
Abstract
We propose a new method for determining the surface energy of paper and its components using the contact angle technique. Calendered sheets and model liquids were used in this study. A rapid evolution of the probe/handsheet contact angle with time was observed for all tested conditions. A suitable method for choosing the contact angle and a new model to determine the surface energy and its components is proposed. The total surface energy of paper hansheets (47.2–50.4 mN m−1) and its components (dispersive γd ~ 19–20, polar γp ~ 4–5 and hydrogen bond γh ~ 25–32) are in good agreement with literature data.
Graphic abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The surface energy of a solid is often determined by Contact Angle (CA) or Inverse Gas Chromatography (IGC). Nowadays, IGC is increasingly used to overcome the delicate conditions required by CA. It has been used in the evaluation of various plant and wood fibers (Jacob and Berg 1994; Papirer et al. 2000; Steele et al. 2008; Cordeiro et al. 2011; Guo et al. 2013; Gamelas 2013). High speed image acquisition techniques allow the analysis of the evolution of the contact angle of a microdrop deposited on the surface of a material, despite the very fast absorption of the liquid probe. It has become possible to measure CA on problematic materials, such as paper, needing several approximations related to flatness, porosity, and heterogeneity (Gellerstedt and Gatenholm 1999; Tze and Gardner 2001; Gómez et al. 2012; Hubbe et al. 2015).
Paper surface energy depends on several parameters that must be taken into account to predict good wetting. For example, pulping and bleaching processes, either alkaline or peroxide-based, can extract lignin increasing the hydrophilicity and consequently the surface energy (Chaiarrekij et al. 2011). Another important factor is the refining process that can make fibers more conformable and consequently denser, smoother and less porous. Increasing the ratio of fines, particles with a high surface energy, can also decrease the contact angle (Vainio 2007; Mirvakili 2011). The surface roughness decreases the contact angle by the presence of hydrophobic air bubbles (Marmur 2006; Nosonovsky and Bhushan 2007; Quéré 2008). For a rough surface, the measured angle is only an average of a distribution of contact angles reflecting the heterogeneity of the surface (Akinli-Kocak 1997; Nosonovsky and Bhushan 2007). The sheet forming technique (laboratory or industrial scale) and the presence of salts in water (demineralized/carbonated water) can yield various levels of roughness (Moutinho et al. 2007). Finally, probe properties like viscosity, molar volume and cohesion energy density (solubility parameters) also influence on the measured contact angle. The diffusion of the drop in the paper and the wetting time increase with the increase of the viscosity of the liquid (Wågberg 2009).
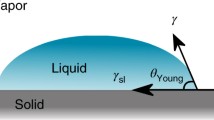
Since the early work of Thomas Young (1805), different models have been proposed for the determination of the surface energy of a solid. Among them, the most used are Zisman (1964), Owens and Wendt (1969), and Young-Good-Girifalco-Fowkes et al. (Van Oss et al. 1988). For a weakly polar, smooth and non-porous surface, the representation of cosθ as a function of the surface energy of the different liquid probes (γl) can be used to calculate the surface energy (γs) of the solid material using Eq. (1) (Fox and Zisman 1950; Ministère des Affaires Sociales et de la santé 2013):
where γsl is the solid–liquid interfacial tension and θ the contact angle.
For polar surfaces with weak hydrogen bonds, Owens and Wendt (1969) have proposed a model for determining γs and its dispersive (\( \gamma_{s}^{d} \)) and polar (\( \gamma_{s}^{p} \)) components where \( \gamma_{s} = \gamma_{s}^{d} + \gamma_{s}^{p} \). The equation connecting these components to the contact angle is:
Equation 2 can be rearranged in a linear form:
By plotting \( y = \frac{{\gamma_{l} \left( {1 + \cos \theta } \right)}}{{2\sqrt {\gamma_{l}^{d} } }} \) as a function of \( x = \frac{{\sqrt {\gamma_{l}^{p} } }}{{\sqrt {\gamma_{l}^{d} } }} \), we obtain \( \sqrt {\gamma_{s}^{d} } \) and \( \sqrt {\gamma_{s}^{p} } \).
In recent years, the most successful model is Van Oss, Chaudhury, and Good known as VOCG (Van Oss et al. 1988). It takes into account the nature of chemical groups present at an interface. In this model, all van der Waals forces, including the polar component (γp), are grouped into a single variable γlw. Forces due to acid–base interactions (hydrogen bonds) are denoted γab, which in turn is a combination of electron donor (γ−) and acceptor (γ+). Thus, the surface energy can be described as the sum of γlw + γab. The equation connecting these components to the contact angle is:
Equation (4) has three unknowns: \( \gamma_{s}^{lw} ,\gamma_{s}^{ + } \) and \( \gamma_{s}^{ - } \). To obtain \( \gamma_{s}^{lw} \), the Fowkes formula for an apolar probe (Eq. 5) is used, where the values of x and y defined above are equal to zero (Fowkes 1964).
Finally, \( \gamma_{s}^{ + } \) and \( \gamma_{s}^{ - } \) are calculated by solving a system of equations obtained by measurements with two polar liquids.
Several authors consider the acid–base approach to be the most reliable for the determination of surface energy and its components. However, as described in the literature, this approach is very sensitive to the numerical values of the components of the surface tension of the liquids used and of the contact angle measurements (Żenkiewicz 2007).
The solubility parameter δ is also used in the evaluation of the surface energy and its components for a solid material. For weakly polar materials, where only dispersion forces are significant, the theory of regular solutions developed by Hildelbrand can be applied (Hansen 1967). This theory is not valid for materials likely to contain hydrogen bonds, such as cellulose. To avoid this drawback, parametric models have been proposed with, in most cases, two- or three-dimensional graphic representations. Among these models, the Hansen model is used to determine the nature and type of interactions between different materials (Hansen 1967). According to Hansen, the density of the cohesion energy (δ) of a liquid includes all intermolecular forces: δd dispersion force, δp polarity force and δh hydrogen bond strength. By representing δd, δp and δh in a tridimensional system and doubling the axis of δd, all solvents that solubilize the polymer would be in a solubility sphere of radius RA. The solubility parameter of a polymer is represented by a point situated in the center of this sphere and its coordinates are (\( \delta_{{d_{0} }} ,\delta_{{p_{0} }} ,\delta_{{h_{0} }} \)).
Since the appearance of Hansen’s model, hundreds of articles have confirmed its validity. It has allowed the selection of the best solvent, swelling or wetting liquid for polar polymers, the replacement of a toxic solvent with a non-toxic liquid mixture, the determination of surface energy, the dispersibility of solid particles in a liquid medium, and the explanation of the adsorption and interactions of the many ingredients in a formulation (paints, cosmetics) (Belfkira and Montheard 1994).
Wetting and solvation of lignocellulosic fibers by organic liquids has been extensively studied. Mantanis et al. (1995) observed that liquids capable of establishing hydrogen bonds will wet lignocellulosic fibers better. Thode and Guide found a linear relationship between the solubility parameters of polar liquids, their retention and the increase in the volume of lignocellulosic fibers (Thode and Guide 1959). Philipp et al. concluded that three parameters influence the swelling of lignocellulosic fibers by organic solvents: (i) the hydrogen bonding fraction δh and the polar fraction δp and the synergetic effect between them, (ii) the solvent volume molar and (iii) the structure of cellulosic fibers, especially their width and pore distribution (Philipp et al. 2007).
A good wetting agent will be a pure liquid, or a mixture, having a solubility parameter with the same components as the fibers constituting the paper sheet. Thus, we were inspired by the Zisman model to determine the components (δd, δp and δh) of the best wetting agent by representing cosθ versus δi (with i = d, p and h). For this, we have chosen polyols (glycerin, ethylene glycol and propylene glycol), liquids that can establish strong interactions with cellulosic fibers (Thode and Guide 1959; Philipp et al. 2007). The point of intersection of each line corresponding to cosθ = 1 will allow to determine the coordinates of the best wetting liquid for each type of cellulosic fibers. From the values found graphically, we used the Hansen and Beerbower Eqs. (6) and (7) to determine the surface energy components (γd, γp and γh) as well as the total energy γ (Hansen and Beerbower 1971).
This work is devoted to the determination of contact angle, surface energy and its components (dispersive, polar and hydrogen bonding) of paper sheets using the sessile drop technique. The method used for pulping and fiber characterization and the effect of fiber size and shape on the physicochemical properties of paper sheets have been widely discussed in our previous paper (El Omari et al. 2017). In consequence, we will limit our study to the effect of roughness and porosity (before and after calendering), two characteristics of the surface of the sheet that have an effect on adsorption and absorption. More emphasis will be given to the determination of the contact angle and the surface energy and its components by the proposed method.
Materials and methods
Lignocellulosic fibers properties and handsheets preparation
Five different lignocellulosic fiber pulps were used in this study. Three were extracted from plants growing on Moroccan soil [Agave (Agave americana), Diss (Ampelodesmos mauritanius), and Typha (Typha latifolia)]. The two other samples were industrial pulps: bleached softwood (kraft) and thermomechanical (TMP) pulps provided by Northeastern Canada mills. In a previous work, we have detailed: (i) the pulping process, (ii) the analysis of these fibers by Fiber Quality Analyzer (FQA), and (iii) the determination of the degree of crystallinity by XRD (El Omari et al. 2017). In addition, this work contains the determination of pH on the five pulp samples. These measurements were made using the method described by Douglas J. Gardner (Segal et al. 1959): 1 g of freshly ground fiber was placed in 100 cm3 distilled water and mixed until complete wetting of the fiber. The pH was measured with a glass electrode pH-meter after a 5 min stabilization time.
The handsheet preparation method, the physical characterization of the paper samples (strength tests, porosity, and roughness) and the study of the effect of fiber morphology on each property have been widely discussed in our previous study. Scanning electron microscopy (SEM) images of the sheet structure were taken using a JSM-5500 system (JEOL Ltd.). All handsheets were calendered at a pressure of 450 PSI at 80 °C before measuring the contact angle.
Probes
Five model probes were used: α-bromonaphtalene (95.0% GC grade, Fluka Chemie GmbH), N,N-dimethylformamide (DMF, 99%, VWR Chemicals), glycerin (99% + , Acros Organics), ethylene glycol (99,5%, Sigma-Aldrich) and propylene glycol (99%, Panreac). All products were used as received. Measurements were done at room temperature.
Contact angle measurement
Contact angles were measured using a Krüss DSA1 (Software for Drop Shape Analysis) tensiometer. The device consists of a CCD camera, a horizontally moving sample holder, a vertical and horizontal displacement syringe holder and a diffuse light source. The solid sample was glued to the sample holder so that the surface is as flat as possible. A 4 μl volume of each probe was deposited on the surface using a micro-syringe. The image of the drop, assimilated to a spherical cap, was acquired by the camera, and processed by the instrument software to determine the value of the contact angle with Eq. (8):
where θ is the contact angle, h is the height of the drop and D is the length of the base of the drop. For each liquid, five measurements of the evolution of the angle as a function of time were carried out. The contact angle, determined by our method, is an average of these measurements.
Crystallinity index
The crystallinity of each sample was determined by X-ray diffraction using a Bruker AXS D8 Advance diffractometer with a scintillation detector and a cobalt tube, with the PANalytical X’Pert HighScore sofware. The crystallinity index of various cellulose fibers was evaluated according to the Segal method (Segal et al. 1959; French 2014).
Results and discussion
Characteristics of pulps and handsheets
A pulp is often characterized by its percentage of fines, the fiber mean length, pH and the degree of crystallinity. The values obtained with our samples are close to those found by other authors for annual and perennial plants (Table 1). The effect of fiber size and morphology, bleaching and fines on handsheet properties such as density, porosity, roughness, moisture adsorption and mechanical properties have been discussed before (El Omari et al. 2017).
The high speed of the evolution of the probe/handsheet contact angle with time is explained by the possibility of establishing strong interactions between the two materials (hydrogen bonds, dipoles). Indeed, pulping makes the surface hydroxyl groups accessible to polyhydroxylated molecules. In consequence, the high fines content, combined with high energy and surface area, will increase the draining properties (Vainio and Paulapuro 2007). The acidic pH of pulp samples indicates that fibers have been oxidized during the bleaching step creating carboxylic groups. The presence of polar groups (OH and COOH) and amorphous zones increases the hydrophilicity of the material. As it can also be seen in Table 2, the amount of water adsorbed by bleached fibers decreases as the degree of crystallinity increases because of the involvement of hydroxyl groups in the crystalline structure (Nakamura et al. 1981). Agave fibers, having the lowest crystallinity and the highest fines content, are the most absorbing (Lewin and Pearce 1998).
Also, fibers are subjected to various chemical and mechanical treatments during the preparation of handsheets. Consequently, substantial changes in fiber configuration and conformation, fiber chemical composition, fines content, formation, density and porosity of the sheet will occur. All these factors will in turn influence the topography and the amplitude of the roughness of the sheet surface and consequently, as it can be seen in Table 4, the contact angle.
In view of the results obtained, it is clear that despite the fact that plant fiber pulps have undergone the same treatment, paper sheets have different properties depending on the plant. However, commercial fibers do not follow the same trend. As expected, handsheets have a rough surface and a porous structure regardless of fiber origin. Differences between handsheets can be explained mainly by the distribution of fiber size in terms of length and thickness, the fines ratio, the degree of crystallinity and the conformability of fibers.
SEM images of handsheets (Fig. 1) show variations in the fiber shape which confirm the porosity and roughness of samples: straight or flattened, presence of curls and kinks, entanglements and empty spaces.

SEM images (×500) of calandered paper from different fiber of fibers: a Kraft, b Typha, c TMP, d Diss and e Agave
Paper surface energy
Results obtained by the sessile drop technique should be treated with caution. Indeed, most conditions required by this technique are not met by our samples. They are characterized by high porosity, rough surface, high polarity, and the use of polar probes. Handsheets are far from an isotropic material. Surface roughness and porosity are two parameters to be considered when analyzing and interpreting the surface energy data evaluated from contact angle measurements. Making measurements on highly polar handsheets, rough and porous fibers is very difficult. Therefore, calendering is necessary to attenuate roughness and porosity. The calendering operation, which consists in passing a sheet through the nip between smooth rolls at high pressure and temperature, strongly affect the smoothness of paper surfaces. Even after calendering, the porosity and roughness of handsheet surfaces remain high making them very difficult to study (Table 3).
In addition, the pulping process increases the availability of surface hydroxyl groups making fiber surfaces very energetic with respect to hydroxylated probes (Fig. 2). Thus, wetting angle measurements will be very difficult on this type of surface. Indeed, the speed of absorption will be very large requiring an ultra-fast measurement system.

Evolution of the contact angle of each probe as a function of time for each fiber type
The choice of the contact angle
For each probe and fiber type, the evolution of the angle θ with time is composed of two decreasing segments. Figure 3 shows an example of a typical curve: the glycerin probe on kraft pulp fibers. Theoretically, the surface energy of the material cannot be determined from this curve since each point is far from steady state (no balance). In fact, once deposited on the surface of the sample, the liquid spreads and penetrates into the material making it impossible to form a spherical cap.

Evolution of the contact angle of glycerin on the kraft pulp handsheet as a function of time
In this situation, which angle should be chosen to determine the surface energy? Three different options can be considered: (i) extrapolating the first segment (S1) to t → 0, i.e. when the drop contacts the surface of the handsheet; (ii) taking the intersection point (I) between the two linear segments (S1) and (S2) or (iii) extrapolating the second segment (S2) to t → 0.
During the first step, as soon as there is contact between the probe microdrop and the surface of the material, we estimate that there is contact only between probe molecules, water molecules adsorbed on the surface of the fibers, and the air trapped in the pores of the material (Fig. 4, First step).

Evolution of the angle of the probe drop as a function of time
Then, depending on the viscosity of the probe and the probe/water affinity, there will be a more or less rapid exchange between probe and water molecules and a displacement of the air in the pores of the material. This will result in a cross-diffusion of probe and water molecules adsorbed on the surface of fibers. This exchange will continue until probe molecules achieve close contact with functional groups covering the fibers (OH and COOH). We propose to use the contact angle value observed at this point for further calculations. After this point, probe molecules will only continue to adsorb on fiber surfaces and to migrate inside the fibrous network by wetting the underlying fibers. The extrapolation of the second segment (S2) to t → 0 makes it possible to obtain the most probable and representative contact angle. We consider that the intersection point (I) of the two segments represents a fairly advanced state of wetting, given the rather high absorption rate of the various selected probes.
Our proposition is confirmed by the evolution of the volume of the drop of each probe for each fiber type (Fig. 5). Here again, two decreasing segments, parallel to those of the contact angle against time curves, are observed. At the time corresponding to the intersection point of the two segments, there is a slight increase in volume (marked by a red circle in Fig. 5) followed by the onset of a decrease in volume corresponding to the beginning of the second segment. This phenomenon is recurring with all probes and for all fiber types. It can be explained by taking into account changes that fibers will undergo upon contact with the solvent: (i) an initial absorption of a part of the liquid which causes a decrease in the volume of the drop; (ii) after a few seconds, the first fibers having absorbed the probe see their volume increase, this increase is a very advanced wetting of the fibers; and then (iii) a final decrease in volume due to the wetting of the fibers underneath their wetted fraction. Hence, we propose to take the angle found by the extrapolation of the second segment to t → 0. Average angles (θ) for the different probes for each fiber types are listed in Table 4.

Evolution of the volume of the deposited drop as a function of time for Kraft and Agave fibers using glycerin and ethylene glycol probes
Calculation of the surface energy and its components by different models
For the determination of the surface energy and its components, we will use the two models that take into account both polarity forces and hydrogen bonds: Owens and Wendt (1969) and VOCG (Van Oss et al. 1988). Then, we present the results obtained by applying our model.
Owens and Wendt model
The dispersive and polar components of the surface energy were calculated with the linear form of the Owens and Wendt model (Eq. 3). The y versus x plot is shown in Fig. 6. The values obtained for γd and γp as well as the total surface energy γ are given in Table 5.

Determination of γd and γp using the Owens–Wendt plot
Considering the non-ideal nature of studied surfaces, the curves obtained are almost straight lines with correlation coefficients close to unity (Table 5). Values of γ are slightly higher than those of treated lignocellulosic fibers with Diss being the only exception (Peršin et al. 2004). This can be explained by the presence of larger amorphous zones in studied fibers. The low degree of crystallinity of Agave (highest energy) and the high crystallinity of kraft pulp fibers (lowest energy) are consistent with these results and those reported by other authors (Peršin et al. 2004). The γd and γp components are also in good agreement with our assumptions. The increase in amorphous zones is generally accompanied by an increase of the polar and a decrease of the dispersive component.
Van Oss, Chaudhury and Good (VOCG) model
The VOCG method requires a minimum of three liquid probes, since Eq. (5) contains three unknowns. In the present work glycerin, ethylene glycol and α-bromonaphthalene were used as test liquids of known energy components: surface tension, dispersive and acid–base components (Wu et al. 1995; Hansen 2000; Moutinho et al. 2007). To solve Eq. (5), \( \gamma_{s}^{lw} \) was calculated with Eq. (4) using the cosθ value found with α-bromonaphthalene (apolar). The values of surface tension components and parameters are found in Table 6.
It is very interesting to note the similarity of our values with those found by other authors using other contact angle and IGC techniques (Shen et al. 1999; Kontogeorgis and Kiil 2016). This shows the relevance of the method we proposed for selecting the contact angle value (Table 7).
Hybrid model
Table 8 contains the value of the solubility parameter of each solvent used, as well as their components (δd: dispersion force, δp: polarity force and δh: hydrogen bond strength) according to Hansen. Figure 7 shows the plot of cos θ against each component of the Hansen solubility parameter (δd, δp and δh) for each probe. These plots are used to determine the coordinates of the best wetting liquid for each fiber type.

Plot of cosθ against solubility parameter components: δd, δp, δh and δΤ
The examination of Fig. 7 shows that there are some straight lines with acceptable correlation coefficients (R2), especially for the representations of δp and δh. Intersects with cosθ = 1 and the correlation coefficients are given in Table 9. Values found by other authors as well as those of the best solvents for cellulosic fibers are also presented.
Results in Table 9 are in good agreement with the literature (Kontogeorgis and Kiil 2016). Discrepancies between the results are not surprising, the opposite would be the case, for well known reasons like the nature of matrices and instrumental uncertainty. However, it can be noted that values found here are almost equal to those of the best solvents for cellulose, [BMIM]PF6, [OMIM]PF6 and NMNO, as well as their components.
Surface energy components were calculated using the Hansen–Berower Eqs. (6) and (7). The molar volume was calculated according to the method of Fedors (1974). This method was already used to calculate the solubility parameters of chitin, another type of polysaccharide (Ravindra et al. 1998). The value of the correction factor l is, on average, equal to 0.8 for many homologous series of compounds. It is interesting to note that the values of γ, determined by our model, are close to those found with other models (Table 10). However, there is a large decrease in dispersion (γd) at the expense of hydrogen bonding (γh) forces. Given the strongly hydroxylated nature of the matrices (bleached lignocellulosic fibers), we believe that the values found reflect a certain reality.
The analysis of the results grouped in Table 11, obtained by the various models including the one proposed here, shows that globally the values found for the total surface energy are close. These results are sufficiently promising to investigate further our approach by the determination of the contact angle and the application of the Zisman–Hansen combined model to determine the surface energy and its components.
Conclusions
Difficulties encountered in the determination of surface energy by the contact angle technique are essentially due to the non-equilibrium state often achieved when wetting a support with liquids. The use of an apparatus allowing the capture of images showing the evolution of the callote of the wetting liquid as a function of time and the choice of suitable probes are essential. The proposed method to select the angle corresponding closely to the state of contact is relevant. Surface energy values, and their components, determined by our model (Zisman–Hansen) are in good agreement with those found by other authors using other techniques.
References
Akinli-Kocak S (1997) The influence of fiber swelling on paper wetting. Ankara University, Ankara
Belfkira A, Montheard J-P (1994) Solubility parameters of poly(4-substituted α-acetoxystyrenes) and alternating copolymers of vinylidene cyanide with substituted styrenes. J Appl Polym Sci 51:1849–1859. https://doi.org/10.1002/app.1994.070511102
Chaiarrekij S, Apirakchaiskul A, Suvarnakich K, Kiatkamjornwong S (2011) Kapok I: characteristics of kapok fiber as a potential pulp source for papermaking. BioResources 7:0475–0488. https://doi.org/10.15376/biores.7.1.0475-0488
Cordeiro N, Gouveia C, Moraes AGO, Amico SC (2011) Natural fibers characterization by inverse gas chromatography. Carbohydr Polym 84:110–117. https://doi.org/10.1016/j.carbpol.2010.11.008
El Omari H, Belfkira A, Brouillette F (2017) Paper properties of typha latifolia, pennisetum alopecuroides, and agave americana fibers and their effect as a substitute for kraft pulp fibers. J Nat Fibers 14:426–436. https://doi.org/10.1080/15440478.2016.1212766
Fedors RF (1974) A method for estimating both the solubility parameters and molar volumes of liquids. Polym Eng Sci 14:147–154. https://doi.org/10.1002/pen.760140211
Fowkes FM (1964) Attractive forces at interfaces. Ind Eng Chem 56:40–52. https://doi.org/10.1021/ie50660a008
Fox H, Zisman W (1950) The spreading of liquids on low energy surfaces. I. Polytetrafluoroethylene. J Colloid Sci 5:514–531. https://doi.org/10.1016/0095-8522(50)90044-4
French AD (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21:885–896. https://doi.org/10.1007/s10570-013-0030-4
Gamelas JAF (2013) The surface properties of cellulose and lignocellulosic materials assessed by inverse gas chromatography: a review. Cellulose 20:2675–2693. https://doi.org/10.1007/s10570-013-0066-5
Gellerstedt F, Gatenholm P (1999) Surface properties of lignocellulosic fibers bearing carboxylic groups. Cellulose 6:103–121. https://doi.org/10.1023/A:1009239225050
Gómez C, Zuluaga R, Putaux J-L et al (2012) Surface free energy of films of alkali-treated cellulose microfibrils from banana rachis. Compos Interfaces 19:29–37. https://doi.org/10.1080/09276440.2012.687978
Guo C, Zhou L, Lv J (2013) Effects of expandable graphite and modified ammonium polyphosphate on the flame-retardant and mechanical properties of wood flour-polypropylene composites. Polym Polym Compos 21:449–456. https://doi.org/10.1177/096739111302100706
Hansen CM (1967) The dimensional solubility parameter and solvent diffusion coefficient. Danish Technical Press, Copenhagen
Hansen CM (2000) Hansen solubility parameters
Hansen CM (2007) Hansen solubility parameters: a user’s handbook. CRC Press, Boca Raton
Hansen CM, Beerbower A (1971) Solubility parameters. Kirk-Othmer Encyclopedia of Chemical Technology, New York
Hubbe MA, Gardner DJ, Shen W (2015) Contact angles and wettability of cellulosic surfaces: a review of proposed mechanisms and test strategies. BioResources 10:8657–8749. https://doi.org/10.15376/biores.10.4.Hubbe_Gardner_Shen
Jacob PN, Berg JC (1994) Microcrystalline cellulose and two wood pulp fiber. Adsorpt J Int Adsorpt Soc 49:3086–3093
Kontogeorgis GM, Kiil S (2016) Introduction to applied colloid and surface chemistry. Wiley, Chichester
Lewin M, Pearce EM (1998) Handbook of fiber chemistry. Marcel Dekker, New York
Mantanis GI, Young RA, Rowell RM (1995) Swelling of compressed cellulose fiber webs in organic liquids. Cellulose 2:1–22. https://doi.org/10.1007/bf00812768
Marmur A (2006) Soft contact: measurement and interpretation of contact angles. Soft Matter 2:12–17. https://doi.org/10.1039/B514811C
Ministère des Affaires Sociales et de la santé (2013) Stratégie Nationale de Santé—Feuille de route. In: Ministère des Aff Soc la santé
Mirvakili MN (2011) Superhydrophobic fibre networks loaded with functionalized fillers. University of British Columbia
Moutinho I, Figueiredo M, Ferreira P (2007) Evaluating the surface energy of laboratory-made paper sheets by contact angle measurements. Peer-reviewed Handsheets 6:26–32
Nakamura K, Hatakeyama T, Hatakeyama H (1981) Studies on bound water of cellulose by differential scanning calorimetry. Text Res J 51:607–613. https://doi.org/10.1177/004051758105100909
Nosonovsky M, Bhushan B (2007) Lotus effect: roughness-induced superhydrophobicity. Springer, Berlin, pp 1–40
Owens DK, Wendt RC (1969) Estimation of the surface free energy of polymers. J Appl Polym Sci 13:1741–1747. https://doi.org/10.1002/app.1969.070130815
Papirer E, Brendle E, Balard H, Vergelati C (2000) Inverse gas chromatography investigation of the surface properties of cellulose. J Adhes Sci Technol 14:321–337. https://doi.org/10.1163/156856100742627
Peršin Z, Stana-Kleinschek K, Sfiligoj-Smole M et al (2004) Determining the surface free energy of cellulose materials with the powder contact angle method. Text Res J 74:55–62. https://doi.org/10.1177/004051750407400110
Philipp B, Schleicher H, Wagenknecht W (2007) The influence of cellulose structure on the swelling of cellulose in organic liquids. J Polym Sci Polym Symp 42:1531–1543. https://doi.org/10.1002/polc.5070420356
Quéré D (2008) Wetting and roughness. Annu Rev Mater Res 38:71–99. https://doi.org/10.1146/annurev.matsci.38.060407.132434
Ravindra R, Krovvidi KR, Khan AA (1998) Solubility parameter of chitin and chitosan. Carbohydr Polym 36:121–127. https://doi.org/10.1016/S0144-8617(98)00020-4
Rinaldi R, Reece J (2013) Solution-based deconstruction of (Ligno)-cellulose. Wiley, Aachen, pp 435–462
Segal L, Creely JJ, Martin AE, Conrad CM (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the x-ray diffractometer. Text Res J 29:786–794. https://doi.org/10.1177/004051755902901003
Shen W, Sheng YJ, Parker IH (1999) Comparison of the surface energetics data of eucalypt fibers and some polymers obtained by contact angle and inverse gas chromatography methods. J Adhes Sci Technol 13:887–901. https://doi.org/10.1163/156856199X00730
Steele DF, Moreton RC, Staniforth JN et al (2008) Surface energy of microcrystalline cellulose determined by capillary intrusion and inverse gas chromatography. AAPS J 10:494–503. https://doi.org/10.1208/s12248-008-9057-0
Thode E, Guide R (1959) A thermodynamic interpretation of the swelling of cellulose in organic liquids. Tappi J 42:35–39
Tze WT, Gardner DJ (2001) Contact angle and IGC measurements for probing surface-chemical changes in the recycling of wood pulp fibers. J Adhes Sci Technol 15:223–241. https://doi.org/10.1163/156856101743427
Vainio AY (2007) Interfibre bonding and fibre segment activation in paper observations on the phenomena and their influence on paper strength properties. Helsinki University of Technology
Vainio A, Paulapuro H (2007) Interfiber bonding and fiber segment activation in paper. BioResources 2:442–458. https://doi.org/10.15376/biores.2.3.442-458
Van Oss CJ, Chaudhury MK, Good RJ (1988) Interfacial Lifshitz-van der Waals and polar interactions in macroscopic systems. Chem Rev 88:927–941. https://doi.org/10.1021/cr00088a006
Wågberg L (2009) Paper chemistry and technology. Walter de Gruyter, Berlin
Wu W, Giese RFJ, van Oss CJ (1995) Evaluation of the Lifshitz-van der Waals/acid-base approach to determine surface tension components. Langmuir 11:379–382. https://doi.org/10.1021/la00001a064
Young T (1805) An essay on the cohesion of fluids. Philo Trans R Soc Lond 95:65–87. https://doi.org/10.1098/rstl.1805.0005
Żenkiewicz M (2007) Methods for the calculation of surface free energy of solids. J Achiev Mater 24:137–145
Zisman WA (1964) Relation of the equilibrium contact angle to liquid and solid constitution, pp 1–51
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
El Omari, H., Ablouh, Eh., Brouillette, F. et al. New method for determining paper surface energy per contact angle. Cellulose 26, 9295–9309 (2019). https://doi.org/10.1007/s10570-019-02695-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-019-02695-4