
Abstract
Sugarcane bagasse was selected as residual feedstock to investigate the effects of an acid/peroxide-alkali (APA) combined pretreatment on the extraction of cellulose and on its applicability for producing cellulose acetate. The pretreatment was effective in disrupting the recalcitrant lignocellulose structure and isolating cellulose (SBC), which reached 85.67% purity. High performance liquid chromatography, 1H and 13C nuclear magnetic resonance, Fourier transform infrared spectroscopy, thermogravimetry, calorimetry, X-ray diffractometry (XRD), scanning electron microscopy and elemental analysis were applied to characterize the materials. Crystallinity and accessibility were evaluated by Segal and deconvolution crystallinity indexes, full-width at half maximum, crystallite size (L), total crystalline index and lateral order index parameters. SBC cellulose presented lower crystallinity and was classified after XRD peak deconvolution as a mixture of cellulose II polymorph with considerable amorphicity and some features of remaining cellulose Iβ from sugarcane bagasse, while CC was labeled type Iβ, indicating that the sustainable material is more susceptible to chemical modifications. The degrees of substitution of acetylation products showed that both celluloses have satisfactory reactivity and reached mono, di and triacetylation in different reaction conditions. SBC cellulose is presented as an efficient and renewable alternative to commercial cellulose for producing chemicals, and it is proposed that the APA pretreatment had a significant influence on the low crystallinity and high accessibility of this material.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lignocellulosic biomass is a renewable, abundant and widely distributed source composed of cellulose, lignin and hemicellulose, which may replace petroleum derivates in the production of some petrochemical-based products and is the main raw material used in biorefineries (Wright et al. 2006). Therefore, this sustainable material can be applied to generate power, fuels, chemicals and new materials (Diep et al. 2012). The main sources used in the studies of lignocellulose are wood, cotton, jute, hemp, corn, flasks, rice, wheat straw, sisal and sugarcane bagasse (Lavanya et al. 2011).
Cellulose is a polymer whose repeating unities (the monomers) consist of two anhydroglucose rings linked by a β-1,4 glycosidic bonding (Ciolacu et al. 2012). Its fibers are composed of crystalline regions where cellulose chains are aligned parallel to the axis, along with amorphous regions where cellulose chains are unordered. Cellulose is one of the most abundant materials on earth and it is found as cellulose I (parallel chains) form naturally, while cellulose II (antiparallel chains) is found after chemical modifications (Cheng et al. 2011). Recently, lignocellulosic residues are being investigated as cellulose sources in order to reduce industrial costs when compared to the use of other raw materials (El Nemr et al. 2015; Fan et al. 2013). Sugarcane bagasse stands out as an abundant, eco-friendly and low-cost cellulosic source, since the sugar and alcohol industry generates a huge amount of this residue. Although some of this bagasse is burned to produce energy for the sugarcane mills, a significant amount is still not used (Cerqueira et al. 2007). Moreover, bagasse is composed of 20–30% lignin, 40–45% cellulose and 30–35% hemicellulose (Cardona et al. 2010), being substances that may be used as raw materials for biorefinery.
Researchers have recently focused on the fractionation of residual biomass from sugarcane bagasse (Kalogiannis et al. 2015), where the main problem is the very low reactivity of cellulose stemming from its complex and rigid crystalline structure. Cellulose pretreatment is necessary, and various techniques to disrupt the recalcitrant lignocellulose structure have been reported to separate the cellulose, lignin and hemicellulose fractions. Some examples are steam explosion (Vochozka et al. 2016), acid pretreatment (Kumar et al. 2014), ionic liquid (Endo et al. 2017), organosolv (Kautto et al. 2013), acid/peroxide-alkali process (Assumpção et al. 2016) and soda/anthraquinone (soda/AQ) pulping (Jahan et al. 2015). The combined method using acid and alkaline peroxide is an emerging technique and a potential candidate for improving pretreatment of lignocellulose. The diluted sulfuric acid is advantageous for removing the hemicellulosic fraction with low degradation of cellulose (Chandel et al. 2014); the sodium hydroxide (Ramadoss and Muthukumar 2015) and hydrogen peroxide (Testova et al. 2014), respectively, present selectivity for the removal and oxidation of lignin, thus reducing the formation of solid residues (Rabelo et al. 2014). These fractions can be separately converted into industrial derivatives such as cellulose acetate (Cerqueira et al. 2007), ethanol (Kautto et al. 2013), carboxymethyl cellulose (Singh and Singh 2013) and 5-hydroxymethylfurfural (Wei and Wu 2017), among several other value-added chemicals.
Instrumental analysis such as the high performance liquid chromatography (HPLC), nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy (FTIR) are performed in order to determine the purity of the cellulose (Brodeur-Campbell et al. 2012; Popescu et al. 2012). A methodology developed by the National Renewable Energy Laboratory (NREL) and optimized by the National Bioethanol Science and Technology Laboratory (Laboratório Nacional de Ciência e Tecnologia do Bioetanol—CTBE) involves HPLC analysis, among other techniques, in order to infer the detailed chemical composition of sugarcane bagasse and straw by determining monomers and other products concentrations in the hydrolyzed sample (Rocha et al. 2014). Moreover, some techniques allow for calculating the crystallinity index (CrI) of cellulose, of which the X-ray diffractometry (XRD) is the most simple and available (Agarwal et al. 2017).
The crystalline cellulose fraction is insoluble in most traditional organic solvents and presents low reactivity due to the chain alignment (Endo et al. 2017). In order to increase its applicability, an alternative path is chemical conversion and incorporating chemical groups into the hydroxyl groups of the glycosidic unit. A chemical modification of cellulose is typically obtained by acetylation reaction, replacing the hydrogens of the hydroxyl groups with acetyl substituents to produce cellulose acetate (Cerqueira et al. 2007). This product is widely applied in many industrial fields such as oil absorption, metal adsorption, matrices for controlled incorporation of drugs, cigarette filters and raw materials for the textile industry (Rodrigues Filho et al. 2000).
This work aimed to investigate the effects of an acid/peroxide-alkali (APA) combined pretreatment on sugarcane bagasse (SB), followed by optimized soda/AQ pulping and bleaching processes on isolating the cellulosic fraction and on the characteristics that affect the accessibility of the obtained sugarcane bagasse cellulose, named SBC. Microcrystalline Avicel commercial cellulose (CC) was used as a model cellulosic material. The reactivity of SBC and CC celluloses were evaluated in terms of accessibility and acetylation susceptibility, as observed by the obtained acetylation degrees under several reaction conditions. Elemental analysis (CHNS), thermogravimetry and calorimetry (TG/DTG/DSC), scanning electron microscopy (SEM), NMR, FTIR, HPLC and XRD were applied to identify the properties of cellulose and cellulose acetates, thus elucidating experimental results using purity and crystallinity index data. HPLC analysis was performed on the hydrolyzed cellulosic samples, with their concentrations of sugar, acids and other substances being inferred and mathematically correlated with mass percentages of cellulose, hemicellulose and lignin (NREL/CTBE methodology). XRD parameters such as crystallinity index by Segal and deconvolution methods, full-width at half maximum (FWHM) and crystallite size (L), as well as FTIR ratios of total crystalline index (TCI) and lateral order index (LOI) were determined to evaluate SBC crystallinity and accessibility. This information is crucial to discuss the reactivity and applicability of the cellulose isolated in this study.
Materials and methods
Feedstock data
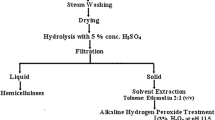
Microcrystalline cellulose Avicel PH-101 (CC) was purchased from Sigma-Aldrich®. The sugarcane bagasse (SB) used in this work was kindly donated by LCD Bioenergia SA, a subsidiary of the Estivas sugar plant (Ares/RN, Brazil). Drying was performed at 100 °C for 24 h. A knife mill (Willy TE-648 – TECNAL) was used to grind the dried biomass to an appropriate range of sizes. A screening apparatus (BERTEL) was used in the particle size differentiation. The particle size used in these experiments was between 42 and 60 mesh (Tyler Mesh). A simplified process flow diagram for the overall methodology described in this work is found below in Fig. 1.

Process flow diagram of this study
Isolation of SBC cellulose by APA combined pretreatment
After washing and drying at 100 °C for 3 h to constant weight the in natura SB, 20 g of the material were added into a flask with 1.45% (v/v) H2SO4 solution in 1:10 (w/v) ratio (fiber:liquor), at 105 °C for 75 min of constant stirring in a 802 D Fisatom rotary evaporator. Then, the bagasse was transferred to another flask with 7.5% (v/v) H2O2 in 1:20 (w/v) ratio, at 80 °C for more 3 h of stirring. During this second step, the pH was adjusted to 11.5 by adding a 4.0% (v/v) NaOH solution. Finally, the remaining solid fraction was washed until neutrality with distilled water and dried at 100 °C in a stove until its weight was stabilized. After this procedure approximately 10 g of pretreated bagasse (PB) was obtained (54.48% mass yield).
Next, the pulping with soda/AQ was performed in a Teflon covered reactor with stainless steel autoclave (Phoenix) containing 10 g of pretreated bagasse and an alkaline load solution with 10% (w/v) NaOH and 0.1% (w/v) AQ in 1:13 ratio (w/v) (fiber:liquor), heated for 1 h at 160 °C. After the reaction time, the cellulosic pulp was filtered and washed with 80 mL of NaOH 1% (w/v) aqueous solution, obtaining 45.45% mass yield. The pure SBC cellulose was obtained after the bleaching step of the cellulosic pulp using a methodology where crude pulp and distilled water were mixed at a 1:40 (w/v) ratio under stirring at 70 °C. The product was treated with sodium chlorite (NaClO2) and glacial acetic acid under stirring and heating for 1 h. The SBC obtained after cooling was then filtered and washed up to neutral pH. Finally, the solid material was dried at 80 °C to a constant weight (41.51% mass yield).
Chemical composition analysis
Sample composition analyses were performed in accordance with NREL/TP-510-42618 procedure, adapted by CTBE (Rocha et al. 2014). Initially, the samples were hydrolyzed with 72% H2SO4, at 45 °C for 7 min. Afterwards, the acid was diluted to 4% (w/w) with distilled water, and the mixture was autoclaved for 1 h at 121 °C. The residual material was cooled and filtered. Solids were dried to constant weight at 105 °C and classified as insoluble lignin. Soluble lignin was determined by measuring the absorbance of the solution at 280 nm, using UV spectroscopy (Agilent CARY 60) (Sluiter et al. 2016). Total lignin was calculated as the sum of insoluble and soluble lignin.
Cellulose and hemicellulose contents were calculated as a weighted sum between the concentrations of specific hydrolysis products such as sugars and organic acids. Celobiose, glucose, xylose and arabinose sugars, besides acetic, galacturonic and formic acids quantification was performed using a Chromaster 600 Merck-Hitachi HPLC with refractive index detector (RI 5450- Hitachi) on an Aminex® column (HPX-87H, Bio-Rad, USA), with 0.5 mL/min flow rate and the oven at 35 °C, using H2SO4 (5 mM) mobile phase. Other substances such as hydroxymethylfurfural (HMF) and furfural are also formed by the subsequent degradation of sugar monomers (Gouveia et al. 2009). The quantification of these products was performed by the same HPLC cromatograph coupled with UV–VIS detector (UV-5410 – Hitachi) on a C18 column (RP-18 Purospher® STAR, Merck, USA) with 0.8 mL/min flow rate and the oven at 35 °C, using acetonitrile:water 1:8 (v/v) mobile phase. All HPLC analysis were performed in triplicate. Calibration data, sugar content and equations used to calculate the mass content of lignocellulosic fractions are described in the Online Resource 1.
Synthesis and degree of substitution (DS) of cellulose acetates
Pure SBC and CC were used as raw material for the synthesis of acetates. The reaction system was composed of 2 g of cellulose, 40 mL of glacial acetic acid and a solution of 0.3 mL of concentrated H2SO4 and 17.5 mL of acetic acid. The cellulose-acetic acid system was initially stirred for 30 min at 25 °C, and then for 15 more min when the solution was added. The mixture was filtered under vacuum and 40 mL of acetic anhydride was added to the filtrate. This solution was placed back into the recipient containing cellulose and stirred for 30 min in order to obtain a homogenous system. After this time, the reaction mixture was kept for different time intervals (8, 12, 16, 20 and 24 h) and temperatures (25 and 50 °C) for evaluating the influence of these variables in the acetylation process. Water was added in each system to stop the reaction and to precipitate the cellulose acetate. The cellulose acetates were washed with distilled water up to a neutral pH and dried at 45 °C.
The acetylation degree, or DS, was used to discuss the accessibility of celluloses and to characterize the cellulose acetates in mono, di or triacetates. DS is the average value of acetyl groups (AG) which replaced the hydroxyl groups in the glycosidic units (Puleo et al. 1989). The DS was determined by a saponification reaction by adding 5 mL of NaOH (0.25 mol L−1) and 5 mL of ethanol to about 0.1 g of cellulose acetate. This mixture was kept for 24 h. Next, 10 mL of HCl (0.25 mol L−1) was added to the system, which was left undisturbed for 30 more min. Finally, the mixture was titrated using a standard 0.25 mol L−1 NaOH solution and phenolphthalein as indicator. This procedure was repeated in triplicate and the average values were used in Eqs. 5 and 6, respectively, to calculate the percentage of AG and the DS of CCA (from CC) and BCA (from SBC) cellulose acetates (Samios et al. 1997; Daud and Djuned 2015).
where %AG is the percentage of acetyl groups; A is the volume of NaOH (mL) added to the sample; B is the volume of NaOH (mL) added to the control; C is the volume of HCl (mL) added to the sample; D is the volume of HCl (mL) added to the control; Na is the HCl normality; Nb is the NaOH normality; W is the weight of cellulose acetate sample (g) and DS is the degree of substitution. Solubility tests were used to confirm the classification of the produced cellulose acetates.
Material characterizations
The samples were dried in a stove at 40 °C to constant weight before all characterizations performed in this research. The FTIR spectra were recorded using a Spectrum 65 FTIR spectrometer with a universal attenuated total reflectance (ATR) sampling accessory (Perkin Elmer). The samples were blended with KBr at 2% (w/w) concentration and the obtained material was hydraulically pressed for producing pellets. Twelve scans were carried out for each pellet of cellulose and cellulose acetate in the 650–4000 cm−1 range.
The thermal properties were analyzed by thermogravimetry (TG), derivative thermogravimetry (DTG) and differential scanning calorimetry (DSC) tests performed in SDTQ 600 equipment (TA Instruments) in a simultaneous analysis (TG/DTG/DSC). Five milligrams of each sample were placed in a 90 μL alumina crucible and heated from 25 to 600 °C at a rate of 20 °C min−1 under 100 mL min−1 nitrogen flow. Materials morphology was investigated using a Shimadzu SSX-550 scanning electron microscope (SEM). The samples were fixed in a support with a double-sided adhesive carbon tape and submitted to metallic coating with gold before analysis.
The 1D and 2D NMR spectra were acquired in an Avance DRX-500 spectrometer (Bruker), using DMSO-d6 as solvent at 343 K. All standard pulse sequences were provided by TopSpin software (Bruker). All samples (20 mg) were dissolved in 0.5 mL of DMSO-d6 and inserted into 5 mm NMR tubes. The 1H and 13C chemical shifts were expressed in δ scale and were referenced to residual DMSO-d6 at δ 2.49 for proton and at δ 39.5 for carbon. The scalar coupling constants are reported in hertz. The DS was calculated by Eq. 3, as described by Goodlett et al. (1971).
where ICH3,H is the integration of the resonances assigned to the three methyl protons of acetyl group (δ ~ 1.6–2.3 ppm) and IAGU,H is the integration of the resonances assigned to the seven protons in the glycose ring (δ ~ 2.8–5.5 ppm, except the residual water signal at around 3.2 ppm). The distribution of acetyl unities among the three OH groups in the cellulose acetate chains was calculated by integrating the spectra of a cellulose ester solution in DMSO-d6 from the 13C NMR spectra.
Crystallinity of samples was determined by one-dimensional wide-angle X-ray diffraction (XRD) patterns in a 1°–60° 2θ range and 2° min−1 rate. The patterns were obtained using a Bragg–Brentano geometry X-ray diffractometer XRD-6000 (Shimadzu) with CuKα radiation (λ = 1.5406 Å), 30 kV filament, 30 mA current and Ni filter. Deconvolution was performed with PeakFit 4.12 software (Systat Software, Inc.) assuming Lorentzian functions for each characteristic peak of the samples. Moreover, broad bands at around 21° 2θ were considered as the amorphous contributions to peak broadening, according to Park et al. (2010) paper (except for BCA). Iterations were repeated until high F numbers (> 10,000) and R2 values (> 0.993) were obtained without overfitting the models. The deconvolutions for CC, SB, PB and BCA samples are depicted in the Online Resource 2. The crystallinity index by deconvolution method (CrId) was determined for the celluloses and the lignocellulosic feedstock by Eq. 4 (Park et al. 2010):
where Acryst is the sum of deconvoluted crystalline band areas and Atotal is the total area under the diffractogram.
Crystallinity index following the method proposed by Segal et al. (1959) (CrIs) was calculated as shown in Eq. 5:
where It represents the maximum intensity of the crystalline reflection at ~ 22.7° 2θ for cellulose I and ~ 21.7° 2θ for cellulose II, and Ia is the diffraction intensity of the amorphous band at 18° 2θ for cellulose I and 16° 2θ for cellulose II (Nam et al., 2016).
The full-width at half maximum (FWHM) and crystalline peak position for the (200) lattice plane of cellulose I and the (020) lattice plane of cellulose II were inferred using XRD data plotted with PeakFit 4.12 software. The crystallite size L (nm), was calculated by the Scherrer equation (Scherrer, 1918):
where K is the correction factor (0.9), λ is the wavelength of the X-ray radiation (1.54056 A), β is the FWHM of the diffraction peak in radians, and θ is the half of the Bragg diffraction angle in radians.
Elemental chemical analysis was performed with a LECO CHNS 932 analyzer to determine the carbon, hydrogen, nitrogen, sulfur and oxygen contents present in the materials through the combustion of 2 mg of sample placed in a silver capsule at 1100 °C in pure oxygen.
Results and discussion
Composition analysis by HPLC
Components mass percentages were measured for bagasse, celluloses and the cellulosic pulp which are summarized in Table 1, along with the estimated compositions of lignin, cellulose and hemicellulose. The yield of substances was seen to vary according to the use of pretreatment in the biomass. The total sum of lignocellulosic fractions is below 100% due to the content of extractives and the cumulative errors in the characterization process. The mass percentage of cellulose in the pretreated bagasse increases in comparison with the untreated material due to the decrease in Klason lignin and hemicellulose fractions, showing that the APA combined pretreatment is efficient for removing the recalcitrant barrier and exposing the cellulose structure, thus making it more susceptible to the chemical attack promoted by the hydrolysis. Therefore, it is proposed that this procedure is going to increase the yield of extracted cellulose in the subsequent pulping and bleaching processes.
Moreover, SBC presented lower concentration of hemicellulose than CC, and the percentage of cellulose in the bagasse cellulose is higher than that in SB and cellulosic pulp. The results indicate that due to the high content of cellulose present in the sugarcane bagasse, it can be used to obtain the cellulose acetate. This sustainable cellulosic source provides cellulose with purity close to that of the commercial material. However, the quality and applicability of cellulose is not only associated to its cellulosic yield, but also to the susceptibility of its chains to chemical modifications for the production of value-added products, which is a factor mainly related to the crystalline structure (Endo et al. 2017).
Cellulose assessment
The micrographs of the materials used in this work are associated with the respective XRD spectra in Fig. 2 in order to evaluate the effects of the APA combined pretreatment on the morphology and crystallinity of SB bagasse and its SBC cellulose and BCA cellulose acetate derivates.

SEM micrographs for in natura sugarcane bagasse (a), pretreated bagasse (b), sugarcane bagasse cellulose (c) and bagasse cellulose acetate (d) samples
In natura SB bagasse micrograph presented a typical structure for this material, in which a well-organized vegetable structure with smooth, compact fibers with no rupture is observed, as described in the literature (Corrales et al. 2012). After the combined acid/alkali pretreatment, PB presented two types of modifications: the opening of cracks or channels and the formation of a stratified cell wall. The various layers formed indicate a fiber with greater surface area, while the cracks characterize greater porosity, contributing to greater physical disorganization of the fibers (Rezende et al. 2011). This behavior could improve the reactivity of this material during a chemical attack and the subsequent extraction of cellulose (Assumpção et al. 2016). Then, the SBC cellulose obtained after soda/AQ pulping and bleaching processes showed an organized fibrillar structure. After the acetylation reaction, BCA acetate presents fine and short fibers of different lengths, as disclosed by Abdelwahab et al. (2015), and unorganized structure with network arrangements, forming a spongy material.
The X-ray diffractograms for the cellulosic materials shown in Fig. 3, associated to CrId, CrIs, FWHM and L parameters obtained from its XRD patterns depicted in Table 2, indicate high amorphicity of the SBC cellulose extracted in this work.

XRD spectra and Segal CrI parameters for the cellulosic materials
The deconvoluted XRD pattern of SBC (Fig. 4) reveals a mixture of two cellulose polymorphs: the predominant cellulose II and remaining cellulose Iβ from in natura sugarcane bagasse. Moreover, unlike what is seen in an ideal cellulose II XRD pattern (French 2014), the cellulose Iβ (200) peak contribution makes the cellulose II (020) peak look more intense than the (110) peak, thus being an indicative for the existence of a mixture of polymorphs. The native crystalline structure of cellulose I found in biomass was changed due to the chemical attacks of the extraction and purification steps (Cheng et al. 2011), and SBC presented mainly characteristic cellulose II polymorph crystallographic planes (1–10), (110) and (020) at 12.3°, 20.3° and 21.9° 2θ, respectively, which is more reactive than cellulose I (French 2014; Ciolacu and Popa 2010). The deviation at around 37.5° 2θ can be related to some interference due to the sample preparation process before analysis.

Deconvolution of SBC cellulose XRD pattern
According to French and Santiago Cintrón (2013), the Segal CrI method by peak height measurement cannot be applied to mixtures of I and II since the crystalline peaks (1–10) and (110) of polymorph I are superposed to the 16° 2θ, and the (110) cellulose II peak is close to the ~ 18° 2θ point for the amorphous cellulose I intensity. Thus, the CrI deconvolution method (CrId) is more adequate for these calculations, considering crystalline and amorphous contributions for both polymorphs and producing a more accurate crystallinity evaluation of chemically treated renewable celluloses.
The results of CrI and the broad band at 21.1° 2θ related to amorphous phase contribution (Agarwal et al. 2010; Park et al. 2010), confirm its low crystallinity in comparison to CC, which presented higher crystallinity indexes and crystallite size, as well as XRD patterns corresponding to the crystal structure planes (1–10), (110) and (200) of cellulose Iβ with peaks at 14.1°, 15.7° and a sharp peak at 21.9° 2θ seen in Fig. 3 (French 2014). As described in literature, the peaks at ~ 14° and 16° often cannot be dissociated due to the presence of the amorphous phase in a typically crystalline material (El Oudiani et al. 2011). Cellulose accessibility usually follows the trend amorphous > II > I (Ciolacu and Popa 2010), therefore SBC could have the highest accessibility based on its lower crystallinity (Table 2). It is proposed that the chemical attack promoted by the APA pretreatment system is able to interfere in the polymer crystalline structure and make the produced SBC more amorphous
SBC and CC celluloses presented deviations from the characteristic crystalline peak position for celluloses I (~ 21.7° 2θ) and II (~ 22.7° 2θ) reported in literature (Nam et al. 2016). This displacement occurs since both celluloses – and the other cellulosic materials evaluated in this work—are not 100% pure (see Table 1), and not only the crystallite size and conformation are responsible for these results, but the overall contribution of species contained in the samples—hemicellulose, lignin, amorphous cellulose—influences the XRD parameters (Agarwal et al. 2016; Carrillo et al. 2018). Furthermore, some studies in literature suggest that FWHM is inversely related to CrI and crystallite size (L) (Nam et al. 2016), but also can be taken as a measure of the disorder of cellulose chains in a sample, considered as the degree of lateral order (DOLO) (Agarwal et al. 2016), meaning that the other non-crystalline aggregates can affect cellulose diffraction data.
XRD parameters (crystallinity indexes, FWHM and L) presented in Table 2 also depict the increasing crystallinity of sugarcane bagasse after the APA pretreatment (PB) due to the removal of recalcitrant hemicellulose and lignin fractions with exposure of cellulosic chains. These data were applied to evaluate the efficiency of the pretreatment by comparing SB and PB, since the diffractogram parameters of these materials may present several interferences, and therefore are not comparable with isolated cellulose.
The peak at 8.6° in the XRD patterns of BCA (Fig. 3) is the main characteristic of the amorphicity generated by adding acetyl groups, causing an increase in interfibrillar distance and the rupture of the micro fibrillar structure (Shaikh et al. 2009). In addition, the appearance of some small peaks between 10° and 20° 2θ confirms the formation of triacetate polymorph II in the modified product (Kono et al. 1999). The peak around 17.5° is known as the Van der Waals area, corresponding to the presence of amorphous regions containing clusters of parallel chain segments (Daud and Djuned 2015). Since BCA XRD pattern have lost any contribution of the initial SBC cellulose, its XRD crystallinity parameters were not calculated.
FTIR spectra for the materials are shown in Fig. 5. The SB and PB bagasse spectra presented the same vibration bands, with absorption features referring to functional groups of cellulose, hemicellulose and lignin. The bands that characterize the presence of cellulose in the materials remained after APA pretreatment and are presented in 3414 cm−1, referring to OH axial deformation; 2900, 1372 and 897 cm−1 corresponding to CH2 and CH3 axial deformation; and at 1121 cm−1, attributed to the stretching of glycosidic bonds. The bands of hemicellulose at 1663 cm−1 related to C=O axial deformation, and 1135 cm−1 assigned to acetate groups, were significantly reduced after the pretreatment due to the removal of hemicellulose after the pretreatment. Likewise, the lignin characteristic spectral band ascribed to the aromatic skeleton vibrations of C=C at 1520 cm−1 was reduced, indicating that there was also effective removal of this contaminant and cellulose was mostly isolated (Silverstein et al. 2005; Cheng et al. 2011).

FTIR spectra for the cellulosic materials and respective bands used in TCI and LOI
The FTIR spectra of SBC and CC celluloses also showed similar absorption features. The broad absorption band at 3400 cm−1 corresponds to OH stretching, and the absorptions at 2910 cm−1 and 1375 cm−1 are related to CH deformations, as seen in SB and PB spectra. The band at 1160 cm−1 refers to the C–O–C asymmetric vibration, and at 903 cm−1 there is a band related to the stretching of β-glycosidic linkages between glycan units, typical of cellulose (Popescu et al. 2012; Horikawa 2017). These features and the absence of bands referent to hemicellulose and lignin structures demonstrate that the APA pretreatment followed by soda/AQ pulping and bleaching processes was effective in extracting these materials to produce high purity cellulose, according to the compositions observed in Table 1 (HPLC). The main features in BCA and CCA spectra are at 1756 cm−1, attributed to C=O stretching, and at 3490 cm−1 corresponding to OH stretching. The intensity of the OH band significantly reduces in comparison with the SBC spectra, showing the substitution of hydroxyl groups by acetyl groups in the reaction (Sun et al. 2004).
The infrared ratios of total crystalline index (TCI) and lateral order index (LOI) were determined (O’Connor et al. 1958; Nelson and O’Connor 1964). TCI is proportional to the crystallinity degree of cellulose and is calculated as the ratio between the absorbance of the bands at 1372 and 2900 cm−1 (A1372/A2900). LOI is correlated with the overall degree of order in cellulose and is taken as the ratio between the absorbance of the bands at 1430 and 897 cm−1 (A1430/A897). These results and their respective absorbances are reported in Table 3. Usually, less crystalline celluloses present lower TCI and LOI values; however, CC presented lower LOI than SBC due to the more intense absorbance at 897 cm−1 observed in its spectra. This can be related to the amorphous cellulose phase identified in XRD patterns (Fig. 3) and the higher mass percentage of hemicellulose of this material (Table 1), since both of these species may present vibrations at this wavenumber and interfere in the measurements (Thakur et al. 2017). Although TCI and LOI parameters can undergo interference referent to sample composition and crystallite size (Nelson and O’connor 1964), this empirical method is useful to provide qualitative information for discussing cellulose crystallinity in conjunction with XRD CrI by evaluating its chemical composition (Carrillo et al. 2018).
Thermogravimetric and calorimetric analysis (Fig. 6) were used to study the thermal properties of celluloses. TG and DTG profiles obtained from SBC and CC are shown in Fig. 6a. Water loss was observed at the range 25–100 °C for both samples. The first step of thermal degradation took place at 267–380 °C, when cellulose structures undergo depolymerization, dehydration and decomposition reactions. Then, the second step of weight loss occurred at 380–530 °C and corresponds to the formation of gases and a carbonized residue (Barud et al. 2008). In cellulose DSC curves (Fig. 6b), the shoulder related to water loss is an endothermic step with a peak at 70 °C. The two-step degradation shows the first endothermic step with a peak at 322 °C, while the second exothermic step is seen with a peak at 358 °C (Rodrigues Filho et al. 2008). The TG/DTG and DSC for SBC cellulose presented similar results of thermal resistance in comparison with commercial cellulose.

TG/DTG (a) and DSC (b) analysis for SBC and CC celluloses
Table 4 shows the elemental contents measured by CHNS analysis. It is seen that SBC and CC presented similar elemental composition and molar ratios to another commercial cellulose found in the literature (Kono et al. 2015). This result, as well as other results mentioned above, confirms the high purity of SBC cellulose after all treatments used in its extraction. The content of N and S in the samples was negligible.
Acetylation process
The main results of degree of substitution (DS) for the acetates produced in the acetylation reactions of SBC and CC are depicted in Table 5. These values show the influence of the reaction conditions and the nature of cellulose in the accessibility and reactivity. Moreover, the solubility of cellulose acetates in acetone and dichloromethane was investigated and then associated to DS data in order to obtain a more accurate classification for the products.
The DS value increases by increasing time and temperature conditions, where the highest DS values are seen in BCA-VI and CCA-VI triacetates (20 h, 50 °C). Furthermore, it was possible to produce triacetates with softer temperature conditions in the case of BCA-III and CCA-III (20 h, 25 °C). The BCAs and CCAs presented similar acetylation degrees, presenting satisfactory DS values according to literature (Cerqueira et al. 2007; Fan et al. 2013); however, in tests V and VI it is seen that SBC reached tri-substituted acetate at the same reaction conditions, while CC was converted into a diacetate, which may be related to the lower CrI and higher accessibility of SBC cellulose.
In the solubility tests, the replacement of hydroxyl groups by acetyl groups caused a decrease in the polarity of these compounds and consequent variations in solubility (Fan et al. 2014). Thus, triacetates are more readily solubilized in non-polar solvents as dichloromethane, and diacetates are soluble in solvents with medium polarity such as acetone. In the case that the acetylation product does not solubilize in any of these solvents, it can be classified as a monoacetate (Cerqueira et al. 2007). The DS values had a significant influence on the solubility of the cellulose acetates and the classifications provided by both tests are in accordance.
The FTIR spectra of the BCA and CCA acetates are shown in Fig. 7. It is seen that the characteristic C=O band of acetates at 1756 cm−1 is more intense according to the increase in the acetylation degree of the sample, since more OH groups are replaced by acetyl groups. The spectra confirm the classifications attributed to the acetates in Table 5.

FTIR spectra of the main BCA (a) and CCA (b) acetates
The thermal characteristics of the CCAs were used as standard for comparison with BCAs. The DSC analysis data depicted in Fig. 8 shows that the DSC steps essentially had similar characteristics for BCAs and CCAs triacetates III and VI. The endothermic peak at 55 °C is associated with water loss (Rodrigues Filho et al., 2000) and the exothermic step around 200 °C is associated to the crystallization of the acetates (He et al., 2009). The melting point of the triacetate chains is around 300 °C, and the degradation process through decomposition and deacetylation reactions takes place between 380 and 600 °C (Candido et al. 2017; Shaikh et al. 2009). The conversion of both CC and SBC celluloses into cellulose acetates is observed with the extinction of the peaks at 322 and 355 °C ascribed to cellulose structure in the DSC of the acetylation products (Fig. 8a, b).

DSC analysis for BCA (a) and CCA (b) triacetates III and VI
1H and 13C NMR spectra of BCA-VI and CCA-VI shown in Fig. 9 were used in order to determine the structures and DS of the samples. The resonance lines for BCA and CCA acetates 13C NMR assigned to three C=O (168.5, 168.9 and 169.8 ppm) and three CH3 (20.1, 19.8 and 19.7 ppm) carbons sugest the triacetate structure. In addition, the methyl carbons signals present correlation with the hydrogens at 2.06, 1.94 and 2.89 ppm in the 1H NMR spectra. The spectra for acetylated samples showed a set of peaks that confirms the formation of triacetates in comparison to the results reported by Kono et al. (2015) (Table 6), and the NMR profiles for the samples evaluated in this work were compatible with 2,3,6-tryacetyl-cellulose derivatives.

13C (a) and 1H (b) NMR spectra for BCA-VI and CCA-VI celluloses
The DS values were also calculated using Eq. 3 with 1H NMR data for the resonance lines ascribed to the protons of the glucose ring and to the methyl protons of the acetate group, described in Table 6. The calculated DS values for BCA-VI and CCA-VI were 2.87 and 2.76, respectively, confirming the production of cellulose triacetates and corroborating the results discussed for FTIR and DSC analysis, along with DS and solubility tests for these samples in Table 5.
Conclusion
Cellulose was isolated from sugarcane bagasse (SBC) by a combined acid/peroxide-alkali pretreatment, followed by soda/AQ pulping, and then evaluated along with a standard commercial cellulose (CC). The correlations applied to the yield of components measured by HPLC analysis were able to provide the cellulose, hemicellulose and lignin contents in each cellulosic material, and the data showed that lignin and hemicellulose were considerably removed after the combined pretreatment, thus providing SBC cellulose with high purity (85.67% cellulose). The removal of these contaminants from the sugarcane bagasse (SB) is also seen in the SEM, XRD and FTIR results.
The cellulose obtained from a sustainable source presented lower crystallinity than the commercial material according to the crystallinity parameters of CrId, CrIs, FWHM, L, LOI and TCI, thus being classified after XRD peak deconvolution as a mixture of cellulose II polymorph with some features of remaining cellulose Iβ from sugarcane bagasse, with significant amorphous characteristics. These data indicate higher accessibility of the chains and susceptibility to chemical modifications for generating several value-added products. CrI deconvolution method (CrId) made possible a more accurate crystallinity assessment of the chemically treated SBC cellulose, considering crystalline contributions for both polymorphs.
SBC and CC were further acetylated by homogeneous acid reaction in order to evaluate the reactivity of these materials. The DS and solubility test data for the acetylated samples presented variations due to the different reaction conditions, as expected, and the celluloses presented satisfactory reactivity results, reaching triacetylation at various conditions. The acetylation degrees for the main acetates were confirmed by FTIR, DSC and RMN analyses, and were found to be in accordance with the literature.
Therefore, it is possible to state that the combined acid/peroxide-alkali pretreatment was efficient in removing the recalcitrant lignocellulose and for optimizing the isolation of highly pure and low crystallinity cellulose from residual sugarcane bagasse. SBC cellulose presented great thermal resistance, purity and accessibility properties in comparison to the Avicel commercial cellulose, showing that it has satisfactory quality and applicability, in addition to potentially being used to totally or partially replace the non-sustainable material with a renewable and efficient alternative.
References
Abdelwahab NA, Ammar NS, Ibrahim HS (2015) Graft copolymerization of cellulose acetate for removal and recovery of lead ions from wastewater. Int J Biol Macromol 79:913–922. https://doi.org/10.1016/j.ijbiomac.2015.05.022
Agarwal UP, Reiner RS, Ralph SA (2010) Cellulose I crystallinity determination using FT-Raman spectroscopy: univariate and multivariate methods. Cellulose 17:721–733. https://doi.org/10.1007/s10570-010-9420-z
Agarwal UP, Ralph SA, Reiner RS, Baez C (2016) Probing crystallinity of never-dried wood cellulose with Raman spectroscopy. Cellulose 23:125–144. https://doi.org/10.1007/s10570-015-0788-7
Agarwal UP, Ralph SA, Baez C, Reiner RS, Verrill SP (2017) Effect of sample moisture content on XRD-estimated cellulose crystallinity index and crystallite size. Cellulose 24:1971–1984. https://doi.org/10.1007/s10570-017-1259-0
Assumpção SMN, Pontes LAM, Carvalho LS, Campos LMA, Andrade JCF, Silva EG (2016) Pré-tratamento combinado H2SO4/H2O2/NaOH para obtenção das frações lignocelulósicas do bagaço da cana-de-açúcar. Revista Virtual de Química 8:803–822. https://doi.org/10.5935/1984-6835.20160059
Barud HS, Araújo AM Jr, Santos DB, Assunção RMN, Meireles CS, Cerqueira DA, Rodrigues Filho G, Ribeiro CA, Messaddeq Y, Ribeiro SJL (2008) Thermal behavior of cellulose acetate produced from homogeneous acetylation of bacterial cellulose. Thermochim Acta 471:61–69. https://doi.org/10.1016/j.tca.2008.02.009
Brodeur-Campbell M, Klinger J, Shonnard D (2012) Feedstock mixture effects on sugar monomer recovery following dilute acid pretreatment and enzymatic hydrolysis. Biores Tech 116:320–326. https://doi.org/10.1016/j.biortech.2012.03.090
Candido RG, Godoy GG, Gonçalves AR (2017) Characterization and application of cellulose acetate synthesized from sugarcane bagasse. Carbohyd Polym 167:280–289. https://doi.org/10.1016/j.carbpol.2017.03.057
Cardona CA, Quintero JA, Paz IC (2010) Production of bioethanol from sugarcane bagasse: status and perspectives. Biores Tech 101:4754–4766
Carrillo I, Mendonça RT, Ago M, Rojas OJ (2018) Comparative study of cellulosic components isolated from different Eucalyptus species. Cellulose 25:1011–1029. https://doi.org/10.1007/s10570-018-1653-2
Cerqueira DA, Filho GR, Meireles CS (2007) Optimization of sugarcane bagasse cellulose acetylation. Carbohydr Polym 69:579–582. https://doi.org/10.1016/j.carbpol.2007.01.010
Chandel AK, Antunes FAF, Anjos V, Bell MJV, Rodrigues LN, Polikarpov I, Azevedo ER, Bernardinelli OD, Rosa CA, Pagnocca FC, Silva SS (2014) Multi-scale structural and chemical analysis of sugarcane bagasse in the process of sequential acid– base pretreatment and ethanol production by Scheffersomyces shehatae and Saccharomyces cerevisiae. Biotechnol Biofuels 7:1–17. https://doi.org/10.1186/1754-6834-7-63
Cheng G, Varanasi P, Li C, Liu H, Melnichenko YB, Simmons BA, Singh S (2011) Transition of cellulose crystalline structure and surface morphology of biomass as a function of ionic liquid pretreatment and its relation to enzymatic hydrolysis. Biomacromology 12:933–941. https://doi.org/10.1021/bm101240zf
Ciolacu D, Popa VI (2010) Cellulose allomorphs—overview and perspective. In: Lejeune A, Deprez T (eds) Cellulose: structure and properties, derivatives and industrial uses. Nova Science Publishers, New York, pp 1–38
Ciolacu D, Pitol-Filho L, Ciolacu F (2012) Studies concerning the accessibility of different allomorphic forms of cellulose. Cellulose 19:55–68. https://doi.org/10.1007/s10570-011-9620-1
Corrales RCNR, Mendes FMT, Perrone CC, Sant’Anna C, Souza W, Abud Y, Bom SEP, Ferreira-Leitão V (2012) Structural evaluation of sugar cane bagasse steam pretreated in the presence of CO2 and SO2. Biotechnol Biofuels 5:1–8. https://doi.org/10.1186/1754-6834-5-36
Daud WWR, Djuned FM (2015) Cellulose acetate from oil palm empty fruit bunch via a one-step heterogeneous acetylation. Carbohyd Polym 132:252–260. https://doi.org/10.1016/j.carbpol.2015.06.011
Diep NQ, Sakanishi K, Nakagoshi N, Fujimoto S, Minowa T, Tran XD (2012) Biorefinery: concepts, current status, and development trends. Int J Biomass Renew 1:1–8
El Nemr A, Ragab S, El Sikaily A, Khaled A (2015) Synthesis of cellulose triacetate from cotton cellulose by using NIS as a catalyst under mild reaction conditions. Carbohyd Polym 130:41–48. https://doi.org/10.1016/j.carbpol.2015.04.065
El Oudiani A, Chaabouni Y, Msahli S, Sakli F (2011) Crystal transition from cellulose I to cellulose II in NaOH treated Agave americana L. fibre. Carbohyd Polym 86:1221–1229. https://doi.org/10.1016/j.carbpol.2011.06.037
Endo T, Aung EM, Fujii S, Hosomi S, Kimizu M, Ninomiya K, Takahashi K (2017) Investigation of accessibility and reactivity of cellulose pretreated by ionic liquid at high loading. Carbohydr Polym 176:365–373. https://doi.org/10.1016/j.carbpol.2017.08.105
Fan G, Wang M, Liao C, Fang T, Li J, Zhou R (2013) Isolation of cellulose from rice straw and its conversion into cellulose acetate catalyzed by phosphotungstic acid. Carbohyd Polym 94:71–76. https://doi.org/10.1016/j.carbpol.2013.01.073
Fan G, Liao C, Fang T, Luo S, Song G (2014) Amberlyst 15 as a new and reusable catalyst for the conversion of cellulose into cellulose acetate. Carbohyd Polym 112:203–209. https://doi.org/10.1016/j.carbpol.2014.05.082
French AD (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21:885–896. https://doi.org/10.1007/s10570-013-0030-4
French AD, Santiago Cintrón M (2013) Cellulose polymorphy, crystallite size, and the Segal Crystallinity Index. Cellulose 20:583–588. https://doi.org/10.1007/s10570-012-9833-y
Goodlett VW, Dougherty JT, Patton HW (1971) Characterization of cellulose acetates by nuclear magnetic resonance. J Polym Sci A 9:155–161. https://doi.org/10.1002/pol.1971.150090114
Gouveia ER, Nascimento RT, Souto-Maior AM, Rocha GJM (2009) Validation of methodology for the chemical characterization of sugar cane bagasse. Química Nova 32:1500–1503. https://doi.org/10.1590/S0100-40422009000600026
He J, Zhang M, Cui S, Wang S (2009) High-quality cellulose triacetate prepared from bamboo dissolving pulp. J Appl Polym Sci 113:456–465. https://doi.org/10.1002/app.30008
Horikawa Y (2017) Assessment of cellulose structural variety from different origins using near infrared spectroscopy. Cellulose 24:5313–5325. https://doi.org/10.1007/s10570-017-1518-0
Jahan MS, Sarkar M, Rahman MM (2015) Sodium carbonate pre-extraction of bamboo prior to soda-anthraquinone pulping. Biomass Convers Biorefin 5:417–423. https://doi.org/10.1007/s13399-015-0160-z
Kalogiannis KG, Stefanidis S, Marianou A, Michailof C, Kalogiann A, Lappas A (2015) Lignocellulosic biomass fractionation as a pretreatment step for production of fuels and green chemicals. Waste Biomass Valoriz 6:781–790. https://doi.org/10.1007/s12649-015-9387-y
Kautto J, Realff MJ, Ragauskas AJ (2013) Design and simulation of an organosolv process for bioethanol production. Biomass Convers Biorefin 3:199–212. https://doi.org/10.1007/s13399-013-0074-6
Kono H, Numata Y, Nagai N, Erata T, Takai M (1999) CPMAS 13C NMR and X-ray studies of cellooligosaccharide acetates as a model for cellulose triacetate. J Polym Sci A Polym Chem 37:4100–4107. https://doi.org/10.1002/(SICI)1099-0518(19991115)37:22%3c4100:AID-POLA8%3e3.0.CO;2-D
Kono H, Hashimoto H, Shimizu Y (2015) NMR characterization of cellulose acetate: chemical shift assignments, substituent effects, and chemical shift additivity. Carbohyd Polym 118:91–100. https://doi.org/10.1016/j.carbpol.2014.11.004
Kumar A, Negi YS, Choudhary V, Bhardwaj NK (2014) Characterization of cellulose nanocrystals produced by acid-hydrolysis from sugarcane bagasse as agro-waste. J Mater Phys Chem 2:1–8
Lavanya D, Kulkarni PK, Dixit M, Raavi PK, Krishna LNV (2011) Sources of cellulose and their applications—A review. Int J Drug Form Res 2:19–38
Nam S, French AD, Condon BD, Concha M (2016) Segal crystallinity index revisited by the simulation of X-ray diffraction patterns of cotton cellulose Iβ and cellulose II. Carbohyd Polym 135:1–9. https://doi.org/10.1016/j.carbpol.2015.08.035
Nelson ML, O’Connor RT (1964) Relation of certain infrared bands to cellulose crystallinity and crystal lattice type. Part I. Spectra of types I, II, III and of amorphous cellulose. J Appl Polym Sci 8:1311–1324. https://doi.org/10.1002/app.1964.070080322
O’Connor RT, DuPré EF, Mitcham D (1958) Applications of infrared absorption spectroscopy to investigations of cotton and modified cottons: Part I—Physical and crystalline modifications and oxidations. Text Res J 28:382–392. https://doi.org/10.1177/004051755802800503
Park S, Baker JO, Himmel ME, Parilla PA, Johnson DK (2010) Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels 3:1–10
Popescu CM, Larsson PT, Olaru N, Vasile C (2012) Spectroscopic study of acetylated kraft pulp fibers. Carbohyd Polym 88:530–536. https://doi.org/10.1016/j.carbpol.2011.12.046
Puleo AC, Paul DR, Kelley SS (1989) The effect of degree of acetylation on gas sorption and transport behavior in cellulose acetate. J Memb Sci 47:301–332. https://doi.org/10.1016/S0376-7388(00)83083-5
Rabelo SC, Andrade RR, Maciel Filho R, Costa AC (2014) Alkaline hydrogen peroxide pretreatment, enzymatic hydrolysis and fermentation of sugarcane bagasse to ethanol. Fuel 136:349–357. https://doi.org/10.1016/j.fuel.2014.07.033
Ramadoss G, Muthukumar K (2015) Influence of dual salt on the pretreatment of sugarcane bagasse with hydrogen peroxide for bioethanol production. Chem Eng J 260:178–187. https://doi.org/10.1016/j.fuel.2014.07.033
Rezende CA, Lima MA, Maziero P, Azevedo ER, Garcia W, Polikarpov I (2011) Chemical and morphological characterization of sugarcane bagasse submitted to delignification process for enhanced enzymatic digestibility. Biotechnol Biofuels 4:1–18. https://doi.org/10.1186/1754-6834-4-54
Rocha GJM, Nascimento VM, Rossell CEV (2014) Caracterização físico-química do bagaço da cana-de-açúcar. Technical Memorandum, CTBE. http://8k5sc3kntvi25pnsk2f69jf1.wpengine.netdna-cdn.com/wp-content/uploads/2016/08/MeT-102014-port.pdf
Rodrigues Filho G, Cruz SF, Pasquini D, Cerqueira DA, Prado VDS, Assunção RMN (2000) Water flux through cellulose triacetate films produced from heterogeneous acetylation of sugar cane bagasse. J Memb Sci 177:225–231. https://doi.org/10.1016/S0376-7388(00)00469-5
Rodrigues Filho G, Monteiro DS, Meireles CS, Assunção RMN, Cerqueira DA, Barud HS, Ribeiro SJL, Messadeq Y (2008) Synthesis and characterization of cellulose acetate produced from recycled newspaper. Carbohyd Polym 73:74–82. https://doi.org/10.1016/j.carbpol.2007.11.010
Samios E, Dart RK, Dawkins JV (1997) Preparation, characterization and biodegradation studies on cellulose acetates with varying degrees of substitution. Polymer 38:3045–3054. https://doi.org/10.1016/S0032-3861(96)00868-3
Scherrer P (1918) Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nach Gesel Wis Göt 26:98–100. https://doi.org/10.1007/978-3-662-33915-2_7
Segal L, Creely JJ, Martin AE Jr, Conrad CM (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text Res J 29:786–794. https://doi.org/10.1177/004051755902901003
Shaikh HM, Pandare KV, Nair G, Varma AJ (2009) Utilization of sugarcane bagasse cellulose for producing cellulose acetates: Novel use of residual hemicellulose as plasticizer. Carbohyd Polym 76:23–29. https://doi.org/10.1016/j.carbpol.2008.09.014
Silverstein RM, Webster FX, Kiemle DJ (2005) Spectrometric identification of organic compounds, 8th edn. Wiley, New York
Singh RK, Singh AK (2013) Optimization of reaction conditions for preparing carboxymethyl cellulose from corn cobic agricultural waste. Waste Biomass Valoriz 4:129–137. https://doi.org/10.1007/s12649-012-9123-9
Sluiter JB, Chum H, Gomes AC, Tavares RPA, Azevedo V, Mello LAM, Pimenta MTB, Rabelo SC, Marabezi K, Curvelo AAS, Scolfaro M, Alves AR, Garcia WT, Carvalho W, Esteves PJ, Mendonça S, Oliveira PA, Ribeiro JAA, Mendes TD, Vicentin MP, Duarte CL, Mori MN (2016) Evaluation of Brazilian sugarcane bagasse characterization: an interlaboratory comparison study. J AOAC Int 99:579–585. https://doi.org/10.5740/jaoacint.15-0063
Sun JX, Sun XF, Sun RC, Su YQ (2004) Fractional extraction and structural characterization of sugarcane bagasse hemicelluloses. Carbohyd Polym 56:195–204. https://doi.org/10.1016/j.carbpol.2004.02.002
Testova L, Nieminena K, Penttilä PA, Serimaa R, Potthast A, Sixta H (2014) Cellulose degradation in alkaline media upon acidic pretreatment and stabilisation. Carbohydr Polym 100:185–194. https://doi.org/10.1016/j.carbpol.2013.01.093
Thakur VK, Thakur MK, Kessler (2017) Handbook of composites from renewable materials: polymeric composites, vol 1, 6th edn. Wiley, Hoboken. https://doi.org/10.1002/9781119441632
Vochozka M, Marousková A, Váchal J, Straková J (2016) Economic and environmental aspects of steam-explosion pretreatment. Waste Biomass Valoriz 7:1549–1554. https://doi.org/10.1007/s12649-016-9555-8
Wei WQ, Wu SB (2017) Conversion of eucalyptus cellulose into 5-hydroxymethylfurfural using Lewis acid catalyst in biphasic solvent system. Waste Biomass Valoriz 8:1303–1311. https://doi.org/10.1007/s12649-016-9676-0
Wright L, Boundy B, Perlack B, Davis S, Saulsbury B (2006) Biomass Energy Data Book, 1st edn. Oak Ridge National Laboratory, Tennessee
Acknowledgments
The authors would like to thank CAPES for the financial support, as well as the Northeastern Center of NMR Application and Use (CENAUREMN) for providing NMR analysis, the Chemistry Post-Graduate Program of the UFRN and the Green Chemistry Nucleus (NQV) of Salvador University (Bahia, Brazil).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Moura, H.O.M.A., Campos, L.M.A., da Silva, V.L. et al. Investigating acid/peroxide-alkali pretreatment of sugarcane bagasse to isolate high accessibility cellulose applied in acetylation reactions. Cellulose 25, 5669–5685 (2018). https://doi.org/10.1007/s10570-018-1991-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-018-1991-0