni et al. 2008) and they subsist in a small proportion in adult rat or mouse CCs (Carabelli et al. 2007). Nonetheless, these channels are up-regulated by membrane-permeant cAMP, adrenergic stimulation (Novara et al. 2004), chronic hypoxia, and adrenal gland denervation (Levitsky and López-Barneo 2009). It has been argued that the re-expression of T-type channels after chronic stimulation of β-adrenoreceptors could enhance excitability and secretion in CCs during sustained sympathetic stimulation or elevated catecholamine plasma levels (Novara et al. 2004). Both conditions are known to occur in SHR (Pak 1981).
The kinetics and activation threshold of the transient ICa2+ component in CCs from WKY and SHR is reminiscent of the T-type Ca2+ current (Carbone and Lux 1984). Moreover, V a1/2 and V i1/2 values reported for the cAMP-recruited T-type current in CCs (−27.4 mV and −45.9 mV, respectively; Novara et al. 2004) are comparable to those of the transient ICa2+ component in WKY–SHR CCs: V a1/2 = −19.6 and −20.1 mV; V i1/2 = −46.8 and −46.9 mV, respectively. If T-type channels contribute significantly to the transient ICa2+ in CCs from WKY–SHR, it should be blocked almost entirely with micromolar concentrations of Ni2+ (Carabelli et al. 2007). Figure 3 illustrates mean ICa2+ I–V relationships obtained in WKY and SHR CCs before and after bath application of 50 μM Ni2+. While a fraction of the transient component is blocked by Ni2+, the shape of the I–V curve in the presence of Ni2+ remains unchanged (Fig. 3a, b). Thus, Ni2+ did not abolish the “low-threshold shoulder” of the T-type current (Carbone and Lux 1984; Carabelli et al. 2007). Ni2+ blocks 31.7 ± 3.2% of the peak transient current (0 to −5 mV) in WKY CCs, and 30.1 ± 7.3% in SHR CCs. (inter-strain differences not significant P = 0.85). As shown in Fig. 3c, a large proportion (~50 to 60%) of the transient component is blocked by Ni2+ in the voltage range between −40 and −25 mV and less (~25 to 40%) in the voltage range between −10 and +10 mV. As shown in Fig. 3d, e Ni2+ also inhibits the sustained component (29.0 ± 3.6% in WKY and 21.6 ± 4.6% in SHR CCs; peak current at 0 to −5 mV). This result is unexpected, since high voltage-activated Ca2+ currents typically are not affected by low concentrations of Ni2+ (cf. Carabelli et al. 2007). Ni2+ blockade of the sustained component is also voltage dependent (see Fig. 3f): the current activated at low voltages is preferentially blocked by Ni2+. This assessment is inconclusive regarding the contribution of T-type channels to the transient component in WKY–SHR. More thorough experiments are required to properly address this question.

Ni2+ blockade of ICa2+. a, b I–V relationships for the transient ICa2+ component of WKY and SHR CCs, respectively, before (black symbols) and after (grey symbols) bath application of 50 μM Ni2+. c Inhibition of the transient component at different step potentials in WKY (black bars) and SHR (white bars). The transient component activated at low voltages is strongly blocked by Ni2+. d, e I–V relationships of the sustained ICa2+ component before (black symbols) and after (grey symbols) Ni2+ application in WKY and SHR CCs, respectively (n = 7). f Voltage-dependent inhibition of the sustained component in WKY and SHR. The sustained component activated at low voltages is also preferentially blocked by Ni2+
Nifedipine Blockade of ICa2+
In cultured venous muscle cells, peak Ca2+ current between WKY and SHR was unchanged, but the contribution of the L-type Ca2+ current increased from 42% in WKY to 62% in SHR cells (Rusch and Hermsmeyer 1988). In another study in smooth muscle cells from mesenteric arteries, the density of L-type Ca2+ current increased by 47% in SHR compared to WKY rats (Cox and Lozinskaya 1995). These changes explain in part the augmented vascular tone that characterizes hypertension. Here we examined the contribution of L-type Ca2+ channels in CCs from SHR and WKY rats.
In rat CCs about 50% of the total Ca2+ current is abolished by nifedipine, which selectively blocks L-type Ca2+-channels (Gandia et al. 1995; see García et al. 2006, for a review). Nifedipine 5 μM blocked 43.8 ± 6.5 and 45.0 ± 2.3% of the transient component at −5 mV (WKY and SHR, respectively, see Fig. 4), and shifted the I–V curve of the remaining current ~5 to 10 mV toward more positive voltages (see Fig. 4a, b). Nifedipine inhibition of the transient component is strongly voltage-dependent (see Fig. 4c); i.e. 89.0 ± 5.3 and 83.2 ± 4.6% inhibition at −40 mV (WKY and SHR, respectively). At +5 mV, nifedipine blocks 33.3 ± 7.8% and 41.6 ± 4.1%, respectively, of the sustained component (WKY and SHR, respectively) and the I–V curve of the nifedipine-insensitive current shifted ~5 mV in the positive direction in both cases (see Fig. 4d, e). Nifedipine block of the sustained Ca2+ current component is also voltage-dependent (see Fig. 4f); i.e. inhibition is 80.7 ± 12.5 and 96.7 ± 2.0% at −40 mV (WKY and SHR, respectively). The conclusions from these experiments are: (1) L-type Ca2+ channels contribution to the total ICa2+ in WKY and SHR CCs is similar to that reported in other rat strains: 43–45% and 33–41% (transient and sustained components, respectively) and (2) unlike vascular muscle cells, the contribution of L-type Ca2+ channels to the total Ca2+ current in CCs from WKY rats and SHR remains the same.

Effects of nifedipine on ICa2+. a, b I–V relationships of the transient ICa2+ component in CCs from WKY and SHR, respectively, before (black symbols) and after (grey symbols) bath application of 5 μM nifedipine (n = 7). c Percent inhibition of the transient component by nifedipine in WKY and SHR as a function of step potentials. The transient component activated at low voltages is more strongly blocked by nifedipine. d, e I–V relationships of the sustained ICa2+ component in WKY and SHR CCs, respectively, before and after bath application of nifedipine (n = 7). f Percent inhibition of the sustained component at different step potentials in WKY and SHR. Nifedipine blockade of the sustained component is stronger at negative voltages
Autocrine Modulation of ICa2+
We have demonstrated that Ca2+ current density, voltage dependence of activation and inactivation, and the contribution of L-type Ca2+ channels in CCs from WKY rats and SHR are indistinguishable. This would apparently rule out inter-strain differences in ICa2+ as the cause of catecholamine hypersecretion in SHR. Nonetheless, enhanced catecholamine release is expected if Ca2+ entry through VGCC is subject to less inhibition by locally released neurotransmitters. Autocrine modulation of ICa2+ by purinergic and opioidergic receptors has been demonstrated in cultured CCs (Hernández-Guijo et al. 1998), and in mouse CCs from adrenal slices (A. Hernández et al., submitted). Figure 5a shows how autocrine modulation of Ca2+ channels is evaluated. After a period of stable ICa2+ recording under flow conditions, bath perfusion was discontinued (flow-stop), and consequently the amplitude of ICa2+ diminished gradually, presumably because endogenous agonists spontaneously released by CCs accumulate locally. ICa2+ inhibition in flow-stop reached steady-state in about 4 min and recovered after flow restoration (Fig. 5a). Example recordings are shown in Fig. 5b. Maximal ICa2+ inhibition during flow-stop was 20.2 ± 1.0% in WKY rats and 22.7 ± 2.4% in SHR, respectively (P = 0.4). A pre-pulse step depolarization to +80 mV was used to reveal the fraction of voltage-dependent ICa2+ inhibition. Under flow conditions, a small voltage-dependent tonic inhibition of ICa2+ is uncovered with this maneuver (4.8 ± 1.4% and 5.5 ± 1.0%; WKY and SHR, respectively; see Fig. 5c, d; Control). Nonetheless, voltage-dependent modulation nearly doubles (9.2 ± 1.4% and 10.4 ± 1.4%; WKY and SHR, respectively) during the flow-stop condition. Figure 5c, d illustrates total ICa2+ inhibition produced by the flow-stop condition, as well as their voltage-dependent (open bar) and voltage independent (grey bar) fractions. Differences in total ICa2+ inhibition or in the voltage-dependent and independent fractions in WKY and SHR CCs are not statistically significant (P = 0.6).

Voltage-dependent and -independent autocrine inhibition of ICa2+ in WKY and SHR chromaffin cells. a Time course of ICa2+ inhibition by the flow-stop condition. In this example, a WKY CC was held at −80 mV and step pulses to 0 mV were applied every 10 s. b Representative Ca2+ current records from the experiment shown in a, at data points labeled a, b, and c. Dotted lines control and recovery ICa2+ peak amplitudes. c, d Autocrine modulation of ICa2+ in WKY and SHR CCs, respectively. Open bars percentage of voltage-dependent modulation both under flow and flow-stop conditions. Grey bar percentage of voltage-independent ICa2+ inhibition during the flow-stop condition. Insets superimposed ICa2+ traces with and without a prepulse step to +80 mV to reveal the fraction of voltage-dependent modulation (see “Methods” section). a flow condition; b flow-stop condition
Our study demonstrates that Ca2+ activation and inactivation kinetics, current density, voltage dependence of activation and inactivation, participation of L-type Ca2+ channels, and autocrine modulation of ICa2+ in CCs from normotensive and hypertensive rats are indistinguishable. This rules out an altered Ca2+ influx through voltage-gated Ca2+ channels as the sole cause for the reported catecholamine hypersecretion in CCs from SHR (Miranda-Ferreira et al. 2008, 2009). Cardiac ventricular myocytes from SHR exhibit stronger contractions and larger intracellular Ca2+ transients compared to normotensive WKY myocytes (Brooksby et al. 1993), and yet, the amplitude of ICa2+ recorded from SHR myocytes was unchanged compared to WKY myocytes. These contradictory results are explained by a prolongation of the action potentials and elevated sarcoplasmic reticulum Ca2+ content in SHR myocytes (Brooksby et al. 1993). Action potential duration and frequency of spontaneous action potential firing should be compared in CCs from SHR and WKY rats. Also, intracellular Ca2+ dynamics and handling by CCs from both strains need to be explored. A study now in progress indicates that in SHR slices, more CCs display spontaneous [Ca2+]i fluctuations than in WKY slices. These fluctuations are larger in amplitude and more frequent in SHR than in WKY CCs. These differences require confirmation and their origin needs to be established. Alterations of intracellular Ca2+ signaling and homeostasis in CCs are likely candidates for the enhanced catecholamine secretion in the adrenal gland, elevated catecholamine plasma levels and high blood pressure which characterize the SHR strain.
References
Anderson EA, Sinkey CA, Lawton WJ, Mark AL (1989) Elevated sympathetic nerve activity in borderline hypertensive humans. Evidence from direct intraneural recordings. Hypertension 14:177–183
Aoki K, Takikawa K, Hotta K (1973) Role of adrenal cortex and medulla in hypertension. Nature 241:122–123
Barbara JG, Poncer JC, McKinney RA, Takeda K (1998) An adrenal slice preparation for the study of chromaffin cells and their cholinergic innervation. J Neurosci Methods 80:181–189
Borkowski KR, Quinn P (1983) The effect of bilateral adrenal demedullation on vascular reactivity and blood pressure in spontaneously hypertensive rats. Br J Pharmacol 80:429–437
Brooksby P, Levi AJ, Jones JV (1993) Investigation of the mechanisms underlying the increased contraction of hypertrophied ventricular myocytes isolated from the spontaneously hypertensive rat. Cardiovasc Res 27:1268–1277
Carabelli V, Marcantoni A, Comunanza V, de Luca A, Diaz J, Borges R, Carbone E (2007) Chronic hypoxia up-regulates α1H T-type channels and low threshold catecholamine secretion in rat chromaffin cells. J Physiol 584:149–165
Carbone E, Lux HD (1984) A low-voltage-activated, fully inactivating calcium channel in vertebrate sensory neurons. Nature 310:501–502
Cox RH, Lozinskaya IM (1995) Augmented calcium currents in mesenteric artery branches of the spontaneously hypertensive rat. Hypertension 26:1060–1064
Gandia L, Borges R, Albillos A, Garcıa AG (1995) Multiple calcium channel subtypes in isolated rat chromaffin cells. Pflügers Arch Eur J Physiol 430:55–63
García AG, de Diego AM, Gandía L, Borges R, García-Sancho J (2006) Calcium signaling and exoctytosis in chromaffin cells. J Physiol Rev 86:1093–1131
Goldstein DS, Horwitz D, Keiser HR, Polinsky RJ, Kopin IJ (1983) Plasma l-[3H] norepinephrine, d-[14C] norepinephrine, and d, l-[3H] isoproterenol kinetics in essential hypertension. J Clin Invest 72:1748–1758
Hernández-Guijo JM, Pascual R, García AG, Gandía L (1998) Separation of calcium channel current components in mouse chromaffin cells superfused with low- and high-barium solutions. Pflügers Arch Eur J Physiol 436:75–82
Hernández-Guijo JM, Carabelli V, Gandía L, García AG, Carbone E (1999) Voltage independent autocrine modulation of L-type channels mediated by ATP, opioids and catecholamines in rat chromaffin cells. Eur J Neurosci 11:3574–3584
Lee RM, Borkowski KR, Leenen FH, Tsoporis J, Coughlin M (1991) Combined effect of neonatal sympathectomy and adrenal demedullation on blood pressure and vascular changes in spontaneously hypertensive rats. Circ Res 69:714–721
Levitsky KL, López-Barneo J (2009) Developmental change of T-type Ca2+ channel expression and its role in rat chromaffin cell responsiveness to acute hypoxia. J Physiol 587.9:1917–1929
Lim DY, Jang SJ, Park DG (2002) Comparison of catecholamine release in the isolated adrenal glands of SHR and WKY rats. Auton Autacoid Pharmacol 22:225–232
Marcantoni A, Carabelli V, Comunanza V, Hoddah H, Carbone E (2008) Calcium channels in chromaffin cells: focus on L and T types. Acta Physiol 192:233–246
Miranda-Ferreira R, de Pascual R, de Diego AM, Caricati-Neto A, Gandía L, Jurkiewicz A, García AG (2008) Single-vesicle catecholamine release has greater quantal content and faster kinetics in chromaffin cells from hypertensive, as compared with normotensive, rats. J Pharmacol Exp Ther 324:685–693
Miranda-Ferreira R, de Pascual R, Caricati-Neto A, Gandía L, Jurkiewicz A, García AG (2009) Role of the endoplasmic reticulum and mitochondria on quantal catecholamine release from chromaffin cells of control and hypertensive rats. J Pharmacol Exp Ther 329:231–240
Novara M, Baldelli P, Cavallari D, Carabelli V, Giancippoli A, Carbone E (2004) Exposure to cAMP and β-adrenergic stimulation recruits CaV3 T-type channels in rat chromaffin cells through Epac cAMP-receptor proteins. J Physiol 558:433–449
Pak CH (1981) Plasma adrenaline and noradrenaline concentrations of the spontaneously hypertensive rat. Jpn Heart J 22:987–995
Pratt PF, Bonnet S, Ludwig LM, Bonnet P, Rusch NJ (2002) Upregulation of L-type Ca2+ channels in mesenteric and skeletal arteries of SHR. Hypertension 40:214–219
Rusch NJ, Hermsmeyer K (1988) Calcium currents are altered in the vascular muscle cell membrane of spontaneously hypertensive rats. Circ Res 63:997–1002
Sakaguchi A, LeDoux JE, Reis DJ (1983) Sympathetic nerves and adrenal medulla: contributions to cardiovascular-conditioned emotional responses in spontaneously hypertensive rats. Hypertension 5:728–738
Acknowledgments
Supported by grant IN227910 from DGAPA-UNAM. IMPULSA-UNAM grant on “Stem Cell Research” and CONACyT (Consejo Nacional de Ciencia y Tecnología, México) grants 42662, 79763 and 102085 to A.H–C and by Instituto de Salud Carlos III (grant PI080227 to J.M.H-G). We also acknowledge support from the cooperation agreement between the National Autonomous University of México (UNAM) and the Autonomous University of Madrid (UAM), and CEAL-UAM-Banco de Santander. A. Hernández-Cortés was a postdoctoral fellow from CONACyT and P. Segura-Chama received a Ph. D. fellowship from CONACyT, and support from the Ph. D. Program of Biomedical Sciences (UNAM). This work was performed by P. Segura-Chama as partial fulfillment of the requirements to obtain the PhD in Biomedical Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
A commentary to this article can be found at doi:10.1007/s10571-010-9611-z.
Rights and permissions
About this article
Cite this article
Segura-Chama, P., Hernández, A., Jiménez-Pérez, N. et al. Comparison of Ca2+ Currents of Chromaffin Cells from Normotensive Wistar Kyoto and Spontaneously Hypertensive Rats. Cell Mol Neurobiol 30, 1243–1250 (2010). https://doi.org/10.1007/s10571-010-9566-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-010-9566-0