
Abstract
Reducing chain ends in various hydrolyzed cellulose samples were quantitatively analyzed by solid-state 13C NMR. Cellulose di- and tetramers served as model systems to demonstrate that distinct signals at 97 ppm and 92.7 ppm can be detected quantitatively and assigned unambiguously to reducing chain ends in β and α anomers, respectively. In standard Avicel microcrystalline cellulose, the same signals were detected at the expected low intensity; their strength increased significantly upon further hydrolysis. The assignment of the 97 and 92.7 ppm signals to reducing chain ends was confirmed by their absence after ethanolysis, which instead produced an α-ethyl-ether end group signal at 99 ppm. Due to moderate-amplitude chain-end mobility, chain-end signals were relatively enhanced in direct-polarization 13C NMR with heteronuclear Overhauser enhancement. In 13C-enriched hydrolyzed cellulose, the assignment of the C–OH chain-end signals was further corroborated by hydroxyl-proton selection and two-dimensional 13C-13C NMR. From the fractional chain-end signal intensity, the number-average degree of polymerization (DPn) was determined. For Avicel, this yielded DPn = 43 (+ 50, -6), consistent with gel-permeation chromatography but significantly lower than deduced from a more indirect optical method likely hampered by limited chain-end accessibility. After 60 min of hydrolysis of ball-milled Avicel or cellulose from maize, highly reliable values of DPn = 18 ± 3 and 15 ± 3, respectively, were obtained. Solid-state NMR completely avoids the potential loss of low-molar-mass chains in solution-based approaches. The accurate, solvent-free solid-state NMR method introduced here can serve as a primary standard to calibrate other methods for molar-mass determination in hydrolyzed cellulose.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose, the main component of lignocellulosic plant biomass and the most abundant biopolymer on Earth, has attracted significant interest as a renewable feedstock for the production of fuels, chemicals, and materials (Shlieout et al. 2002; Zhang and Lynd 2004; Rubin 2008). Rational design of cellulose-based structural materials or processes for targeted applications of cellulose depends on a fundamental understanding of the molecular structure of cellulose and its relationship with properties (Shlieout et al. 2002; Trache et al. 2016).
Cellulose is a linear homopolymer of glucose monomers connected via β-1,4 glycosidic bonds (Crawford 1981). One of its fundamental structural characteristics as a polymer is the degree of polymerization (DP), i.e. the number of covalently linked glucose rings. Mechanical properties and susceptibility to depolymerization have been reported to depend on DP (Shlieout et al. 2002; Trache et al. 2016). Due to the implications of molecular weight in cellulose applications (Shlieout et al. 2002), DP is an important parameter in theoretical models attempting to unravel the mechanisms of cellulose depolymerization via hydrolysis and pyrolysis (Gupta and Lee 2009; Matsuoka et al. 2014). For instance, chain ends, whose concentration is inversely proportional to DP, are considered the active points for pyrolysis (Lu et al. 2018) and cellobiohydrolase processivity (Gupta and Lee 2009). Therefore, the ability to accurately quantify the degree of polymerization and chain ends in cellulose and its hydrolysis products is crucial for the theoretical description of cellulose chemistry (Puri 1984).
Various solution methods aimed at measuring DP of cellulose have been developed. In principle, as for other polymers, the number-average (DPn), weight-average (DPw), and viscosity-average (DPv) degrees of polymerization can be estimated by membrane or vapor pressure osmometry, light scattering, and viscometry, respectively (Oberlerchner et al. 2015). However, since cellulose is difficult to dissolve, complex methods of cellulose solubilization are required, typically based on derivatization, e.g. with bulky phenyl carbamate groups, or using targeted solvent mixtures. During this complex sample preparation lower-molecular-weight species may be washed out, leading to overestimate of DP (Evans et al. 1989; Engel et al. 2012). Size-exclusion or gel-permeation chromatography, GPC, gives the molecular weight distribution but requires not only full dissolution (Röder et al. 2001) but also external calibration, which typically relies on polystyrene standards that are not directly representative of the cellulosic oligomers (Evans et al. 1989), or on careful light-scattering analysis (Evans et al. 1989; Saalwächter et al. 2000). At low molecular weights, a transition of cellulose tricarbanilate (Evans et al. 1989; Wood et al. 1989) or cellulose-metal complexes (Saalwächter et al. 2000), common derivatized forms of cellulose, from a random coil to a rigid rod (Wood et al. 1989; Saalwächter et al. 2000) may complicate the analysis. A polysaccharide-specific method for determining DPn is the 2,2’-bicinchoninate (BCA) method (Waffenschmidt and Jaenicke 1987; Zhang and Lynd 2005; Gupta and Lee 2009), where the concentration of reducing (C–OH) chain ends of cellulose in aqueous suspension is evaluated from the concentration of the resulting Cu(I)-BCA complex with strong light absorption at 560 nm. Since cellulose is not fully dissolved under these conditions, limited chain-end accessibility may result in an underestimate of chain ends and therefore an overestimate of DPn. Recently, it was found that ionic liquid electrolytes can directly dissolve cellulose samples (Holding et al. 2016; King et al. 2018), which enables estimation of DPn of low- and moderate-molar-mass cellulose via solution NMR (Holding et al. 2016; Koso et al. 2020).
For a meaningful comparison of DPn from different techniques, results from a reproducibly prepared model cellulose are needed. Avicel PH-101, a widely used and comprehensively characterized (Evans et al. 1989; Schelosky et al. 1999; Saalwächter et al. 2000; Zhang and Lynd 2005; Zhang et al. 2006; Pala et al. 2007; Engel et al. 2012) microcrystalline cellulose (MCC) material produced by hydrolysis of wood cellulose and spray drying (Chaerunisaa et al. 2020), can be used for this purpose, assuming that batch-to-batch variations are minor. A fairly wide range of DPn values, from 56 to 212 ± 7, has been reported for Avicel PH-101. GPC of Avicel tricarbanilate carefully calibrated with light scattering gave DPn = 56 (Evans et al. 1989). GPC of Avicel PH-101 in a mix of DMF and an ionic liquid yielded a similar DPn of 57 (Engel et al. 2012). A less detail-oriented GPC study reported a twice-higher DPn = 129 (Pala et al. 2007). An even higher DPn = 212 ± 7 was confidently reported from the BCA method, which has been touted as an efficient technique for determination of DPn of cellulose (Zhang and Lynd 2004, 2005; Zhang et al. 2006; Yang et al. 2019). Given this wide scatter of results, a reliable technique for direct DPn determination of microcrystalline cellulose is of interest.
Here we introduce solid-state 13C nuclear magnetic resonance (ssNMR) spectroscopy as a direct, solvent-free primary method for determining DPn in hydrolyzed cellulose. Without the necessity to dissolve or otherwise pretreat cellulose, solid-state NMR can directly and quantitatively determine DPn as the ratio of the number of anomeric carbons to the number of reducing chain ends, from the intensities of the corresponding NMR signals. The approach is quite accurate and no external standards are needed. It is validated here by measurements on cellulose (i.e., β-glucose) oligomers.
The reducing chain ends are OC1OH groups, which are resolved from the regular OC1OC ether carbons in cellulose by the systematic ~ 7-ppm chemical shift difference between alcohols and ethers (Reich, 2021). We analyze the backbone C1 carbons resonating at 100–110 ppm and the α- and β-C1 carbons of reducing chain ends resolved at ~ 93 and 97 ppm in cellulose oligomers and in hydrolyzed cellulose. The mobility of chain ends is assessed by direct polarization with heteronuclear Overhauser enhancement (Schmidt-Rohr and Spiess 1991). Ethanolyzed ball-milled cellulose (Tyufekchiev et al. 2020) provides a reference of moderate DPn without reducing chain ends.
The ssNMR method is demonstrated on Avicel PH-101 and Avicel recrystallized after ball milling and hydrolysis, which provides particularly clear chain-end signals. The analysis is validated in a different sample, uniformly 13C-enriched cellulose from maize recrystallized after ball milling and hydrolysis, where the signal-to-noise ratio is excellent and advanced characterization of chain-end signals, for instance by 2D 13C-13C exchange (Johnson et al. 2013; Kang et al. 2019) and hydroxyl-proton selection (HOPS) NMR (Duan and Schmidt-Rohr 2019), becomes possible. The DPn of Avicel is determined and compared with literature values.
Experimental
Materials
Avicel PH-101 cellulose was purchased from Sigma-Aldrich (catalog number 11365), D(+)-cellobiose from Fisher Scientific (catalog number AC108460050). Microcrystalline cellulose was purchased from Aldrich (catalog number 43,523–6). Cellotetraose (> 90% purity, 25 mg) was obtained from ThermoFisher. Ethyl α-D-glucopyranoside was purchased from Carbosynth. The samples were packed into NMR rotors and measured as received.
The uniformly 13C-labeled cellulose (u-13C-cellulose, 97% 13C) from maize was purchased from IsoLife (Wageningen, The Netherlands). It contains ~ 68 wt.% glucan with a high degree of polymerization, ~ 20 wt.% of hemicellulose, and ~ 10 wt.% impurities.
Ball milling
Ball milling was used to decrystallize cellulose samples before hydrolysis or ethanolysis with recrystallization (Tyufekchiev et al. 2019, 2020). Briefly, 5 g of Avicel or 0.4 g of 13C enriched cellulose and three stainless-steel balls (two 9.5 mm in diameter and one 15.9 mm in diameter) were placed in a stainless-steel cylinder 18 mm in diameter and 55.5 mm in length with a total volume of 10 mL. The cylinder was clamped in a Retsch MM2000 vibratory shaker and shaken for 50 min to yield amorphous cellulose (Avicel-bm and u-13C-cell-bm).
Hydrolysis and ethanolysis
Procedures of hydrolysis and ethanolysis were followed as detailed previously (Tyufekchiev et al. 2019, 2020). For the Avicel series, 0.25 g of Avicel-bm samples were hydrolyzed for 5 min and for 60 min at 150 °C and 0.1 M HCl to produce Avicel-bm-HCl5 and Avicel-bm-HCl samples, respectively. Double ethanolysis of 0.25 g Avicel-bm, each time for 90 min at 140 °C and 0.1 M HCl, produced Avicel-bm-Eth.
Of the ball-milled uniformly 13C-enriched cellulose, 0.10 g was hydrolyzed for 60 min to produce u-13C-cell-bm-HCl (conversion 57%), and 0.15 g was ethanolyzed for 120 min to give u-13C-cell-bm-Eth (conversion 54%).
General NMR parameters
Solid-state NMR experiments were performed on a Bruker Avance Neo 400WB NMR spectrometer at 1H and 13C resonance frequencies of 400 and 100 MHz, respectively. Most of the measurements were conducted using a Bruker double resonance magic-angle-spinning (MAS) probe with 4-mm zirconia rotors. Samples were loaded into the rotor directly, except for the u-13C-cellulose and u-13C-cell-bm-HCl samples, which were each first packed into a Kel-F HR-MAS rotor insert. The 90° pulse lengths for 1H and 13C were 3.6 μs and 4.0 μs, respectively. Two-pulse phase modulation (TPPM) (Bennett et al. 1995) 1H decoupling at a field strength of γB1/2π = 95 kHz was used for 1H dipolar decoupling during the Hahn echo (Hahn 1950) or total suppression of sidebands (TOSS) (Dixon et al. 1982) for dead-time-free detection, while decoupling by SPINAL-64 (Fung et al. 2000) at γB1/2π = 85 kHz was used during signal acquisition. 13C chemical shifts were referenced to TMS via the carboxyl resonance of 1-13C-glycine in the α-modification at 176.49 ppm as a secondary reference.
An acquisition time of 19 ms was used for all the 1D 13C NMR experiments. The 13C B1 field strength used in cross-polarization was optimized for each MAS frequency. The typical contact time for ramped (90–100%) CP was 1.1 ms. CP 13C NMR spectra of cellobiose, Avicel, Avicel-bm-HCl, and Avicel-bm-Eth samples were acquired at 9 kHz MAS for DPn determination, with total signal averaging times ranging from one day for cellobiose to four days for Avicel. The resulting quantitative spectra were directly integrated and also deconvoluted using the MultiPeakFit package in iGor 5.3. Quantitative composite-pulse multiCP (Duan and Schmidt-Rohr 2017) 13C NMR spectra were all acquired at 12 kHz MAS for u-13C-cellulose, u-13C-cell-bm-HCl and u-13C-cell-bm-Eth samples. Five 1.1 ms CP blocks separated by 4 1H repolarization periods of 4 s each were used and 64 scans were accumulated for the spectra shown. Direct polarization of 13C with selective enhancement of chain-end signals by (hetero)nuclear Overhauser enhancement (NOE) was achieved with a 5-s recycle delay and saturation of 1H by inversion pulses spaced by 0.2 s.
HOPS 13C NMR
The structure near the C1 carbons of reducing chain ends is distinct from that of C1 sites in the backbone of cellulose, in that the former are O–CH-OH groups while the latter are O–CH-O-C ethers. In 13C-enriched organic materials, the signals of C–OH protons and carbons can be selectively observed by hydroxyl proton selection (HOPS) (Duan and Schmidt-Rohr 2019). HOPS relies on dipolar dephasing of 1H magnetization in 13C-1H groups by the strong dipolar coupling of the 13C spin, while OH proton magnetization remains mostly unaffected. The experiment can be implemented conveniently at 13.5 kHz with the one-bond dipolar dephasing occurring in one rotation period with a recoupling 1H π-pulse at its center. Cross polarization of moderate duration (< 400 μs) transfers the surviving OH proton magnetization to nearby carbons, most prominently in C–OH groups.
While HOPS works well for C not directly bonded to H (Duan and Schmidt-Rohr 2019), in the (CH–OH)-rich structure of polysaccharides additional challenges arise. Even ether carbons are surrounded by and cross-polarized from OH groups bonded to neighboring carbons. In order to suppress the ether signal, we take advantage of the dipolar evolution of 1H in 13C-1H groups partially inverting the magnetization of these methine protons. Accordingly, the carbon magnetization is negative at short cross polarization times < 70 μs. However, at longer times the negative C-H contribution is compensated by the large positive magnetization of OH protons. For HC–OH groups, the positive contribution is stronger than for HC–O–C ethers and therefore the 13C magnetization of the former is already positive when that of the latter passes through zero, after cross polarization times of ca. 115 μs. This makes it possible to obtain a spectrum with only minimal O-C–O–C ether signals. Since the spectrum contains known C–OH peaks between 70 and 80 ppm and known C–O–C ether peaks near 105 ppm and 89 ppm, the cross-polarization time can be fine-tuned to null the ether signal. The HOPS (Duan and Schmidt-Rohr 2019) spectra were collected with a recycle delay of 2 s, and a typical spectrum was averaged overnight.
2D 13C-13C NMR
A two-dimensional 13C-13C exchange NMR spectrum (Jeener et al. 1979) of u-13C-cell-bm-HCl was measured at 7.5 kHz MAS with high power dipolar decoupling and TOSS before detection. The mixing time was set to 10 ms to allow for dipolar magnetization transfer over a few bonds and thus probe the local environment of each carbon. A recycle delay of 1.8 s was used for 13 h of signal averaging.
Results
Quantitative 13C NMR of cellulose oligomers
Cellulose oligomers contain reducing chain ends at known concentrations and are therefore useful reference materials for developing the chain-end measurement methodology in this study. The spectra of cellobiose (a glucose or cellulose dimer) and cellotetraose are shown in Fig. 1. They exhibit well-resolved signals of reducing chain ends near 97 and 93 ppm. The spectrum of cellotetraose shows broad bands characteristic of an amorphous carbohydrate; nevertheless, the chain-end signals are sufficiently well resolved for a quantitative analysis. Based on corresponding signals in glucose, chemical shift trends, and peak intensity ratios, the assignment of these O–CH–OH signals is completely certain. The greater intensity of the β-anomer peak at 97 ppm relative to the α-anomer signal at 93 ppm is consistent with the predominance of the β-anomer in the monomer, glucose (Roslund et al. 2008). The ratio of the two end-group C1 signals to the total peak integrals of all C1 signals (91.5–110 ppm) is 1/(2.0 ± 0.05) for cellobiose, and 1/(4.06 ± 0.2) for cellotetraose. This is consistent with the expected chain end signal fraction of 1/DPn, where DPn is the number of repeat units in the oligosaccharide, and confirms that the determination of low DPn by ssNMR is quantitative.

Quantitative solid-state 13C NMR spectra of a cellobiose and b cellotetraose. Peak integrals, shown in red, are accurate to within ± 2% of each peak area for resolved CH signals. The signals of reducing chain ends near 97 and 93 ppm are clearly and quantitatively detected. Note that the peak-to-peak integral of the chain-end signals in b gives only half of their total area and therefore needs to be multiplied by two
Quantitative 13C NMR of hydrolyzed cellulose
Extending the analysis to representative cellulose substrates, Fig. 2 displays the spectra of standard Avicel PH-101 cellulose before and after ball milling plus hydrolysis or ethanolysis. The highlighted signals at 97 and 92.7 ppm are in a ~ 2:1 intensity ratio. In the hydrolyzed sample, the peaks clearly arise from reducing chain ends. The absence of the 97 and 92.7 ppm signals in Fig. 2c after ethanolysis, which produces ethyl ethers at the reducing chain ends, confirms the assignment, as does advanced NMR applied to a corresponding ball-milled and hydrolyzed 13C-enriched cellulose, see below.

Quantitative solid-state 13C NMR spectra of a Avicel PH-101 cellulose before hydrolysis; b hydrolyzed Avicel, with arrows highlighting the chain-end peaks and resolved crystalline signals of cellulose I and II (VanderHart and Atalla 1984) confirmed by 2D NMR below; and c ethanolyzed Avicel. Expanded views of the chain-end signals are shown further below
Due to resolved signals of crystalline cellulose, NMR can be used to identify crystal modifications (VanderHart and Atalla 1984) and quantify the degree of crystallinity (Haslinger et al. 2019; Pintiaux et al. 2019). The spectra in Fig. 2 show spectral changes from cellulose-II crystallites produced during hydrolysis (Tyufekchiev et al. 2019), with distinct resonances near 107, 88, and 63 ppm (VanderHart and Atalla 1984); nevertheless, the positions of the chain-end signals have remained unchanged.
Direct polarization with heteronuclear Overhauser enhancement
The chain end signals can be detected more clearly, with better dynamic range, after selective enhancement by direct 13C polarization with a fairly short 5 s recycle delay and pulsed saturation of 1H that generates a heteronuclear nuclear Overhauser enhancement (NOE) of mobile segments. Figure 3 shows the chain-end signals in Avicel and hydrolyzed Avicel increased relative to others when compared with suitably scaled cross-polarization spectra. The greatest enhancement is observed for noncrystalline CH2-OH side groups resonating near 62 ppm, which can evidently perform fast motions of fairly large amplitudes. The relative intensity of the chain-end signals at 97 and 93 ppm is greater than that of the main C1 peak, greater in fact than all other OCH signals, due to faster relaxation and therefore greater NOE. The faster relaxation of the chain ends compared to the other OCH sites, including the noncrystalline surface sites, is confirmed by a Torchia experiment (Fig. S5). This faster relaxation and the NOE can be attributed to faster relaxation driven by moderate-amplitude motions with rates exceeding 1 MHz, which is not unexpected for chain ends since they are less constrained than segments in the interior of a cellulose chain. It is interesting to note that a broad defect signal around 98 ppm in amorphous cellulose, see Fig. S4b, does not show this signal enhancement in the direct-polarization/NOE experiment, which is clearly characteristic of side and end groups.

Direct-polarization solid-state 13C NMR spectra with heteronuclear nuclear Overhauser enhancement (NOE) of mobile segments in a Avicel cellulose before hydrolysis and b ball-milled and hydrolyzed Avicel. Arrows highlight the chain-end signals. The corresponding CP spectrum, matched at the chain-end peaks, is shown dashed for reference
Ethanolyzed chain ends
After ethanolysis, the 97- and 93-ppm signals of the reducing chain ends disappear (see Fig. 2) and instead a 99-ppm signal is observed, which can be convincingly assigned to ethylated α-C1 by comparison with α-ethylglucoside (see Fig. S6) and based on the CH3 cross peak in 1H-13C HetCor NMR (see Fig. S7). In a recent publication (Tyufekchiev et al. 2020), quantitative 13C NMR showed the presence of ethyl ethers in ethanolyzed ball-milled-cellulose samples, indicating that the reducing ends in cellulose chain have been capped by ethyl ethers during the ethanolysis process.
While the close chemical shifts of the chain-end signals at 97 and 99 ppm in the spectra in Figs. 2b and c might suggest similar structures, the similarity is actually a coincidence, resulting from two distinct, well-known down-field-shift effects: (i) Ether formation typically increases the chemical shift by ~ + 7 ppm relative to the corresponding C–OH (Reich, 2021); (ii) the β anomer of glucose and xylose is typically + 5 ppm higher in chemical shift than the α anomer.
Evidence for reducing chain ends in u-13C-cellulose from spectral editing
In the following, we show that the small chain end signals at 97 and 93 ppm are not unique to Avicel. They can be detected with ~ 90-fold improved signal-to-noise ratio in uniformly 13C-enriched cellulose that has been ball-milled and HCl-treated. Thanks to the 13C-enrichment, the molecular environment of the 97- and 93-ppm carbons can be probed by advanced NMR experiments to further confirm the assignment to the reducing chain ends.
The increased signal intensity enables spectral editing even of small peaks. 1H-13C dipolar dephasing and CH selection by dipolar DEPT, see Fig. S3, show that the signals between 93 and 97 ppm arise from CH groups without fast large-amplitude mobility. Their chemical shift requires assignment to O–CH-O segments. Hydroxyl-proton selection (HOPS) can yield selective spectra of C-OH groups in 13C-enriched solids (Duan and Schmidt-Rohr 2019). As explained in the Experimental section, for 13C-labeled polysaccharide samples, initially negative (inverted) signals of CH carbons not bonded to OH groups recover more slowly than the OH-bonded CH, since only the OH protons can provide positive magnetization. With a fine-tuned short CP contact time, the signal of CH not bonded to OH is nulled or slightly negative while that of CH-OH is positive, and thus only signals of CH-OH carbons are significant.
Figure 4 shows the HOPS spectra for u-13C-cell-bm-HCl and u-13C-cell-bm-Eth and compares them to the corresponding quantitative multiCP spectra (dashed). The signals of C1 and C4, which have ether linkages and are not directly bonded to OH groups, are small when the CP contact time was set to 114 μs, while both α and β reducing chain end C1 carbons show distinct positive peaks whose scaling fully agrees with that of the C–OH backbone signals, as demonstrated by comparison with the quantitative unmodulated reference spectrum (dashed line). This confirms that the peaks at 93 ppm and 97 ppm must be assigned to OH-bonded CH groups in the glucan ring. By contrast, in u-13C-cell-bm-Eth the ethoxy-protected α chain ends are not bonded to OH, and therefore their signal intensity at 99 ppm is very small after 115 μs CP, similar to that of the ether C1 and C4 in the backbone but unlike the COH signals between 70 and 80 ppm.

Hydroxyl-proton selection (HOPS) 1H-13C NMR of u-13C-cellulose provides evidence for C1-OH reducing chain ends after ball milling and a hydrolysis, but not after b ethanolysis
Evidence for reducing chain ends in u-13C-cellulose from 2D exchange NMR
Two-dimensional 13C-13C exchange NMR with a moderate spin-exchange mixing time of 10 ms can probe the local environment of the carbons resonating at 97 and 93 ppm. Figure 5a presents a 2D exchange spectrum where the chain ends’ strongest cross peaks, at 75 and 72 ppm, respectively, show that both C1 and C2 chemical shifts are lower for the α- than for the β-anomer, as is also the case in glucose and its various dimers (Roslund et al. 2008).

a Two-dimensional 13C-13C NMR spectrum, with 10 ms spin exchange, of u-13C-cellulose after ball milling and hydrolysis. Some signals of the α and β reducing chain ends are labeled. I: cellulose-I crystals; II: cellulose-II crystals (VanderHart and Atalla 1984); the cross peaks of the latter (on the purple dashed line) confirm the cellulose-II peak assignments in Fig. 2. b Cross section from the 2D spectrum at 97 ppm (blue trace), which is a spectrum of carbons near the 97-ppm β-anomer end group. A spectrum of magnetization originating from ether C1 resonating at 105 ppm is shown dashed (black trace) for reference. S: Surface chains
A comparison of cross sections from the 2D exchange spectrum at the C1 backbone and chain-end peak positions in Fig. 5b also exhibits structurally telling intensity differences. For the ether C1 in the cellulose backbone, the C4 exchange intensity is nearly twice the C6 intensity due to inter- and intra-ring C1-C4 exchange, the former across C1OC4 in the glycosidic linkage. For the chain ends, only intra-ring C1-C4 exchange is significant and matches C1-C6 intra-ring exchange. The peak position and shape of C4 detected near the chain ends in Fig. 5b is that of noncrystalline C4, which indicates that the chain ends are mostly located in the noncrystalline regions.
Discussion
Chain-end peak assignment
We have shown overwhelming evidence that the 97- and 93-ppm signals in hydrolyzed cellulose must be assigned to reducing chain ends, by HOPS, 2D, and direct-polarization/NOE NMR, as well as comparison with an ethanolyzed sample. They match the reducing end groups in oligosaccharides, whose fractional peak areas are quantitatively correct.
Degree of polymerization of Avicel from ssNMR
The relative area of chain-end and backbone C1 NMR signals in cellulose of moderate or low molecular weight can be evaluated to accurately determine the degree of polymerization. Figure 6 demonstrates the integration and deconvolution procedure for DPn estimation. The accurate peak positions and widths for both reducing C1 ends were obtained through deconvolution of the most extensively hydrolyzed Avicel-bm-HCl sample in Fig. 6a. These parameters were adopted for the deconvolution of the other two samples as shown in Figs. 6b and S8b. For the hydrolyzed 13C-enriched cellulose, the chain-end region shown in Fig. 7 exhibits the same peak pattern with excellent signal-to-noise ratio and only slightly larger line widths (due to 13C-13C couplings) in the deconvolution.

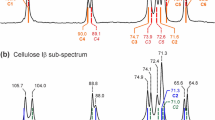
Quantification of cellulose chain-end signals for a Avicel-bm-HCl and b Avicel PH-101. The corresponding analysis for Avicel-bm-HCl5 is shown in Fig. S8. Chain-end fractions (denominator of the integral fractions) were determined based on peak-to-peak integrations to reduce the potential overlap from the main C1 and C4 peaks; since this integral range covers only half of the peak area, a factor of 2 is included in the denominator. More chain-ends are generated by ball milling and hydrolysis. The same parameters for the chain-end peaks were used for both fits. The residuals shown beneath the fits were referenced to the peak near 105 ppm

Deconvolution of the chain-end region of the spectrum of u-13C-cell-bm-HCl. The 13C enrichment greatly improves the signal-to-noise ratio but results in slightly wider signals due to the 13C-13C couplings
All DPn evaluations from the Gaussian deconvolutions agree well with the values from the direct integration of the chain-end signals peak-to-peak, from 97 to 92.7 ppm, a spectral range where overlap with backbone signals is minimal; since this integral covers only half of each peak, its value is multiplied by two to yield the full chain-end peak area. As Table 1 shows, the highest DPn of 43 is found for the Avicel PH-101 sample. A similar line shape was observed for the Aldrich MCC sample as for the Avicel sample, see Fig. S2, and a DPn of 38 (+ 44, -6) was determined by integration. The DPn is significantly reduced to 24 ± 4 after ball milling and 5 min of hydrolysis, as expected, see Figure S8. Prolonged hydrolysis for 60 min further reduced the DPn to 18 ± 3 (see Fig. 6a).
Error margins of DP n from ssNMR
Given the significant discrepancy between DPn of Avicel from NMR and the value of 212 ± 7 from the BCA method (Zhang and Lynd 2005; Zhang et al. 2006; Gupta and Lee 2009), sources of error need to be considered. Poor cross polarization of the chain-end carbons cannot account for the observed discrepancy: If the chain-end signals were artificially too low, the NMR-derived DPn would be artificially too high, which is not the case. The same applies for oxidation of the reducing end groups, which would also decrease the signals between 97 and 92.7 ppm and therefore artificially increase DPn, which is not observed. The oxidation products, such as onic acids or lactones, if present in significant concentrations should be detectable by NMR between 165 and 215 ppm without interference from regular cellulose peaks. Background signals between 97 and 92.7 ppm could artificially decrease the DPn value derived from NMR. However, the deconvolution of the Avicel spectrum with the well-defined chain-end signals of hydrolyzed Avicel, see Fig. 6, shows no indication of a significant background between 97 and 92.7 ppm.
In order to determine a hard upper limit to DPn, we assumed a maximum possible background, based on the signal of cotton balls, see Fig. 8, scaled as high as possible without generating a negative subtraction artifact. Even with such a maximum background assumed as shown in Fig. 8, still DPn ≤ 93, more than a factor of two smaller than from the BCA method (Zhang and Lynd 2005; Zhang et al. 2006; Gupta and Lee 2009). The fact that the NMR analysis is based on two chain-end signals makes it less prone to error from an unexpected impurity signal.

Alternative, maximally aggressive deconvolution of the chain-end region of the spectrum of Avicel, assuming the signal of cotton balls between 100 and 92 ppm to be non-chain-end background in Avicel. The intensity of the resulting difference spectrum (dashed line with green shading) would correspond to a higher but still moderate degree of polymerization of ~ 93 in Avicel
For short chains, e.g. after additional hydrolysis, a given background signal between 97 and 92.7 ppm has a relatively small effect on DPn. The apparent DPn’, true DPn, and background B as a fraction of C1 intensity are related by
For example, a background of B = 1% would result in a strong decrease from true DPn = 74 to apparent DPn’ = 43, since
while true DPn = 18 would correspond to an only slightly smaller apparent DPn’ = 15, since
Thus, the ssNMR method is particularly accurate for aggressively hydrolyzed cellulose.
Comparison with Avicel DP n from other methods
A superficial survey of the literature, in particular relatively recent publications by a prominent biofuels expert (Zhang and Lynd 2004, 2005), may give the impression that the degree of polymerization of Avicel and similar microcrystalline cellulose materials is many hundreds to 1000 (Zhang and Lynd 2004), much greater than the value of 43 reported here. This discrepancy has more than one origin.
The molar mass of MCCs like Avicel is likely batch-dependent to some extent. More importantly, in a system with a distribution of chain lengths, the number-average degree of polymerization, as reported here, is always smaller than the corresponding weight- or viscosity-averages. The polydispersity index or dispersity Đ = DPw/DPn ≥ 1, has values of 2–20 in synthetic polymers unless special synthesis procedures are employed (Odian 2004). GPC measurements on microcrystalline cellulose (Daňhelka et al. 1976; Schelosky et al. 1999; Pala et al. 2007; Engel et al. 2012; Holding et al. 2016), including Avicel (Schelosky et al. 1999; Pala et al. 2007; Engel et al. 2012), have demonstrated that the dispersity of this type of hydrolyzed material is Đ ≈ 3. Thus, the commonly reported DPw values will be three times higher than the DPn measured by NMR.
Even the DPn values of Avicel recently reported by Zhang, Lynd, and coworkers based on the BCA method greatly exceed ours; however, most DPn values of Avicel reported in the literature (Evans et al. 1989; Schelosky et al. 1999; Saalwächter et al. 2000; Engel et al. 2012) actually agree with our result within the experimental uncertainties. Using DMF and an ionic liquid as the mobile phase, Engel et al. achieved complete dissolution of Avicel and reported Mw = 28,400 and Đ = 3.1, which corresponds to DPn = 57 and is compatible with our solid-state NMR result of 43 (+ 50, – 6). Another study (Schelosky et al. 1999) reported DPn = 67 and Đ ≈ 3. Saalwächter et al. (2000) determined DPw ~ 220 for Avicel PH-101, and Shlieout et al. (2002) found a similar value for other MCCs, which with Đ ≈ 3 corresponds to DPn ~ 70, still compatible with our result. A possibly more superficial GPC study (Pala et al. 2007) reported DPn = 129.
The BCA method has yielded higher values of DPn = 212 ± 7 (Zhang and Lynd 2005; Zhang et al. 2006; Gupta and Lee 2009). This may be attributed to limited accessibility of chain ends in dispersed cellulose fibrils, although in a modified version (Zhang and Lynd 2005), the same (relatively high) DPn was obtained both before and after amorphization, which was taken as evidence that the reducing chain ends are fully accessible to Cu(II) in solution. Nevertheless, given all the previous studies (Evans et al. 1989; Schelosky et al. 1999; Saalwächter et al. 2000; Engel et al. 2012) that found a lower DPn for the same cellulose material, this still indirect accessibility evidence is not strong enough.
At 38 (+ 44, -6), the NMR-derived DPn of Aldrich MCC is slightly lower than that of Avicel PH-101; the underlying slightly larger chain-end signals can be directly recognized in Fig. S2b. This DPn value agrees within the error margins with results from solution NMR, 55 ± 5 (Koso et al. 2020), and 78 (Holding et al. 2016); before application of T2 correction, the solution-NMR values were even smaller, while GPC gave 153 (Koso et al. 2020) and 87 (Holding et al. 2016), respectively.
The most likely DPn value of Avicel obtained from our NMR investigation, 43, is still somewhat smaller than all the previously reported values. While background NMR signals might give the impression of more chain ends than are actually present, we can highlight that our solid-state NMR method is the first that is sure to detect chains of all lengths in their correct proportions. All previous methods (Daňhelka et al. 1976; Evans et al. 1989; Wood et al. 1989; Schelosky et al. 1999; Saalwächter et al. 2000; Röder et al. 2001; Shlieout et al. 2002; Zhang and Lynd 2005; Zhang et al. 2006; Pala et al. 2007; Gupta and Lee 2009; Engel et al. 2012; Holding et al. 2016; King et al. 2018; Koso et al. 2020) were solution-based and the required aggressive dissolution procedures may result in loss of low-molecular material (Evans et al. 1989). No pretreatment is required for solid-state NMR, which can comprehensively excite all the chain ends in the system and therefore provide a more accurate DPn estimation. Our study also stands out in that unlike most previous papers (Daňhelka et al. 1976; Evans et al. 1989; Wood et al. 1989; Schelosky et al. 1999; Saalwächter et al. 2000; Röder et al. 2001; Shlieout et al. 2002; Pala et al. 2007; Engel et al. 2012) it reports an experimental uncertainty for DPn of cellulose (note that the dispersity Đ does not quantify the error margin of DPn); this is probably witness to its particularly direct relation between observed signal and DPn, and thus ultimately its scientific reliability.
Upper DP n limit of ssNMR
The upper limit of DPn of cellulose that can be determined by NMR in our laboratory is around 100. It is partly determined by the 13C NMR detection limit, which results in a low signal-to-noise ratio of the small signals of the few chain ends of higher-DPn cellulose. The experiments shown here were performed at 9.4 T (100 MHz 13C frequency). In a higher external magnetic field of 19.8 T, the detection limit could be lowered by at least a factor of two and peak resolution might be somewhat enhanced as well. This suggests that the maximum NMR-accessible DPn of clean cellulose might reach 200 in a high-field NMR instrument.
The lower range of degrees of polymerization of cellulose is arguably the most important for properties, because it corresponds to a significant concentration of reactive chain ends. This is certainly true during cellulose depolymerization for biofuels production, as chain ends are the active sites for enzyme processing (Gupta and Lee 2009). Since cellulose is not soluble under biological conditions and cannot easily be melted, the specific value of its high molecular weight in plants may not affect tissue properties much. While in light-scattering analysis, the lowest degrees of polymerization may pose some special challenges due to rigid-rod behavior, in solid-state NMR chain ends from chains of any length are equally detected.
Conclusions
This paper introduces solid-state 13C NMR as a reliable method for determining the degree of polymerization of hydrolyzed cellulose. The approach is based on quantitative NMR detection of chain-end signals, without solvent and without requiring an external reference. The assignment of the chain-end signals has been validated using cellulose oligomers, ethanolyzed cellulose, and advanced NMR of 13C-enriched hydrolyzed cellulose. In Avicel, the number-average degree of polymerization is at least three times lower than previously estimated using the indirect BCA method, but in agreement with several careful GPC studies. Further hydrolysis is found to progressively reduce the DPn value, as expected. The values of DPn = 18 ± 3 and 15 ± 3 for two cellulose samples that were ball-milled and hydrolyzed for 60 min are highly reliable. The appearance of two chain-end peaks at known chemical shifts, from α- and β-anomers, makes the method less prone to error from an unrelated background signal. The upper limit for cellulose DPn that can be determined by NMR is currently around 100. Unlike other methods, solid-state NMR does not require any sample pretreatment and does not have to assume accessibility of all chain ends to dissolved reporter molecules. It completely avoids the potential loss of low-molecular-weight chains in solution-based methods. For crystalline cellulose of moderate degree of polymerization (< 100), the NMR approach probably provides more accurate DPn values than previous methods. Two main applications of cellulose DPn measurements by NMR can be envisaged: (i) as the most accurate primary method against which other, less direct cellulose DPn measurements are calibrated; (ii) as a convenient and reliable method for determining DPn in hydrolyzed cellulose, for instance in biofuels production.
References
Bennett AE, Rienstra CM, Auger M et al (1995) Heteronuclear decoupling in rotating solids. J Chem Phys 103:6951–6958. https://doi.org/10.1063/1.470372
Bielecki A, Kolbert AC, De Groot HJM et al (1990) Frequency-Switched Lee—Goldburg Sequences in Solids. Adv Magn Opt Reson. https://doi.org/10.1016/b978-0-12-025514-6.50011-3
Caravatti P, Braunschweiler L, Ernst RR (1983) Heteronuclear correlation spectroscopy in rotating solids. Chem Phys Lett 100:305–310. https://doi.org/10.1016/0009-2614(83)80276-0
Chaerunisaa AY, Sriwidodo S, Abdassah M (2020) Microcrystalline Cellulose as Pharmaceutical Excipient. In: Pharmaceutical Formulation Design - Recent Practices. IntechOpen. doi:https://doi.org/10.5772/intechopen.88092
Crawford R (1981) Lignin biodegradation and transformation. Wiley, New York
Daňhelka J, Kössler I, Boháčková V (1976) Determination of molecular weight distribution of cellulose by conversion into tricarbanilate and fractionation. J Polym Sci, Part a: Polym Chem 14:287–298. https://doi.org/10.1002/pol.1976.170140202
Dixon WT, Schaefer J, Sefcik MD et al (1982) Total suppression of sidebands in CPMAS C-13 NMR. J Magn Reson 49:341–345. https://doi.org/10.1016/0022-2364(82)90199-8
Duan P, Schmidt-Rohr K (2017) Composite-pulse and partially dipolar dephased multiCP for improved quantitative solid-state 13C NMR. J Magn Reson 285:68–78. https://doi.org/10.1016/j.jmr.2017.10.010
Duan P, Schmidt-Rohr K (2019) Quick, selective NMR spectra of C-OH moieties in 13C-enriched solids. J Magn Reson 301:80–84. https://doi.org/10.1016/j.jmr.2019.02.007
Engel P, Hein L, Spiess AC (2012) Derivatization-free gel permeation chromatography elucidates enzymatic cellulose hydrolysis. Biotechnol Biofuels 5:77. https://doi.org/10.1186/1754-6834-5-77
Evans R, Wearne RH, Wallis AFA (1989) Molecular weight distribution of cellulose as its tricarbanilate by high performance size exclusion chromatography. J Appl Polym Sci 37:3291–3303. https://doi.org/10.1002/app.1989.070371202
Fung BM, Khitrin AK, Ermolaev K (2000) An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson 142:97–101. https://doi.org/10.1006/jmre.1999.1896
Gupta R, Lee YY (2009) Mechanism of cellulase reaction on pure cellulosic substrates. Biotechnol Bioeng 102:1570–1581. https://doi.org/10.1002/bit.22195
Hahn EL (1950) Spin Echoes Phys Rev 80:580–594. https://doi.org/10.1103/PhysRev.80.580
Haslinger S, Hietala S, Hummel M et al (2019) Solid-state NMR method for the quantification of cellulose and polyester in textile blends. Carbohydr Polym 207:11–16. https://doi.org/10.1016/j.carbpol.2018.11.052
Holding AJ, Makela V, Tolonen L et al (2016) Solution-state one- and two-dimensional nmr spectroscopy of high-molecular-weight cellulose. Chemsuschem 9:880–892. https://doi.org/10.1002/cssc.201501511
Jeener J, Meier BH, Bachmann P et al (1979) Investigation of exchange processes by two-dimensional NMR spectroscopy. J Chem Phys 71:4546–4553. https://doi.org/10.1063/1.438208
Johnson RL, Anderson JM, Shanks BH et al (2013) Spectrally edited 2D 13C–13C NMR spectra without diagonal ridge for characterizing 13C-enriched low-temperature carbon materials. J Magn Reson 234:112–124. https://doi.org/10.1016/j.jmr.2013.06.006
Kang X, Kirui A, Dickwella Widanage MC et al (2019) Lignin-polysaccharide interactions in plant secondary cell walls revealed by solid-state NMR. Nat Commun 10:347. https://doi.org/10.1038/s41467-018-08252-0
King AWT, Makela V, Kedzior SA et al (2018) Liquid-state NMR analysis of nanocelluloses. Biomacromol 19:2708–2720. https://doi.org/10.1021/acs.biomac.8b00295
Koso T, Rico del Cerro D, Heikkinen S et al (2020) 2D Assignment and quantitative analysis of cellulose and oxidized celluloses using solution-state NMR spectroscopy. Cellulose 27:7929–7953. https://doi.org/10.1007/s10570-020-03317-0
Lu Q, Hu B, Zhang Z-x et al (2018) Mechanism of cellulose fast pyrolysis: the role of characteristic chain ends and dehydrated units. Combust Flame 198:267–277. https://doi.org/10.1016/j.combustflame.2018.09.025
Matsuoka S, Kawamoto H, Saka S (2014) What is active cellulose in pyrolysis? An approach based on reactivity of cellulose reducing end. J Anal Appl Pyrol 106:138–146. https://doi.org/10.1016/j.jaap.2014.01.011
Oberlerchner JT, Rosenau T, Potthast A (2015) Overview of methods for the direct molar mass determination of cellulose. Molecules 20:10313–10341. https://doi.org/10.3390/molecules200610313
Odian G (2004) Principles of polymerization. Wiley-Interscience, Hoboken, New Jersey. https://doi.org/10.1002/047147875X
Pala H, Mota M, Gama F (2007) Enzymatic depolymerisation of cellulose. Carbohydrate Polym 68:101–108. https://doi.org/10.1016/j.carbpol.2006.07.015
Pintiaux T, Heuls M, Vandenbossche V et al (2019) Cellulose consolidation under high-pressure and high-temperature uniaxial compression. Cellulose 26:2941–2954. https://doi.org/10.1007/s10570-019-02273-8
Puri VP (1984) Effect of crystallinity and degree of polymerization of cellulose on enzymatic saccharification. Biotechnol Bioeng 26:1219–1222. https://doi.org/10.1002/bit.260261010
Reich HJ Hans Reich's Collection. NMR Spectroscopy. https://organicchemistrydata.org/hansreich/resources/nmr/?index=nmr_index%2F13C_shift#cdata01. Accessed 05/17 2021
Röder T, Morgenstern B, Schelosky N et al (2001) Solutions of cellulose in N N -Dimethylacetamide/lithium Chloride Studied by Light Scattering Methods. Polymer 42:6765–6773. https://doi.org/10.1016/s0032-3861(01)00170-7
Roslund MU, Tahtinen P, Niemitz M et al (2008) Complete assignments of the 1H and 13C chemical shifts and JH, H coupling constants in NMR spectra of D-glucopyranose and all D-glucopyranosyl-D-glucopyranosides. Carbohydr Res 343:101–112. https://doi.org/10.1016/j.carres.2007.10.008
Rubin EM (2008) Genomics of cellulosic biofuels. Nature 454:841–845. https://doi.org/10.1038/nature07190
Saalwächter K, Burchard W, Klüfers P et al (2000) Cellulose solutions in water containing metal complexes†. Macromol 33:4094–4107. https://doi.org/10.1021/ma991893m
Schelosky N, Roeder T, Baldinger T (1999) Molecular mass distribution of cellulosic products by size exclusion chromatography in DMAc/LiCl Das Papier (Germany):728–738
Schmidt-Rohr K, Mao JD (2002) Efficient CH-group selection and identification in 13C solid-state NMR by dipolar DEPT and 1H chemical-shift filtering. J Am Chem Soc. 124:13938–13948. https://doi.org/10.1021/ja027362m
Schmidt-Rohr K, Spiess HW (1991) Chain diffusion between crystalline and amorphous regions in polyethylene detected by 2D exchange 13C NMR. Macromolecules. 24:5288–5293. https://doi.org/10.1021/ma00019a011
Shlieout G, Arnold K, Muller G (2002) Powder and mechanical properties of microcrystalline cellulose with different degrees of polymerization. AAPS PharmSciTech 3:E11. https://doi.org/10.1208/pt030211
Trache D, Hussin MH, Hui Chuin CT et al (2016) Microcrystalline cellulose: Isolation, characterization and bio-composites application-A review. Int J Biol Macromol 93:789–804. https://doi.org/10.1016/j.ijbiomac.2016.09.056
Tyufekchiev M, Kolodziejczak A, Duan P et al (2019) Reaction engineering implications of cellulose crystallinity and water-promoted recrystallization. Green Chem 21:5541–5555. https://doi.org/10.1039/c9gc02466b
Tyufekchiev M, Ralph K, Duan P et al (2020) Rapid Depolymerization of decrystallized cellulose to soluble products via ethanolysis under mild conditions. Chemsuschem 13:2634–2641. https://doi.org/10.1002/cssc.201903446
VanderHart DL, Atalla RH (1984) Studies of microstructure in native celluloses using solid-state 13C NMR. Macromolecules 17:1465–1472. https://doi.org/10.1021/ma00138a009
Waffenschmidt S, Jaenicke L (1987) Assay of reducing sugars in the nanomole range with 2,2’-bicinchoninate. Anal Biochem 165:337–340. https://doi.org/10.1016/0003-2697(87)90278-8
Wood BF, Conner AH, Hill CG (1989) The heterogeneous character of the dilute acid hydrolysis of crystalline cellulose. J Appl Polym Sci 37:1373–1394. https://doi.org/10.1002/app.1989.070370520
Yang J, Tu M, Xia C et al (2019) Effect of fenton pretreatment on C1 and C6 oxidation of cellulose and its enzymatic hydrolyzability ACS sustainable. Chem Eng 7:7071–7079. https://doi.org/10.1021/acssuschemeng.8b06850
Zhang YH, Lynd LR (2004) Toward an aggregated understanding of enzymatic hydrolysis of cellulose: noncomplexed cellulase systems. Biotechnol Bioeng 88:797–824. https://doi.org/10.1002/bit.20282
Zhang YH, Lynd LR (2005) Determination of the number-average degree of polymerization of cellodextrins and cellulose with application to enzymatic hydrolysis. Biomacromol 6:1510–1515. https://doi.org/10.1021/bm049235j
Zhang YHP, Cui J, Lynd LR et al (2006) A transition from cellulose swelling to cellulose dissolution byo-phosphoric acid: evidence from enzymatic hydrolysis and supramolecular structure. Biomacromolecules 7:644–648. https://doi.org/10.1021/bm050799c
Funding
The solid-state NMR spectrometer used in this work was funded by the NSF MRI program (Award No. 1726346). Partial student support for MVT was provided by the U.S. National Science Foundation (ENG/#1554283).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yuan, S., Tyufekchiev, M.V., Timko, M.T. et al. Direct quantification of the degree of polymerization of hydrolyzed cellulose by solid-state NMR spectroscopy. Cellulose 29, 2131–2144 (2022). https://doi.org/10.1007/s10570-022-04433-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-022-04433-9