
Abstract
In order to guarantee better conditions for competition, the nervous system has developed not only mechanisms controlling muscle effectors, but also retrograde systems that, starting from peripheral structures, may influence brain functions. Under such perspective, physical activity could play an important role in influencing cognitive brain functions including learning and memory. The results of epidemiological studies (cross-sectional, prospective and retrospective) support a positive relationship between cognition and physical activities. Recent meta-analysis confirmed a significant effect of exercise on cognitive functions. However, the biological mechanisms that underlie such beneficial effects are still to be completely elucidated. They include supramolecular mechanisms (e.g. neurogenesis, synaptogenesis, and angiogenesis) which, in turn, are controlled by molecular mechanisms, such as BDNF, IGF-1, hormone and second messengers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The complex functions of the central nervous system (CNS) of all animals include the ability to elaborate differentiated motor responses, ranging from the very simple ones such as the movement of cilia to the extremely intricate ones such as human speech. All these kinds of motor activities are aimed at reaching better conditions in the competition for survival, keeping in mind that in humans, competition does not only mean to obtain food, but also to elaborate multifaceted behavioural responses in order to guarantee adaptability to very complex social interactions for which cognitive functions are important components. In order to provide such an important function, it is possible that the nervous system has developed not only mechanisms controlling muscle function, but also feedback systems that influence brain activity through signaling systems starting from peripheral structures such as muscles or joints. Based on this proposal, it is possible that behaviours such as physical activity (PA) may have developed simultaneously and interdependently during evolution to ultimately influence brain functions, including cognition, learning and memory (Vaynman and Gomez-Pinilla 2006).
In the last two decades both epidemiological and experimental studies have accumulated showing that PA may act as primary prevention for cognitive decline in elderly. The results of cross-sectional, prospective and retrospective epidemiological studies generally support a positive relationship between the cognitive activity of older adults and PA (Friedland et al. 2001; Weuve et al. 2004; Taaffe et al. 2008). However, several authors failed to observe such a relationship (Tsutsumi et al. 1997; Verghese et al. 2003; Sturman et al. 2005). There are many reasons that can explain such scattered exceptions including the use of self-report versus more objectively measured PA; the difficulties to evaluate the relative contribution of social, intellectual and physical factors to different everyday activities; the failure to distinguish between different PA training protocols (aerobic versus non-aerobic, exercise versus fitness); the difficulty of eliminating subjects with subclinical signs of dementia; and finally the role played by genetic factors.
Recently, Hamer and Chida (2009) conducted a very large meta-analysis of 16 prospective studies which included 163,797 non-demented subjects at baseline with 3,219 cases at follow-up. The authors concluded that PA is inversely associated with risk of dementia. Over the past years several clinical trials and cohort studies have been conducted reporting significant effect in preserving cognition in older adults (Blumenthal and Maden 1988; Kramer et al. 1999; Williamson et al. 2009) and preventing dementia (Verghese et al. 2009; Middleton et al. 2008; Lautenschlager et al. 2008). In order to determine whether the positive effect of PA on the cognitive functions across the literature is reliable, several meta-analyses have been conducted. Colcombe and Kramer (2003) observed a significant effect of aerobic exercise training in those studies with a randomized design of an aerobic fitness training group and a control group. Moreover, the effects were greatest for those tasks involving executive control processes. Further studies, also recently, largely confirmed these results (Heyn et al. 2004; Angevaren et al. 2008; van Uffelen et al. 2008).
Although the ability of PA in reducing the risk of cognitive decline is generally accepted, the biological mechanisms that result in such effect are only poorly understood. The purpose of the present work is to provide an up-to-date overview on the biological mechanisms which lie behind the benefits of PA on cognitive functions. We will first address the controversial topic of the experimental paradigms used to study the effect of environment on nervous system and the biological relationship between periphery and CNS. We will then report the experimental studies focused to understand the cellular (supramolecular) and molecular mechanisms responsible for exercise effects on cognitive functions. Finally, we will critically discuss the topic focusing on future directions for the research.
Experimental Paradigms to Study the Effect of Environment on CNS
Many of the animal studies addressed to understand the relationship between environment and CNS have used as experimental paradigm the so-called environmental enrichment (EE). EE is an experimental paradigm which includes a variety of objects (toys, tunnel, running wheels) that generally need an increased PA to be used. Although it is not easy to discriminate between the effects of PA per se and those related to cognitive stimulation, many observations suggest that these two manipulations affect CNS through ways only apparently equivalent. Behavioural manipulations such as housing the animals in an EE or allowing them to engage in voluntary exercise (i.e. animals with access to a running wheel for voluntary activity) have shown that cognitive stimulation and PA may influence structures and functions of CNS differently. In the by now classical experiments of van Praag et al. (1999a, b), the authors showed that EE produced increased neurogenesis, but when the single components of EE were removed, only the exercise wheel continued to be associated with enhanced neurogenesis. During these and the following studies it became evident that EE and PA affect CNS differently, whereas PA seems to act prevalently on proliferating precursor cells inducing neurogenesis, EE promotes synaptogenesis. Also at molecular level the two experimental paradigms act differently. While EE increases the expression of two proteins found in the mature synapse such as synaptophysin (Lambert et al. 2005) and Postsynaptic-Density-95 (PSD-95 or SAP-90) (Nithianantharajah et al. 2004), several genes important for synaptic plasticity (e.g. GluR5, NR2A, NR2B and EAAC1) have been found to be increased after PA (Molteni et al. 2002). Furthermore, PA is also associated with both increased long-term potentiation (LTP) in the dentate gyrus (DG) and enhanced performance of hippocampal-dependent learning and memory tasks (van Praag et al. 1999a; Naylor et al. 2005), while EE exposure actually reverses already established LTP (Abraham et al. 2002).
Molecular Crosstalk Between Muscle and CNS
The functional relationship between CNS and its periphery has been traditionally regarded as an unidirectional downstream flow of information controlling muscle function, ultimately the contraction. Considering the crucial role of movement in the adaptability of the animal organisms to the environment, it is plausible to assume the presence of a retrograde, or upstream, flow of information to the CNS, possibly with feedback functions. This possibility is supported by many observations, including that in rats the discharge frequency of hippocampal CA1 pyramidal cells and interneurons result increased as the running velocity increased (Czurko et al. 1999).
The molecular details of the signaling system between periphery and CNS are still incompletely understood. Experimental observations suggest two fundamental mechanisms. Evidence indicates that events associated with energy balance can play a role in nervous functions. Brain metabolic responses to acute PA seem to extend beyond the regions specifically associated with skeletal motor, sensory, and cardiovascular autonomic control (Ide and Secher 2000). In contrast, McCloskey et al. (2001) reported that oxidative capacity after chronic voluntary activity wheel running is only increased in the striatum and limb representations in the motor cortex and not in the hippocampus of rats. Lactate taken up from skeletal muscle seems to act as an intercellular energy shuttle within the brain during high-intensity exercise (Dalsgaard et al. 2004). Exercise increases pyruvate dehydrogenase kinase-4 (PDK-4) transcription in skeletal muscle, thus limiting the use of glucose by the muscle to assure sufficient amounts for the increased brain metabolic needs (Pilegaard et al. 2000).
However, there is also clear evidence for effects of PA on nervous functions that are only indirectly dependent, or independent on energy metabolism. It was found that in the hippocampus, exercise significantly increases the levels of the uncoupling protein 2 (UCP2), a mitochondrial protein which uncouples substrate oxidation from ATP synthesis. Vaynman et al. (2006) proposed a model in which the presence of UCP2 at the pre-synaptic and post-synaptic membranes could allow neuronal mitochondria to limit oxidative stress, increase ATP production and modulate calcium levels. These modulations would subsequently influence vesicular release and transcription, by acting on vesicular release proteins, such as synapsin I, and signal transduction molecules, such as cAMP response element binding (CREB) protein, respectively. These results suggest the presence of fundamental mechanisms through which exercise affects key elements of energy metabolism that modulate substrates of synaptic plasticity underlying learning and memory. Finally, UCP2 could represent a mechanism linking the energy metabolism to the production of neurotrophic factors, such as brain-derived neurotrophic factor (BDNF). Indeed, UCP2 seems to modulate the synthesis of BDNF and its downstream molecular effectors such as CREB and calcium/calmodulin protein kinase II (CAMK-II) (Vaynman et al. 2003).
A possible candidate for the role of reciprocal signaling systems between muscle and CNS may be interleukin-6 (IL-6), a cytokine with primarily immunomodulatory effects. It was demonstrated that the level of circulating IL-6 increases dramatically in response to exercise. Data from marathon runners suggest that there is a correlation between the intensity of exercise and the increase in plasma IL-6 level (Ostrowski et al. 2001). Muscle biopsies obtained before and after exercise in humans (Starkie et al. 2001; Steensberg et al. 2001) and in rats (Jonsdottir et al. 2000) revealed very little IL-6 mRNA in resting muscle, but up to a 100-fold increase in exercising skeletal muscle. Recently, Chennaoui et al. (2008) showed in rats that a training program induced a decrease of IL-6 concentration in the cerebellum, suggesting that this effect could contribute to the positive consequences of PA on the CNS.
The mechanisms responsible for IL-6 production in the contracting skeletal muscle are still not fully understood. It was proposed that IL-6 increase may be linked to glycogen depletion. Steensberg et al. (2001) found that situations in which muscle glycogen concentrations were low, such as exercise, increased the IL-6 mRNA, the transcription rate and the protein release. There is evidence suggesting that IL-6 may be linked to the regulation of glucose homeostasis during exercise and/or that IL-6 may work as a sensor of carbohydrate availability. Steensberg et al. (2001) observed that the increased expression of IL-6 was associated with increased glucose uptake during exercise. Additionally, Helge et al. (2003) demonstrated that the IL-6 release from working skeletal muscle is positively related to work intensity, glucose uptake and plasma adrenaline concentration. This might suggest that IL-6 may be involved, at least in part, in mediating glucose uptake.
Neurobiological Bases
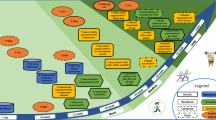
The biological mechanisms responsible for the beneficial effects of PA on cognition are still debated. PA was demonstrated to affect angiogenesis, neurogenesis and synaptogenesis through different molecular mechanisms (Fig. 1).

Potential mechanisms through which physical activity may counteracts cognitive decline. BDNF is a critical modulator of the energy independent effects of physical activity on neurogenesis and synaptogenesis. BDNF increases neurogenesis (directly or through neurotransmitters, especially ACh) and activates a complex presynaptic and postsynaptic molecular cascades that induce synaptogenesis. In this process, CAMK-II and MAP-K pathways seem to be fairly important acting on both LTP and LTM, the latter through the regulation of CREB level. Physical activity also activates IGF-1 production that may lead both to synaptogenesis and angiogenesis. IGF-1 acts on synaptogenesis through a downstream signaling cascade that at presynaptic level includes CAMK-II and MAP-K whereas postsynaptically involves CAMK-II. Angiogenesis is increased by VEGF that is regulated by IGF-1 and several energy-dependent mechanisms such as cellular hypoxia and glucose deficit. Details are given in the text. BDNF brain-derived neurotrophic factor, IGF-1 insulin-like growth factor, VEGF vascular endothelial growth factor, UCP2 uncoupling protein 2, TrKB-R tyrosine kinase receptor B, CAMK-II Ca/calmodulin protein kinase II, MAPK mitogen-activated protein kinase, CREB cAMP response element-binding, LTP long-term potentiation, LTM long-term memory
Supramolecular Mechanisms
Angiogenesis
The brain vascular system is highly plastic throughout life. Any manipulation that increases brain vascularisation, and hence blood flow, might prove to be an effective strategy to minimize or delay the cognitive decline associated with aging. A large body of evidence suggests that angiogenesis can be induced by PA (Black et al. 1991; Isaacs et al. 1992). Increasing evidence supports the observation that manipulating blood flow to the brain alters behavioural performance. For example, administration of erythropoietin has been shown to enhance cognitive performance in rodents (Sadamoto et al. 1998) and humans (Ajmani et al. 2000). Numerous studies have documented transient increases in cerebral blood flow both in animals (Sokoloff et al. 1997) and humans (Fox and Raichle 1986), during the performance of motor tasks as well as for a brief period following the cessation of PA. In addition to these studies that have revealed an increased blood flow, subsequent studies in rats and primates have shown that fitness training can also enhance vascularisation in other regions of the brain than the motor cortex. Churchill et al. (2002) proposed the hypothesis that angiogenesis is likely to occur in any area of the adult brain that is activated and lacks sufficient vascularisation to support chronic levels of elevated neuronal activity requiring increased oxygen. Black et al. (1990) compared the effects of aerobic fitness training to motor skill learning and showed that these two different forms of training had effects on the vasculature and synaptic connectivity of the brain respectively.
The molecular mechanisms for angiogenesis are only partially known. Vascular endothelial growth factor (VEGF) seems to play an important role in this process. Many philological and pathological conditions may affect VEGF production. For example, cellular hypoxia (Ladoux and Frelin 1993) or glucose deficits (Satake et al. 1998) have been reported to increase the production of VEGF in cell cultures.
Neurogenesis
It is now accepted that in the adult brain of various species (i.e. rodents, non-human primates and humans), neurogenesis occurs at least in selected regions, such as the subgranular zone and granule cell layer of the DG in the hippocampus and the subventricular zone of the lateral ventricle (Ming and Song 2005), in response to specific conditions including stroke (Arvidsson et al. 2002), ischemia (Liu et al. 1998), seizures (Parent et al. 1997; Steiner et al. 2008) or injuries (Gould and Tanapat 1997). The focus of neurogenesis investigations has been directed to identify stimuli that can modulate the proliferation and/or survival rate of the newly formed neurons. Many substances, including hormones, growth factors, drugs and neurotransmitters (Ming and Song 2005) affect hippocampal neurogenesis. Behavioural stimuli such as PA (Uda et al. 2006) or EE (Brown et al. 2003) are also able to affect neurogenesis. Research on animals demonstrated that exercise-induced hippocampal cell proliferation and cell survival can occur at many stages of development, including pups (Kim et al. 2007), juvenile (Lou et al. 2008), young adulthood (van Praag et al. 1999b) and old age (van Praag et al. 2005).
Although the functional significance of such constitutive neurogenesis was initially unclear (Eriksson et al. 1998), there is growing evidence that newly formed neurons can be integrated into neural networks and become functionally active (Lledo et al. 2006). Experiments in animal models showed that stimulation of hippocampal neurogenesis by aerobic exercise is accompanied by facilitated induction and maintenance of LTP (van Praag et al. 1999a) and by enhancement of short-term memory in the DG (Kim et al. 2007; Hillman et al. 2008). Regarding the relationship between the effects of PA and EE on neurogenesis, Olson et al. (2006) suggested that the two conditions act differently: PA could increase cell proliferation, and thus more cells are generated that might become neurons, while EE could increase neurogenesis without affecting cell proliferation.
Synaptogenesis
The specific role on synaptogenesis of PA compared to EE is more controversial. Whereas the role of PA on neurogenesis is well established, synaptogenesis seems to be prevalently induced by cognitive stimuli such as those present in the experimental paradigm of EE (Kempermann 2008). Nevertheless, a direct effect of PA on synaptogenesis has been reported. Eadie et al. (2005) showed in rats that spine density on dendrites in DG increased following voluntary exercise which suggests profound changes in synaptic molecules. Using high-density oligonucleotide microarrays, a large number of gene transcripts (synaptotagmin 5, clathrin-associated protein 17, beta prime COP, kinesin light chain C, Vesl) associated with synaptic structure and function has been showed in the hippocampus of rats voluntary running for 3 weeks (Tong et al. 2001). The ability of PA to affect the composition of pre- and postsynaptic compartment was further confirmed (Molteni et al. 2002; Farmer et al. 2004) also very recently (Hu et al. 2009). The mechanisms that underlie the exercise-induced synaptic modifications are still unclear. Recently, it has been suggested that a mitochondrial mechanism related to UCP2 function is essential to induce synaptogenesis in response to PA (Dietrich et al. 2008). Along with the structural changes, there is evidence for modifications in synaptic plasticity following PA. In particular, LTP amplitude appeared enhanced in the DG in running mice as compared to controls (van Praag et al. 1999a). These findings have also been replicated since the initial report (Farmer et al. 2004).
Experimental evidence also suggests that PA might possess therapeutic efficacy for some of pathologies characterized by synaptic loss. Jankowsky et al. (2005) showed that learning and memory deficits observed in a transgenic mouse model of Alzheimer’s disease (AD) can be ameliorated by EE. They conclude that environmental factors can strongly modulate the pathological and behavioural progression of AD in a mouse model. Lazarov et al. (2005) reported that exposure of the AD model-transgenic mice to EE caused a reduction in cerebral Aβ levels and its deposition as compared to controls in standard housing. However, other studies with AD transgenic mouse models, whereby EE and PA were compared, suggested that EE seems to be more beneficial for cognition than PA alone (Wolf et al. 2006; Cracchiolo et al. 2007).
Molecular Mechanisms
Brain-Derived Neurotrophic Factor
For the past few years there have been an increasing number of investigations focused on the molecular bases of PA benefits. A crucial role seems to be played by growth factors such as BDNF (Ang et al. 2003; Neeper et al. 1995). BDNF is involved in neuroplasticity, neuroprotection, growth and differentiation, during development and in adult brain (Lindvall et al. 1994; Xuan et al. 2008). Direct administration of BDNF increases cell proliferation in the hippocampus and blocking BDNF reduces cell proliferation (Vaynman et al. 2004). Besides its traditional trophic function, BDNF has emerged as a critical modulator of many other neuronal functions such as neurotransmitter release (Bolton et al. 2000) and synaptic plasticity (Lo 1995). BDNF has been shown to regulate neurotransmitters, including dopaminergic and cholinergic systems playing an important role in the exercise-induced effects on neurotransmitters. A large number of observations suggest that BDNF plays a crucial role in LTP. For example, BDNF gene deletion or inhibition (Figurov et al. 1996) impairs LTP. Ma et al. (1998) showed that blocking endogenous BDNF reduced LTP. Moreover, replenishing the factor-depleted hippocampus with exogenous BDNF restored the ability to induce LTP (Patterson et al. 1996). Learning increases BDNF gene expression, suggesting that mechanisms inducing BDNF gene expression may enhance learning (Tokuyama et al. 2000). Clinical studies also support the importance of BDNF in learning and memory in humans. A study conducted by Hariri et al. (2003) reported that individuals expressing a specific polymorphism in the BDNF gene exhibit learning impairments.
Neeper et al. (1995) first observed that PA affects BDNF production in the brain. Surprisingly, the authors showed that the greatest effects of exercise on BDNF mRNA occurred in regions not directly related to the motor system but associated with cognitive function such as the hippocampus and caudal cortex. Voluntary exercise in rodents increases both mRNA and protein levels of BDNF in the hippocampus, cerebellum and frontal cortex. Blocking the binding of BDNF to its tyrosine kinase B receptor (TrkB-R) abolishes the exercise-induced performances benefits (Vaynman et al. 2004). Other trophic factors, including NGF (Neeper et al. 1995) and fibroblast growth factor 2 (FGF-2) (Gomez-Pinilla et al. 1997), were also induced in the hippocampus in response to exercise, but their upregulation was transient and less robust than that of BDNF, suggesting that BDNF is a better candidate for mediating the long-term benefits of exercise on the brain.
These findings suggest that PA through the regulation of BDNF synthesis might be neuroprotective and prevent the development of cognitive symptoms associated with neurodegenerative diseases such as AD. BDNF shows large deficit in AD brain (Connor et al. 1997). In AD transgenic mice model (APPswe X PS1ΔE9), it was reported that transcripts encoding BDNF are significantly upregulated in brain of mice exposed to EE and PA (Adlard et al. 2005; Farmer et al. 2004). In contrast, Wolf et al. (2006) in a different AD transgenic mice model (APP23) observed an increased hippocampal production of BDNF in animals expounded to EE. Surprisingly, in animals expounded to PA alone a down-regulation of BDNF was observed. The reasons of such conflicting results might be in both the different animal models used and the not comparable type of PA.
The mechanisms that underlie the exercise-driven increase in the level of BDNF are varied. They include neurotransmitters with their receptors (Blomstrand et al. 1989; Fordyce et al. 1991; MacRae et al. 1987; Poutlton and Muir 2005) and peripheral factors such as estrogen, corticosterone (Berchtold et al. 2001) and possibly insulin-like growth factor 1 (IGF-1) (Carro et al. 2000; Trejo et al. 2001). Finally, it is now well accepted that the PA effects on BDNF modulate the function of intracellular signaling systems.
Insulin-Like Growth Factor-1
Exercise enhances hippocampal neurogenesis most likely by stimulating the systemic production of IGF-1 (Carro et al. 2000). Trejo et al. (2001) reported that blocking the entrance of IGF-1 into the brain repressed exercise-induced neuron proliferation in the DG. Peripheral IGF-1 appears to participate in the neuroprotective effect of exercise, as peripheral infusion of IGF-1 blocking antibodies before a CNS injury reduced the protective effect (Carro et al. 2001).
IGF-1 is also crucial for exercise-induced angiogenesis in the brain. It has been shown that the systemic injection of IGF-1 stimulates angiogenesis and its inhibition reduces the phenomenon. IGF-1 might induce new blood vessel formation indirectly through the synthesis of VEGF, a molecule that plays a pivotal role in blood vessel growth. Lopez-Lopez et al. (2004) reported that blocking IGF-1 abolished the secretion of VEGF, which resulted in a significant reduction in the appearance of new capillaries. Furthermore, it was reported that IGF-1 and VEGF are upregulated following aerobic exercise, leading to the formation of new blood vessels and that this event occurs both in childhood and in the elderly (Ding et al. 2004). Finally, IGF-1 might mediate the effects of BDNF, being an upstream mediator of its gene regulation (Carro et al. 2001; Trejo et al. 2001). Indeed, peripheral administration of IGF-1 induces BDNF mRNA in the brain (Carro et al. 2000).
Neurotransmitters
The neurotransmitter systems are also affected by exercise. Serotonin levels are increased throughout the brain in exercising rats (Blomstrand et al. 1989). Chronic wheel running increased basal levels of serotonin (Dishman et al. 1997), attenuated stress-induced c-Fos induction in the serotoninergic neurons (Greenwood et al. 2005) and increased levels of 5HT1A inhibitory autoreceptor mRNA (Greenwood et al. 2003) in the dorsal rafhe nucleus. Furthermore, serotonin has been shown to enhance neuron proliferation (Brezun and Daszuta 2000), whereas its depletion decreases neuron proliferation (Brezun and Daszuta 1999).
Exercise may also act through noradrenergic system. Wheel running increased basal levels of noradrenalin (Dishman et al. 1997) and mRNA for the noradrenalin modulator, galanin (O’Neal et al. 2001) in the locus coeruleus. The same type of exercise blunted the release of noradrenalin in the frontal cortex (Soares et al. 1999) and its depletion in the locus coeruleus, hippocampus, hypothalamus, and amygdala (Dishman et al. 2000) in response to stress.
It was shown that exercise can reverse age-related cognitive declines through an increased dopamine receptor density in the striatum (MacRae et al. 1987). Animal studies (Poutlton and Muir 2005) suggested that exercise may be a potential intervention for reduction of the onset rate or incidence of Parkinson disease, based on the observation that treadmill running resulted in an attenuation of dopamine depletion in the striatum of hemi-Parkinsonian rats.
Acetylcholine level and muscarinic receptor density are increased in the hippocampus of adult exercising rats (Fordyce et al. 1991). Along with a direct effect of PA on ACh, a large body of evidence supports the idea that an ACh-mediated mechanism regulates BDNF gene expression in the hippocampus (Knipper et al. 1994; Berchtold et al. 2002). Furthemore, Knusel et al. (1991), in a study in culture, have shown that rhBDNF (recombinant human BDNF) stimulates development of basal forebrain cholinergic neurons and increases dopamine uptake in mesencephalic cultures.
Intracellular Pathways
The effect of exercise on signal transduction mechanisms has not been extensively explored. The most important signal transduction mechanisms for mediating exercise effects on hippocampal synaptic plasticity are mitogen-activated protein kinase (MAP-K), CAMK-II, and the N-methyl-d-aspartate receptor (NMDA-R). MAP-K, CAMK-II, and the NMDA-R were found to influence downstream effectors of BDNF action on gene expression and synaptic transmission, such as CREB and synapsin I (Vaynman et al. 2003).
CREB, a well-known stimulus-induced transcriptional regulator, was described as a switch for the activation of transcription of molecules essential for long-term memory (LTM) (Yin et al. 1995) and involved in activity-dependent long-term neuronal plasticity (Tully 1997). In addition, CREB seems to be important in the BDNF-mediated mechanism responsible for the potentiating effects of exercise on learning and memory (Vaynman et al. 2004). With exercise, CREB mRNA and BDNF levels were significantly and positively associated with each other as well as with performance on memory recall in animals. CREB may provide a self-perpetuating loop for BDNF action, since it has been found to regulate BDNF transcription (Tao et al. 1998) and is regulated itself by BDNF (Finkbeiner et al. 1997; Tully 1997). Both MAP-K and CAMK-II have been repeatedly described as conserved signaling pathways that lead to CREB mediated gene transcription (Finkbeiner et al. 1997; Ying et al. 2002). MAP-K targets synaptic potentiation (English and Sweatt 1996), nuclear signaling (Adams et al. 2000), LTP (English and Sweatt 1997), and seems to be especially necessary for learning and memory (Blum et al. 1999; Sweatt 2001). It was suggested that the capacity of MAP-K to induce synaptic plasticity was to be attributed to its ability to regulate (Finkbeiner et al. 1997) and prolong the transcriptionally active state of CREB (Hardingham et al. 2001). Like MAP-K, CAMK-II is believed to be important for mechanisms underlying learning and memory (Yin and Tully 1996). Mice with gene deletions of a CAMK-II isoform show an impaired performance on spatial learning tasks (Silva et al. 1992). Finally, NMDA-R is one more factor able to mediate the effects of exercise on hippocampal synaptic plasticity. BDNF can enhance synaptic transmission through the NMDA-R (Song et al. 1998), providing an alternative pathway to CAMK-II and MAP-K mediated changes. The NMDA-R is critical for modulating LTP (Bliss and Collingridge 1993) and for learning and memory processes (Cammarota et al. 2000). Vaynman et al. (2003) showed that blocking the NMDA-R abolished the exercise-induced increases in BDNF, TrkB-R, CREB and synapsin I mRNAs.
Modulation of the transmission properties at the synapse has been proposed for the beneficial effects of exercise on brain function. It has been shown that BDNF regulates synapsin I and synaptophysin. BDNF gene deletion in mice results in a reduction in synaptic proteins, sparsely docked vesicles, impaired neurotransmitter release, and decreased synapsin I levels (Pozzo-Miller et al. 1999). An important function of synapsin I is to modulate vesicular release by tethering synaptic vesicles to the actin cytoskeleton of the cell (Greengard et al. 1993). Synaptophysin, a major integral protein on synaptic vesicles, may be a key protein in the biogenesis of synaptic vesicles from cholesterol which promotes membrane curvature facilitating vesicular budding and membrane retrieval (Thiele et al. 2000). During exercise, MAP-K and CAMK-II were shown to contribute to the BDNF regulation of synapsin I expression (Vaynman et al. 2003). Exercise may augment the effects of BDNF on synaptic-plasticity, through a positive feedback loop in which it concurrently increases the mRNA levels of both itself and its TrkB-R (Vaynman et al. 2003).
BDNF-mediated regulation of synapsin I may influence synaptic plasticity in additional alternative ways. Besides modulating transmitter release, synapsin I is involved in the formation and maintenance of the presynaptic structure (Melloni et al. 1994) and in axonal elongation (Akagi et al. 1996). An adequate vesicular release pool and adequate and sustainable transmitter release provided by functional levels of synapsin I may provide the level of synaptic communication necessary for learning. Evaluation of an array of genes activated by exercise has corroborated the involvement of these molecules in exercise-induced synaptic plasticity. In the rat hippocampus, 3 weeks of exercise led to changes, both increases (e.g. synaptotagmin, Vesl and AP17) and decreases (e.g. synaptopotin, thrombomodulin precursor gene, cytosolyc sorting protein PACS-1b) in the expression of a number of genes, indicating a direct effect of exercise on synaptic function (Tong et al. 2001).
Conclusions and Critical Topics
A growing body of animal and human studies suggests that PA strongly influences brain functions, including cognition, learning and memory and their pathological counterpart, such as cognitive decline in aging. In recent years, there has also been increased acknowledgement of the biological mechanisms that underlie the beneficial effects of the movement on brain health. Nevertheless, many critical topics remain to be clarified. From a practical point of view, we know little about the optimal amount and kind of PA for sustaining this beneficial effect. We are also largely ignorant of the possible interaction between exercise and other lifestyle factors. Finally, data about the ability of fitness to reduce the ruinous effects of neurodegenerative process are still insufficient.
The challenge lies in understanding the relationships between systemic, cellular, and molecular mechanisms by which PA influences brain functions such as cognition, learning and memory and to possibly recognize the relative contribute of each of them. In order to reach this goal, it will be mandatory to understand not only the single mechanisms but also to put together the single pieces of the puzzle made by the complex interactions between different levels. Several of such interactions are those between different systems (e.g. nervous, cardiovascular, endocrinological etc), between molecular and cellular mechanisms and, finally, between different molecules. For example, are the effects of exercise mediated by the reduction of internal pathologies (e.g. cardiovascular diseases, diabetes, hypertension) or is PA able to act directly on brain function? If a cooperative contribute between these two levels is the most likely hypothesis, recognizing the specific contribute of both mechanisms on each single function could open new pharmacological perspective in preventing cognitive decline. Still, are there molecular mechanisms (e.g. BDNF, IGF-1, IL-6) specifically involved in mediating the neurotrophic effects of PA, as several observations seem to suggest or, on the contrary, PA shares the same mechanisms with other kind of environmental stimulations. Finally, we ignore other aspects that in the future might open exiting scenarios such as the relationship between exercise and genetic polymorphisms. In conclusion, although future research still has many aspects to clarify, PA may represent a simple and inexpensive approach to maintain brain health.
References
Abraham WC, Logan B, Greenwood JM, Dragunow M (2002) Induction and experience-dependent consolidation of stable long-term potentiation lasting months in the hippocampus. J Neurosci 22:9626–9634
Adams JP, Roberson ED, English JD, Selcher JC, Sweatt JD (2000) MAP-K regulation of gene expression in the central nervous system. Acta Neurobiol Exp (Warsz) 60:377–394
Adlard PA, Perreau VM, Cotman CW (2005) The exercise-induced expression of BDNF within the hippocampus varies across life-span. Neurobiol Aging 26:511–520
Ajmani RS, Metter EJ, Jaykumar R, Ingram DK, Spangler EL, Abugo OO et al (2000) Hemodynamic changes during aging associated with cerebral blood flow and impaired cognitive function. Neurobiol Aging 21:257–269
Akagi S, Mizoguchi A, Sobue K, Nakamura H, Ide C (1996) Localization of synapsin I in normal fibers and regenerating axonal sprouts of the rat sciatic nerve. Histochem Cell Biol 105:365–373
Ang ET, Wong PTH, Mochhala S, Ng YK (2003) Neuroprotection associated with running: Is it a result of increased endogenous neurotrophic factors? Neuroscience 118:335–345
Angevaren M, Aufdemkampe G, Verhaar HJ, Aleman A, Vanhees L (2008) Physical activity and enhanced fitness to improve cognitive function in older people without known cognitive impairment. Cochrane Database Syst. Rev. 3:CD005381
Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002) Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 8:963–970
Berchtold NC, Kesslak JP, Pike CJ, Adlard PA, Cotman CW (2001) Estrogen and exercise interact to regulate brain-derived neurotrophic factor mRNA and protein expression in the hippocampus. Eur J Neurosci 14:1992–2002
Berchtold NC, Kesslak JP, Cotman CW (2002) Hippocampal brain-derived neurotrophic gene regulation by exercise and the medial septum. J Neurosci Res 68:511–521
Black JE, Isaacs KR, Anderson BJ, Alcantara AA, Greenough WT (1990) Learning causes synaptogenesis, whereas motor activity causes angiogenesis, in cerebellar cortex of adult rats. Proc Natl Acad Sci USA 87:5568–5572
Black JE, Zelazny AM, Greenough WT (1991) Capillary and mitochondrial support of neural plasticity in adult rat visual cortex. Exp Neurol 111:204–209
Bliss TVP, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39
Blomstrand E, Perrett D, Parry-Billings M, Newsholme EA (1989) Effect of sustained exercise on plasma amino acid concentrations and on 5-hydroxytryptamine metabolism in six different brain regions in the rat. Acta Physiol Scand 136:473–481
Blum S, Moore AN, Adams F, Dash PK (1999) A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci 19:3535–3544
Blumenthal JA, Maden DJ (1988) Effects of aerobic exercise training, age, and physical fitness on memory-search performance. Psychol Aging 3:280–285
Bolton MM, Pittman AJ, Lo DC (2000) Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci 20:3221–3232
Brezun JM, Daszuta A (1999) Depletion in serotonin decreases neurogenesis in the dentate gyrus and the subventricular zone of adult rats. Neuroscience 89:999–1002
Brezun JM, Daszuta A (2000) Serotonin may stimulate granule cell proliferation in the adult hippocampus, as observed in rats grafted with foetal raphe neurons. Eur J Neurosci 12:391–396
Brown J, Cooper-Kuhn CM, Kempermann G, Van Praag H, Winkler J, Gage FH et al (2003) Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur J Neurosci 17:2042–2046
Cammarota M, de Stein ML, Paratcha G, Bevilaqua LR, Izquierdo I, Medina JH (2000) Rapid and transient learning-associated increase in NMDA NR1 subunit in the rat hippocampus. Neurochem Res 25:567–572
Carro E, Nunez A, Busiguina S, Torres-Aleman I (2000) Circulating insulin-like growth factor I mediates effects of exercise on the brain. J Neurosci 20:2926–2933
Carro E, Trejo LJ, Busiguina S, Torres-Aleman I (2001) Circulating insulin-like growth factor 1 mediates the protective effects of physical exercise against brain insults of different etiology and anatomy. J Neurosci 21:5678–5684
Chennaoui M, Drogou C, Gomez-Merino D (2008) Effects of physical training on IL-1beta, IL-6 and IL-1ra concentrations in various brain areas of the rat. Eur Cytokine Netw 19:8–14
Churchill JD, Galvez R, Colcombe S, Rodney AS, Kramer AF, William TG (2002) Exercise, experience and the aging brain. Neurobiol Aging 23:941–955
Colcombe S, Kramer AF (2003) Fitness effects on the cognitive function of older adults: a meta-analytic study. Psychol Sci 14:125–130
Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M (1997) Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Mol Brain Res 49:71–81
Cracchiolo JR, Mori T, Nazian SJ, Tan J, Potter H, Arendash GW (2007) Enhanced cognitive activity—over and above social or physical activity—is required to protect Alzheimer’s mice against cognitive impairment, reduce Aβ deposition, and increase synaptic immunoreactivity. Neurobiol Learn Mem 88:277–294
Czurko A, Hirase H, Csicsvari J, Buzsaki G (1999) Sustained activation of hippocampal pyramidal cells by ‘space clamping’ in a running wheel. Eur J Neurosci 11:344–352
Dalsgaard MK, Quistorff B, Danielsen ER, Selmer C, Vogelsang T, Secher NH (2004) A reduced cerebral metabolic ratio in exercise reflects metabolism and not accumulation of lactate within the human brain. J Physiol Lond 554:571–578
Dietrich MO, Andrews ZB, Horvath TL (2008) Exercise-induced synaptogenesis in the hippocampus is dependent on UCP2-regulated mitochondrial adaptation. J Neurosci 28:10766–10771
Ding Y, Li J, Luan X, Ding YH, Lai Q, Rafols JA et al (2004) Exercise pre-conditioning reduces brain damage in ischemic rats that may be associated with regional angiogenesis and cellular overexpression of neurotrophin. Neuroscience 124:583–591
Dishman RK, Renner KJ, Youngstedt SD, Reigle TG, Bunnel BN, Burke KA et al (1997) Activity wheel running reduces escape latency and alters brain monoamine levels after footshock. Brain Res Bull 42:399–406
Dishman RK, Renner KJ, White-Welkley JE, Bunnell BN (2000) Treadmill exercise training augments brain norepinephrine response to familiar and novel stress. Brain Res Bull 52:337–342
Eadie BD, Redila VA, Christie BR (2005) Voluntary exercise alters the cytoarchitecture of the adult dentate gyrus by increasing cellular proliferation, dentritic complexity, and spine density. J Comp Neurol 486:39–47
English JD, Sweatt JD (1996) Activation of p42 mitogen-activated protein kinase in hippocampal long-term potentiation. J Biol Chem 271:24329–24332
English JD, Sweatt JD (1997) A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem 272:19103–19106
Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn AM, Nordborg C, Peterson DA et al (1998) Neurogenesis in the adult human hippocampus. Nat Med 4:1313–1317
Farmer J, Zhao X, van Praag H, Wodtke K, Gage FH, Christie BR (2004) Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience 124:71–79
Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B (1996) Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381:706–709
Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19:1031–1047
Fordyce DE, Starnes JW, Farrar RP (1991) Compensation of the age-related decline in hippocampal muscarinic receptor density through daily exercise or underfeeding. J Gerontol 46:245–248
Fox PT, Raichle ME (1986) Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc Natl Acad Sci USA 83:1140–1144
Friedland RP, Fritsch T, Smyth KA, Koss E, Lerner AJ, Chen CH et al (2001) Patients with Alzheimer’s disease have reduced activities in midlife compared with healthy control-group members. Proc Natl Acad Sci USA 98:3440–3445
Gomez-Pinilla F, Dao L, So V (1997) Physical exercise induces FGF-2 and its mRNA in the hippocampus. Brain Res 764:1–8
Gould E, Tanapat P (1997) Lesion-induced proliferation of neuronal progenitors in the dentate gyrus of the adult rat. Neuroscience 80:427–436
Greengard P, Valtorta F, Czernik AJ, Benfenati F (1993) Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 259:780–785
Greenwood BN, Foley TE, Day HE, Campisi J, Hammack SH, Campeau S et al (2003) Freewheel running prevents learned helplessness/behavioural depression: role of dorsal raphe serotoninergic neurons. J Neurosci 23:2889–2898
Greenwood BN, Foley TE, Burhans DJ, Maiser SF, Fleshner M (2005) The consequences of uncontrollable stress are sensitive to the duration of prior wheel running. Brain Res 1033:164–178
Hamer M, Chida Y (2009) Physical activity and risk of neurodegenerative disease: a systematic review of prospective evidence. Psychol Med 39:3–11
Hardingham G, Arnold FJ, Bading H (2001) Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci 4:261–267
Hariri AR, Goldberg TE, Mattay VS, Kolachana BS, Callicott JH, Egan MF et al (2003) Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. J Neurosci 23:6690–6694
Helge JW, Stallknecht B, Pedersen BK, Galbo H, Kiens B, Richter EA (2003) The effect of graded exercise on IL-6 release and glucose uptake in skeletal muscle. J Physiol 546:299–305
Heyn P, Abreu BC, Ottenbacher KJ (2004) The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil 85:1679–1704
Hillman CH, Erickson KI, Kramer AF (2008) Be smart, exercise your heart: exercise effects on brain and cognition. Nat Rev Neurosci 9:58–65
Hu S, Ying Z, Gomez-Pinilla F, Frautschy SA (2009) Exercise can increase small heat shock proteins (sHSP) and pre- and post-synaptic proteins in the hippocampus. Brain Res 1249:191–201
Ide K, Secher NH (2000) Cerebral blood flow and metabolism during exercise. Prog Neurobiol 61:397–414
Isaacs KR, Anderson BJ, Alcantara AA, Black JE, Greenough WT (1992) Exercise and the brain: angiogenesis in the adult rat cerebellum after vigorous physical activity and motor skill learning. J Cereb Blood Flow Metab 12:110–119
Jankowsky JL, Melnikova T, Fadale DJ, Xu GM, Slunt HH, Gonzales V et al (2005) Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci 25:5217–5224
Jonsdottir I, Schjerling P, Ostrowski K, Asp S, Richter EA, Pedersen BK (2000) Muscle contractions induces interleukin-6 mRNA production in rat skeletal muscles. J Physiol Lond 528:157–163
Kempermann G (2008) The neurogenic reserve hypothesis: What is adult hippocampal neurogenesis good for? Trends Neurosci 31:163–169
Kim H, Lee SH, Kim SS, Yoo JH, Kim CJ (2007) The influence of maternal treadmill running during pregnancy on short-term memory and hippocampal cell survival in rat pups. Int J Dev Neurosci 25:243–249
Knipper M, Berzaghi M, Blöchl A, Breer H, Thoenen H, Lindholm D (1994) Positive feedback between acetylcholine and the neurotrophins nerve growth factor and brain-derived neurotrophic factor in the rat hippocampus. Eur J Neurosci 6:668–671
Knusel B, Winslow JW, Rosenthal A, Burton LE, Seid DP, Nikolics K et al (1991) Promotion of central cholinergic and dopaminergic neuron differentiation by brain-derived neurotrophic factor but not neurotrophin 3. Proc Natl Acad Sci USA 88:961–965
Kramer AF, Hahn S, Cohen NJ, Banich MT, McAuley E, Harrison CR et al (1999) Ageing, fitness and neurocognitive function. Nature 400:418–419
Ladoux A, Frelin C (1993) Hypoxia is a strong inducer of vascular endothelial growth factor mRNA expression in the heart. Biochem Biophys Res Commun 195:1005–1010
Lambert TJ, Fernandez SM, Frick KM (2005) Different types of environmental enrichment have discrepant effects on spatial memory and synaptophysin levels in female mice. Neurobiol Learn Mem 83:206–216
Lautenschlager NT, Cox KL, Flicker L, Foster JK, van Bockxmeer FM, Xiao J et al (2008) Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: a randomized trial. JAMA 300:1027–1037
Lazarov O, Robinson J, Tang YP, Hairston IS, Korade-Mirnics Z, Lee VM et al (2005) Environmental enrichment reduces Aβ levels and amyloid deposition in transgenic mice. Cell 120:701–713
Lindvall O, Kokaia Z, Bengzon J, Elmér E, Kokaia M (1994) Neurotrophins and brain insults. Trends Neurosci 17:490–496
Liu J, Solway K, Messing RO, Sharp FR (1998) Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci 18:7768–7778
Lledo PM, Alonso M, Grubb MS (2006) Adults neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci 7:179–193
Lo DC (1995) Neurotrophic factors and synaptic plasticity. Neuron 15:979–981
Lopez-Lopez C, LeRoith D, Torres-Aleman I (2004) Insulin-like growth factor I is required for vessel remodeling in the adult brain. Proc Natl Acad Sci USA 101:9833–9838
Lou SJ, Liu JY, Chang H, Chen PJ (2008) Hippocampal neurogenesis and gene expression depend on exercise intensity in juvenile rats. Brain Res 1210:48–55
Ma YL, Wang HL, Wu HC, Wei CL, Lee EH (1998) Brain-derived neurotrophic factor antisense oligonucleotide impairs memory retention and inhibits long-term potentiation in rats. Neuroscience 82:957–967
MacRae PG, Spirduso WW, Walters TJ, Farrar RP, Wilcox RE (1987) Endurance training effects on striatal D2 dopamine receptor binding and striatal dopamine metabolites in presenescent older rats. Psychopharmacology 92:236–240
McCloskey DP, Adamo DS, Anderson BJ (2001) Exercise increases metabolic capacity in the motor cortex and striatum, but not in the hippocampus. Brain Res 891:168–175
Melloni RH Jr, Apostolides PJ, Hamos JE, De Gennaro LJ (1994) Dynamics of synapsin I gene expression during the establishment and restoration of functional synapses in the rat hippocampus. Neuroscience 58:683–703
Middleton L, Kirkland S, Rockwood K (2008) Prevention of CIND by physical activity: different impact on VCI-ND compared with MCI. J Neurol Sci 269:80–84
Ming GL, Song H (2005) Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci 28:223–250
Molteni R, Ying Z, Gomez-Pinilla F (2002) Differential effects of acute and chronic exercise on plasticity-related genes in the rat hippocampus revealed by microarray. Eur J Neurosci 16:1107–1116
Naylor AS, Persson AI, Eriksson PS, Jonsdottir IH, Thorlin T (2005) Extended voluntary running inhibits exercise-induced adult hippocampal progenitor proliferation in the spontaneously hypertensive rat. J Neurophysiol 93:2406–2414
Neeper SA, Gomez-Pinilla F, Choi J, Cotman CW (1995) Exercise and brain neurotrophins. Nature 373:109
Nithianantharajah J, Levis H, Murphy M (2004) Environmental enrichment results in cortical and subcortical changes in levels of synaptophysin and PSD-95 proteins. Neurobiol Learn Mem 81:200–210
O’Neal HA, Van Hoomissen JD, Holmes PV, Dishman RK (2001) Prepro-galanin mRNA levels are increased in rat locus coeruleus after treadmill exercise training. Neurosci Lett 299:69–72
Olson AK, Eadie BD, Ernst C, Christie BR (2006) Environmental enrichment and voluntary exercise massively increase neurogenesis in the adult hippocampus via dissociable pathways. Hippocampus 16:250–260
Ostrowski K, Rohde T, Asp S, Schjerling P, Pedersen BK (2001) Chemokines are elevated in plasma after strenuous exercise in humans. Eur J Appl Physiol 84:244–245
Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH (1997) Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 17:3727–3738
Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER (1996) Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16:1137–1145
Pilegaard H, Ordway GA, Saltin B, Neufer PD (2000) Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. Am J Physiol Endocrinol Metab 279:806–814
Poutlton NP, Muir GD (2005) Treadmill training ameliorates dopamine loss but not behavioral deficits in hemi-parkinsonian rats. Exp Neurol 193:181–197
Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R et al (1999) Impairments in high frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. J Neurosci 19:4972–4983
Sadamoto Y, Igase K, Sakanaka M, Sato K, Otsuka H, Sakaki S et al (1998) Erythropoietin prevents place navigation disability and cortical infarction in rats with permanent occlusion of the middle cerebral artery. Biochem Biophys Res Commun 253:26–32
Satake S, Kuzuya M, Miura H, Asai T, Ramos MA, Muraguchi M et al (1998) Up-regulation of vascular endothelial growth factor in response to glucose deprivation. Biol Cell 90:161–168
Silva AJ, Paylor R, Wehner JM, Tonegawa S (1992) Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science 257:206–211
Soares J, Holmes PV, Renner KJ, Edwards GL, Bunnel BN, Dishman RK (1999) Brain noradrenergic responses to foot-shock after chronic activity-wheel running. Behav Neurosci 113:558–566
Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD et al (1997) The [14C] deoxyglucose method for the measurement of local glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28:897–916
Song DK, Choe B, Bae JH, Park WK, Han IS, Ho WK et al (1998) Brain-derived neurotrophic factor rapidly potentiates synaptic transmission through NMDA, but suppresses it through non-NMDA receptors in rat hippocampal neuron. Brain Res 799:176–179
Starkie RL, Rolland J, Angus DJ, Anderson MJ, Febbraio MA (2001) Circulating monocytes are not the source of elevations in plasma IL-6 and TNF-alpha levels after prolonged running. Am J Physiol Cell Physiol 280:769–774
Steensberg A, Febbraio MA, Osada T, Schjerling P, van Hall G, Saltin B et al (2001) Interleukin-6 production in contracting human skeletal muscle is influenced by pre-exercise muscle glycogen content. J Physiol 537:633–639
Steiner B, Zurborg S, Hörster H, Fabel K, Kepermann G (2008) Differential 24 h responsiveness of prox1-expressing precursor cells in adult hippocampal neurogenesis to physical activity, environmental enrichment, and kainic acid-induced seizures. Neuroscience 154:521–529
Sturman MT, Morris MC, Mendes de Leon CF, Bienias JL, Wilson RS, Evans DA (2005) Physical activity, cognitive activity, and cognitive decline in a biracial community population. Arch Neurol 62:1750–1754
Sweatt JD (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76:1–10
Taaffe DR, Irie F, Masaki KH, Abbott RD, Petrovitch H, Ross GW et al (2008) Physical activity, physical function, and incident dementia in elderly men: the Honolulu–Asia Aging Study. J Gerontol A Biol Sci Med Sci 63:529–535
Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20:709–726
Thiele C, Hannah M, Fahrenholz F, Huttner WB (2000) Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat Cell Biol 2:42–49
Tokuyama W, Okuno H, Hashimoto T, Xin Li Y, Miyashita Y (2000) BDNF upregulation during declarative memory formation in monkey inferior temporal cortex. Nat Neurosci 3:1134–1142
Tong L, Shen H, Perreau VM, Balazs R, Cotman CW (2001) Effects of exercise on gene-expression profile in the rat hippocampus. Neurobiol Dis 8:1046–1056
Trejo JL, Carro E, Torres-Aleman EI (2001) Circulating insulin-like growth factor mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J Neurosci 21:1628–1634
Tsutsumi T, Don BM, Zaichkowsky LD, Delizonna LL (1997) Physical fitness and psychological benefits of strength training in community dwelling older adults. Appl Human Sci 16:257–266
Tully T (1997) Regulation of gene expression and its role in long-term memory and synaptic-plasticity. Proc Natl Acad Sci USA 94:4239–4241
Uda M, Ishido M, Kami K, Masuhara M (2006) Effects of chronic treadmill running on neurogenesis in the dentate gyrus of the hippocampus of adult rat. Brain Res 1104:64–72
van Praag H, Christie BR, Sejnoswski TS, Gage FH (1999a) Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci USA 96:13427–13431
van Praag H, Kempermann G, Gage FH (1999b) Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 2:266–270
van Praag H, Shubert T, Zhao C, Gage FH (2005) Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci 25:8680–8685
van Uffelen JG, Chin A, Paw MJ, Hopman-Rock M, van Mechelen W (2008) The effects of exercise on cognition in older adults with and without cognitive decline: a systematic review. Clin J Sport Med 18:486–500
Vaynman S, Gomez-Pinilla F (2006) Revenge of the “sit”: how lifestyle impacts neuronal and cognitive health through molecular systems that interface energy metabolism with neuronal plasticity. J Neurosci Res 84:699–715
Vaynman S, Ying Z, Gomez-Pinilla F (2003) Interplay between BDNF and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity. Neuroscience 122:647–657
Vaynman S, Ying Z, Gomez-Pinilla F (2004) Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur J Neurosci 20:2580–2590
Vaynman S, Ying Z, Wu A, Gomez-Pinilla F (2006) Coupling energy metabolism with a mechanism to support BDNF mediated synaptic plasticity. Neuroscience 139:1221–1234
Verghese J, Lipton RB, Katz M, Hall CB, Derby CA, Kuslansky G et al (2003) Leisure activities and the risk of dementia in the elderly. N Engl J Med 348:2508–2516
Verghese J, Wang C, Katz MJ, Sanders A, Lipton RB (2009) Leisure activities and risk of vascular cognitive impairment in older adults. J Geriatr Psychiatry Neurol 22:110–118
Weuve J, Kang JH, Manson JE, Breteler MMB, Ware JH, Grodstein F (2004) Physical activity, including walking, and cognitive function in older women. JAMA 292:1454–1461
Williamson JD, Espeland M, Kritchevsky SB, Newman AB, King AC, Pahor M et al (2009) Changes in cognitive function in a randomized trial of physical activity: results of the lifestyle interventions and independence for elders pilot study. J Gerontol A Biol Sci Med Sci 64:688–694
Wolf SA, Kronenberg G, Lehmann K, Blankenship A, Overall R, Staufenbiel M et al (2006) Cognitive and physical activity differently modulate disease progression in the amyloid precursor protein (APP)-23 model of Alzheimer’s disease. Biol Psychiatry 60:1314–1323
Xuan AG, Long DH, Gu HG, Yang DD, Hong LP, Leng SL (2008) BDNF improves the effects of neural stem cells on the rat model of Alzheimer’s disease with unilateral lesion of fimbria-fornix. Neurosci Lett 440:331–335
Yin JC, Tully T (1996) CREB and the formation of long-term memory. Curr Opin Neurobiol 6:264–268
Yin JC, Del Vecchio M, Zhou H, Tully T (1995) CREB as a memory modulator: induced expression of a dCREB2 activator isoform enhances long-term memory in Drosophila. Cell 81:107–115
Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV et al (2002) Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci 22:1532–1540
Acknowledgements
This study was sponsored by grants from MIUR (PRIN); Regione Campania (L.R. 5/02), Italy; Progetto Strategico Alzheimer (Ministero della Salute-Regione Lazio) Italy; and Provincia di Salerno (Assessorato allo Sport, Salute e Qualità della Vita) Italy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lista, I., Sorrentino, G. Biological Mechanisms of Physical Activity in Preventing Cognitive Decline. Cell Mol Neurobiol 30, 493–503 (2010). https://doi.org/10.1007/s10571-009-9488-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-009-9488-x