
Abstract
Poly(vinyl alcohol) (PVA) foams reinforced with cellulose nanocrystals (CNCs) were prepared with formaldehyde as a crosslinking agent. Two initial reaction times (10, 120 s) and the addition of CNCs (0–2 wt% based on total reaction suspension) were found to affect the foam density, water uptake, morphology and mechanical properties. A longer initial reaction time resulted in higher mechanical properties and density, due to the small pore size. The addition of CNCs induced a progressive decrease in the pore diameter and an increase in the foam density, as well as improved mechanical properties. With 1.5 wt% CNC content, the compressive strength of the PVA foams was significantly improved from 7 to 58 kPa for 10 s-initial reaction time and from 65 to 115 kPa for 120 s-initial reaction time. Results showed that the cross-linked PVA foams with CNC had promising properties for use in biomedical applications.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In recent years, the problems of waste management associated with petroleum-based foams have provided opportunity to focus on environmentally compatible and biodegradable materials. The materials based on synthetic polymers or natural polymers from renewable resources, such as starch and polyvinyl alcohol (PVA), have attracted more attention. They can serve as substitutes for petroleum-based polymers in many applications (Liu et al. 2014).
PVA is a well-known synthetic polymer of great industrial value with many desirable characteristics. It is non-toxic, biocompatible, water soluble, semi-crystalline, fully biodegradable and has relatively lower cost than current dominating raw material of foam, polyurethane (De Merils and Schoneker 2003; Peppas and Tennenhouse 2004; Wang et al. 2006; Zhao et al. 2014). PVA is also versatile, with varying average molar mass and degree of deacetylation (Christie and Nikolaos 2000), which makes PVA adaptable for many applications (Răpă et al. 2014). Environmentally friendly PVA foams have been studied as early as the 1950s (Wilson 1952), and today we can find PVA in packaging, medical, and energy-absorption applications (Gottrup et al. 2000; Siró and Plackett 2010; Avella et al. 2011; Karimi et al. 2014; Bai et al. 2015). PVA foams are still studied, for instance, in combination with natural polymers, such as chitosan, starch and also cellulose (Wang et al. 2006; Avella et al. 2012; Li et al. 2012).
Porous PVA foams have been widely and commercially produced by high-speed mixing with and without pore-forming agent since 1952 (Wilson 1952), through PVA and aldehyde hybrid reaction using acid as catalyst, i.e., acetalization. The acetalization reaction has been described by several researchers (Imai et al. 1984; Toncheva et al. 1992, 1994; Gousse and Gandini 1997). Owing to complete biodegradability, starch is usually used as a pore-forming agent. Within the process, starch granules swell in elevated temperature and acid and occupy a space. Under the same conditions the starch is also hydrolysed. When the acetalization of PVA and aldehyde is complete, the hydrolysed starch is leached using water and the pores are formed. Meanwhile, the whole porous structure is solidified by the chemical crosslinks of PVA and aldehyde (Nishimura et al. 1972; Sueoka et al. 1981; Rosenblatt 1996). The foam exhibits good mechanical properties from chemical crosslinking, outstanding water adsorption capacity because of large amounts of hydroxyl groups on the surface of aldehyde PVA foam network (Buchhlz and Graham 1997), and excellent biocompatibility (De Merils and Schoneker 2003). The method is still studied and further developed in order to enhance properties and improve the fabricating process (Iwasaki et al. 1985; Yoshizawa 1990; Wang and Chiu 2003; Wongsuban et al. 2003; Wang et al. 2006; Pan et al. 2014; Karimi and Navidbakhsh 2014).
Currently, nanotechnology and nanocomposites enable opportunities within materials science, including cellulose nanomaterials from renewable bioresources. Cellulose nanocrystals (CNCs), also called cellulose nanowhiskers, are needle-like cellulose crystals of 10–20 nm in width and some hundred nanometers in length, with modulus as high as 172 GPa, determined by X-ray diffraction (Tashiro and Kobayashi 1985). This has led to a great interest in the use of CNCs as reinforcement for polymers and also for PVA. It is reported that the addition of nanocellulose improves the mechanical properties, thermal and moisture stability of PVA films (Lee et al. 2009; Souza et al. 2010; Cho and Park 2011; Zhou et al. 2012; Baheti and Militky 2013; Li et al. 2013; Liu et al. 2013; Lacroix et al. 2014; Lani et al. 2014; Virtanen et al. 2014a, b). However, only a few studies on PVA foams with cellulose nanofibers or crystals can be found (Srithep et al. 2012; Kumar et al. 2013, 2014; Liu et al. 2014), and no research about PVA/nanocellulose foams made by means of the aldehyde crosslinking method has been reported. Research of PVA/nanocellulose foams based on the aldehyde crosslinking method is interesting, since this method is industrial and might be easily utilised for commercial use. Furthermore, chemically cross-linked PVA foams are also expected to have better mechanical properties than PVA-starch-cellulose mixed foams.
The aim of this work was to investigate how the modified aldehyde crosslinking process, with starch as pore-former as well as the addition of cellulose nanocrystals, affects the foam structure, density, water uptake and mechanical properties. PVA foams with improved mechanical properties have potential to find applications, in soft-tissue engineering for example to replace cartilage, kidney and liver (Karimi and Navidbakhsh 2014).
Materials and methods
Materials
Polyvinyl alcohol (PVA) with a degree of hydrolysis of over 99 % and a molecular weight ranging between 89 and 98 kDa was supplied by Sigma-Aldrich (Stockholm, Sweden). The other PVA specifications: viscosity 25–31 mPa·s and max. ash content 0.4 %. Potato starch was supplied by Sigma-Aldrich (Stockholm, Sweden).
Microcrystalline cellulose (MCC, Avicel® PH-101) with a particle size of ~50 μm was supplied by Sigma-Aldrich, Stockholm, Sweden. Formaldehyde solution (ACS reagent, 37 wt% in H2O, 10–15 % methanol), hydrochloride acid (ACS reagent, 37 wt% fuming), sodium hypochlorite solution (ACS reagent, available chlorine 10–15 %), sodium bromide and 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO) were supplied by Sigma-Aldrich (Stockholm, Sweden). Calcium carbonate was supplied by Merck KGaA (Darmstadt, Germany). All chemical reagents were used as received.
Preparation of cellulose nanocrystals
TEMPO-oxidized cellulose nanocrystals (CNCs) were produced in lab scale as follows. Briefly, MCC, was suspended in distilled water containing TEMPO and sodium bromide, according to a procedure described by Saito (Saito et al. 2007) with minor modifications. TEMPO-mediated oxidation of MCC suspension was started by adding NaClO reagent, while the suspension was continuously stirred at room temperature. After the addition of NaClO, the pH remained constant at 10.5 by adding 0.5 M NaOH solution until no more consumption of alkali was observed, indicating that the reaction was completed. This step took approximately 4–5 h, depending on the amount of NaClO added. The sediment of the TEMPO-oxidized CNCs was obtained after thoroughly washing with distilled water by centrifugation four times to remove unreacted chemicals. The final CNC suspension was obtained by ultra-sonication (Isogai et al. 2011). Ultra-sonication was performed on batches of 100 ml CNC suspensions in beakers supported by an ice bath to minimize overheating of suspensions for 10 min. The generated CNC suspension was stored in a refrigerator at 4 °C.
Foaming of PVA
PVA was dispersed in cold distilled water by stirring (300 rpm, Heidolph, Germany), and dissolved by heating (Heidolph MR Hei-End, Germany) with temperature rising slowly to 80 °C for 30–60 min. The PVA solution was cooled to below 50 °C, and the potato starch was subsequently added into the solution while stirring at 50–60 °C until a homogeneous, pasty, aqueous solution was obtained. The PVA-starch solution was cooled down below 50 °C. After that, foaming agent (CaCO3) and formaldehyde solution were gently added while stirring and large amounts of froth were immediately formed after adding HCl. The mixture was vigorously stirred (14,000 rpm) using Ultra-Turrax (IKA T25, Germany) for two different initial reaction times (10, 120 s). The two initial reaction times were chosen based on the experimental trials, when averagely after 10 s the formed froth started to collapse down, and averagely after 120 s the froth did not change further by visual. The final solution was poured into a mold and transferred into oven for 3 h at 55 °C, and a water-insoluble foam was formed. It was then washed by distilled water until the washing water became transparent and the pH of the water reached about 5.5, indicating that most of the remaining HCl, formaldehyde and starch were removed.
The CNC-reinforced foams were prepared similarly to the neat PVA foams by firstly dispersing a certain amount of the CNC gel (3.67 % dry weight) in calculated amounts of distilled water (Table 1). A visible homogeneous and transparent suspension was obtained by stirring after 30 min. After that, PVA was added and dissolved in the CNC suspension with heating and stirring 300 rpm using a shear mixer Heidolph (Germany) for about 60 min. Starch, foaming agent, formaldehyde and HCl were added, and foams containing 10 wt% PVA, 5 wt% potato starch and CNC (0.5, 1, 1.5 and 2 wt%) were formed, as described above for foams without CNC presence.
For the purpose of characterizations, all foams were first frozen at −40 °C, and then lyophilized using a freeze dryer Alpha 2-4 LD plus (Germany) for 72 h. The flow scheme of the foaming process is shown in Fig. 1, and prepared foam combinations are presented in Table 1.

Schematic of the foaming process
Characterization
Flow birefringence is a preliminary and very easy method to confirm the presence and dispersion of CNCs in water. The whole setup includes two cross-polarized filters and a lamp.
CNC diameter distribution was characterized by atomic force microscope (AFM) Veeco Multimode, (Santa Barbara, CA, USA). A drop of diluted CNC suspension was spin-coated on the surface of a clean mica film. The surface of mica was scanned in air. Height, amplitude and phase images were obtained in tapping mode. The CNC diameter was determined from the height images using the Nanoscope V program.
Bulk density of foams was calculated according to BS EN ISO 845:2006 “Cellular plastics and rubbers”. Determination of apparent density, the ratio of the dried weight over the dimensions of each sample were measured with an electronic digital caliper. The average density was obtained by measuring 7 specimens for each sample.
Water uptake (W u) was tested by immersion of freeze-dried cubic specimens into distilled water until the weight remained constant. The specimens were taken out of the water at specific intervals and weighed on balance after removing the excess water by gently tapping the specimens on dry soft tissue paper. The value was calculated through gravimetric determination using the equation:
where W w is the weight of the specimen after uptake of water, and W d is the dried weight before water uptake.
Morphology of foams was investigated using a high-resolution scanning electron microscopy (HR-SEM) (Carl Zeiss, Merlin, Germany). Samples were sputter-coated with a thin layer of gold to avoid electron charging effects.
Mechanical properties of the samples were measured using a TA-DMA Q800 dynamic mechanical analyzer at a compressive strain rate of 10 % min−1 and a preload force of 0.1 N. Prior to testing, samples were incubated in distilled water at 37 °C for 24 h. The test was continued until 70 % compressive strain. All tests were conducted in a submersion compression clamp with distilled water at 37 °C. Average values of compression strength and modulus for each material were obtained from three single measurements.
In order to study the reversible behaviors of the foams, a cyclic loading–unloading experiment was performed. Foam samples were compressed at a constant rate of 10–70 % min−1 compressive strain. After reaching the 70 % compressive strain, the samples were unloaded to a small load of 0.1 N. Relaxation time was 5 min before the beginning of the next cycle. The loading–unloading cycle was continuously repeated three times. The tests were carried out in distilled water at 37 °C.
Results and discussion
Cellulose nanocrystals characteristics
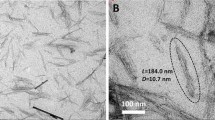
The obtained TEMPO-CNCs form a transparent gel, shown in Fig. 2a, with 3.67 % dry weight. The presence and dispersion of crystals in aqueous suspension obtained from TEMPO-mediated oxidation was confirmed by flow birefringence (Fig. 2b). AFM height (Fig. 2c) and amplitude (Fig. 2d) images showed the presence of well-dispersed CNC in water, and diameter distribution of the crystals, seen in Fig. 2e, ranging between 2 and 5 nm.

a Transparent TEMPO-CNC gel, b flow birefringence of CNCs in water, indicating well-dispersed crystals and AFM images of CNCs c height and d amplitude and e diameter distribution measured from height AFM images
Dispersion of CNCs in PVA
The homogeneous dispersion of nanocellulose will lead to a very large interfacial area between the matrix and the reinforcement, which is critical to enhance the mechanical properties of the final nanocomposites. Well-dispersed CNCs in PVA and in PVA//starch mixture will promote the actual formation of hydrogen bonds between PVA and CNCs and are therefore important for the reinforcement of PVA foams. In order to ensure dispersion of CNCs, optical microscopy images (6.3× magnification) were taken every time immediately after mixing the CNC in dissolved PVA, and images are presented in Fig. 3. The PVA-S-CNC showed similar microcopy images; therefore, only the image of PVA-S-0.5CNC is presented. For comparison, this image is also shown with 40× magnification. Figure 3b, d indicate homogeneous dispersion of CNC in water and in PVA-mixture, respectively. This also confirms that the addition of stable negative or positive electrostatic charges on the surface of CNCs is a simple means of obtaining good dispersion. Few small particles in neat PVA and PVA-CNC suspension are visible (Fig. 3a, d); these are probably small amounts of undissolved PVA granules. When starch was presented in the mixture, large amounts of big particles were seen (Fig. 3e, f). The particles were swollen starch granules formed when temperature was over 50 °C (Wajira and David 2006). Furthermore, in Fig. 3 the PVA-starch-CNC mixture (Fig. 3f, g), there are a lot of slurry-like substances seen between the swollen starch granules. The formation of the slurry can be explained by gelatinization of starch at 60 °C (Wajira and David 2006). At this temperature, starch granules can disperse in the highly sticky PVA-CNC mixture. The same phenomena can also be confirmed in SEM micrographs (Fig. 3h, i). Well-dispersed CNCs in PVA-CNC mixture were also confirmed by flow birefringence, as shown in Fig. 3d. However, flow birefringence cannot be applied after adding starch into PVA-CNC, because of the high viscosity of the suspension.

Microscopy images of a neat PVA, b neat CNC dispersion in water, c neat starch in water, d PVA-CNC, e PVA-starch, f PVA-starch-0.5CNC, g PVA-starch-0.5CNC with ×40 magnification; SEM images of h PVA-starch, and i PVA-starch-0.5CNC
Morphology
SEM images of PVA foams by two initial reaction times are illustrated in Figs. 4 and 5, showing the cross-section (Fig. 4) of the prepared foams, and the surfaces in the pores (Fig. 5). Figure 4a shows that the pore size distribution of 10PVA-starch-CNC foams varied greatly, mostly from about 50 µm up to about 1 mm. Also, some nanosize pores were seen, but only very few. On the other hand, longer time stirring enhanced the overall homogeneity of the 120PVA-starch-CNC foam surface and resulted in decreases in pore size (Fig. 4b). Obviously, more pores with diameters of larger than 200 µm in 10PVA-starch-CNC foams were observed, compared to those in 120PVA-starch-CNC foams. Most of the bigger pores (diameter over 200 µm) were from the CO2 gas entrapped during the foaming process. However, the leaching of big swollen starch granules can also form the pores in this range, since the diameter of potato starch is in the range of approximately 10–100 µm (Baldwin et al. 1998; Fig. 4d). The number of big pores decreased with the increase in CNC content, due to the increase in viscosity, which inhibited the formation of big bubbles from CO2 gas into the mixture, and also the leaching of starch by water. Some remnants of swollen starch granules can be seen on the surfaces of the foams under greater magnification (Fig. 4c), especially those obtained by longer time stirring, and containing PVA and CNCs.

SEM images of external surface of a 10PVA-starch-CNC foam, b 120PVA-starch-CNC foam and c 120PVA-starch-CNC foam with higher magnification, with CNC content 0, 1 and 2 %, as seen from top to bottom in the pictures; SEM images of d potato starch granules; cross-section photos of dry 10PVA-starch-CNC foam and 120PVA-starch-CNC foam

SEM images of inner surface of a 10PVA-starch-CNC foam and b 120PVA-starch-CNC foam, with CNC content of 0, 1 and 2 %, as seen from top to bottom in the pictures
High-magnification images show that the interior of the pores appears to be more homogeneous than cross-sectional pore surface (Fig. 5a, b). Both initial reaction times created similar inner pore surfaces. The pores were interconnected and had an obvious open-cell structure. All pores have a very small size, up to about 20 µm. It is worth mentioning that these small pores were probably mainly from removal of ice crystals in the samples during freeze-drying, and also from CO2 gas, which can only form very small bubbles in the polymer matrix. The leaching of small starch granules could also form pores within 20 µm.
Density
The bulk density of the foams made from two different processes is listed in Table 2. As it is listed in Table 2, with shorter time stirring, the density of the 10PVA-S-CNC foams was increased with increased CNC content. This was caused by the increase in viscosity of 10PVA-S-CNC mixture due to the higher content of CNCs, which resulted in the difficulty for CO2 gas generated from CaCO3 to form big bubbles in the mixture. Furthermore, starch granules inside the foams were difficult to leach into the water bath, which is also expected to increase the density. The previous SEM images (Figs. 4, 5) supported these density results.
However, with longer time stirring, the density of 120-foams was higher than 10-foams and appears almost the same, independent of the CNC content, because all mixtures were homogenized during the process. Most of the large bubbles generated from CO2 were broken and became homogenous. Therefore, there were only a few big bubbles in the mixture, which can be seen from the SEM images shown previously.
Water uptake
Soft tissue (e.g., cartilage, nucleus pulposus, dura mater, synovial fluid, collagen-proteoglycan matrix, etc.) is rich in water and constitutes a significant part of the human body. It is important to investigate the water uptake of the PVA-S-CNC foams. The results of the water uptake are shown in Fig. 6. All foams had very fast water uptake (300–500 %) after immersing them into water for 10 min. 10PVA-S-CNC foams showed a higher water uptake than the 120PVA-S-CNC foams. After 7 days immersion, the water uptake of 10PVA-S-CNC foams was in the range of 400–1000 %, compared to that of 120PVA-S-CNC foams (300–500 %). Most of the 10PVA-S-CNC foams reached an equilibrium water uptake after 24 h, except the 10PVA-S-0CNC foam. It seems that the water uptake ability of 10PVA-S-CNC foams is mainly dependent on the density, corresponding to size and amount of the pores. The foams with lower bulk density have more big pores in the structure (Fig. 6), which results in higher water uptake. Furthermore, in the foams with higher density, there were a lot of starch granules in the structure. This caused fewer pores in the foam structure, which also resulted in lower water uptake. The 120PVA-S-CNC foams almost reached equilibrium of water uptake after 24 h. There was a slight increase of about 50 % after 7 days. Considering 120PVA-S-CNC foams have similar density, the difference in water uptake can be inferred as such by the effect of CNC addition. When CNCs were added, interactions between PVA and CNC free hydroxyl groups to form hydrogen bonding may occur (Peresin et al. 2010). When a small amount of CNCs was added, the interactions resulted in lower amounts of free hydroxyl groups in the foams to interact with water molecules, and further decreased the ability of water uptake. However, when a larger amount of CNCs was added, after interactions of PVA and CNC-free hydroxyl groups, there were still large amounts of free hydroxyl groups on CNC surfaces, which were able to interact with water molecules. In this regard, the water uptake of 120PVA-S-CNC foam increased with the addition of a greater amount of CNC.

Water uptake of 10PVA-S-CNC foams (top) and 120PVA-S-CNC foams (below) with five different CNC contents, and a picture of 10PVA-S-CNC foams with 2 % CNCs (left) and 0 % CNCs (right) after 24 h water absorption
Mechanical properties
Stress–strain behavior of PVA foams with different CNC contents and initial reaction times conducted at 37 °C are shown in Fig. 7. The deformation mechanism of the foam can be separated into three different phases during compression (Ashby and Medalist 1983; Pampolini and Piero 2009). At the early stage a linear-elastic region appears, where the cell walls start to bend. After this, there is a plateau-like state, corresponding to the buckling of cell walls. When the foam is further compressed to a higher strain, its volume decreases and the cell walls begin to collapse, showing a significant increase in the stress. In this work, the foams with a longer initial reaction time showed higher mechanical properties than those with a shorter time. Considering the interaction between PVA and CNC was the same, this is due to the higher density of the foams with smaller pore sizes obtained by longer initial reaction time, as discussed in the previous section. A compressive stress value of 6.7 kPa can be obtained from 10 PVA-s, while that of 65.2 kPa was measured from 120 PVA-s. With the addition of CNCs, compressive strength and modulus of the PVA foams increased, as summarized in Table 3. With 1.5 wt% CNCs, both foams with 10 and 120 s reaction times showed the highest compressive strength. Values of 58.2 and 114.6 kPa were obtained from the compressive strength of PVA foams with 1.5 wt% CNCs with the initial reaction time of 10 and 120 s, respectively, while the compressive strength of the neat PVA foams with the initial reaction time of 10 and 120 s was only 6.7 and 65.2 kPa, respectively. Compressive strength and modulus of the foams decreased when the content of CNCs was higher than 1.5 wt%. Similar decrease was observed for mechanical properties of the PVA foams with the content of hydroxyapatite more than 1.5 wt% (Gonzalez and Alvarez 2014).

Stress–strain compressive curves for CNC reinforced PVA foams with a 10 s and b 120 s initial reaction time
Also, the polyurethane foams reinforced with CNCs showed the same behavior, with increasing CNC contents (Zhou et al. 2015). At low concentration of CNCs, the compressive strength and modulus of the foams were improved due to the good dispersion of CNCs, and strong interaction between PVA and CNC. With the higher CNC content, however, values of the compressive strength and modulus of the foams were reduced. This decrease can be because of aggregated CNCs because of high viscosity and difficulties to disperse the CNCs.
These materials were further studied to understand their reversible properties. Figure 8 shows three compression loading–unloading cycles up to 70 % compressive strain with a 5 min relaxation time between each cycle. It is worth noting that the first loading cycle showed the highest stress, the stress decreased slightly for the subsequent cycle with the same strain. This decrease could be caused by the collapse or the breakage of the pore structure. After the first cycle, similar stress–strain behavior could be observed for all types of foams, meaning no further change of the pore structure (Harrass et al. 2013). This indicates that the foams prepared in this work could withstand the load at 70 % strain and recover to the original point. These deformation results indicate that foams prepared in this study perform like rubber-like materials and the foam behavior under cyclic deformation shows a change in mechanical properties with a number of cycles (Gong et al. 2005; Cantournet et al. 2009; Drozdov 2009). This behavior can also be observed in polyurethane foams (Gong et al. 2005) and hydrogels (Harrass et al. 2013).

Reversible cycles for PVA foams with CNCs with a 10 s and b 120 s initial reaction time (a 5 min relaxation time was provided between each cycle)
Conclusions
PVA foams reinforced with different CNC contents were successfully prepared using crosslinking technology. Two different initial reaction times (10, 120 s) were applied. The initial reaction time was found to have a significant impact on the foam properties. A wide range of pores ranging in size from a couple of micrometers up to about 1 mm was observed. 10PVA-S-CNC foams had obviously more and bigger pores than 120PVA-S-CNC foams; however, 120PVA-S-CNC foams had a more homogenous surface structure.
Because of the formation of bigger pores with shorter initial reaction time, 10PVA-S-CNC foams showed lower bulk density but higher water uptake ability compared to the foams with 120 s reaction time. The addition of CNCs to PVA resulted in the decrease in pore size and, subsequently, also in the decrease in water uptake and increase of the density.
Mechanical properties of foams were also affected by the presence of CNCs. Foams with 1.5 wt% CNCs showed the highest values of compressive strength (58.2 and 114.6 kPa for 10 and 120 s-initial reaction times), while compressive stress values of 6.7 and 65.2 kPa were obtained from the PVA foams without CNCs. However, with higher CNC content than 1.5 wt%, the mechanical properties of the foams decreased due to the aggregation of CNCs. Notably, the compression loading–unloading testing showed that after the foam is compressed to 70 % strain, it can resume its original shape and it can possibly be compressed again.
These results show that biocompatible PVA foams prepared in this study are promising materials for soft-tissue engineering applications because of their improved mechanical properties in wet conditions.
References
Ashby MF, Medalist RFM (1983) The mechanical-properties of cellular solids. Metall Mater Trans A 14:1755–1769
Avella M, Cocca M, Errico M, Gentile G (2011) Biodegradable PVOH-based foams for packaging applications. J Cell Plast 47(3):271–281
Avella M, Cocca M, Errico ME, Gentile G (2012) Polyvinyl alcohol biodegradable foams containing cellulose fibres. J Cell Plast 48(5):459–470
Baheti V, Militky J (2013) Reinforcement of wet milled jute nano/micro particles in polyvinyl alcohol films. Fiber Polym 14(1):133–137
Bai HY, Li YF, Wang W, Chen GL, Rojas OJ, Dong WF, Liu XY (2015) Interpenetrated polymer networks in composites with poly(vinyl alcohol), micro- and nano-fibrillated cellulose (M/NFC) and polyHEMA to develop packaging materials. Cellulose 22:3877–3894
Baldwin PM, Adler J, Davies MC, Melia CD (1998) High resolution imaging of starch granule surfaces by atomic force microscopy. J Cereal Sci 27:255–265
Buchhlz FL, Graham T (1997) Modern superabsorbent polymer technology. Wiley-VCH, New York
Cantournet S, Desmorat R, Besson J (2009) Mullins effect and cyclic stress softening of filled elastomers by internal sliding and friction thermodynamics model. Int J Solids Struct 46:2255–2264
Cho MJ, Park BD (2011) Tensile and thermal properties of nanocellulose-reinforced poly(vinyl alcohol) nanocomposites. J Ind Eng Chem 17:36–40
Christie MH, Nikolaos AP (2000) Structure and applications of poly(vinyl alcohol) hydrogels produced by conventional crosslinking or by freezing/thawing methods. Adv Polym Sci 153:37–65
De Merils CC, Schoneker DR (2003) Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem Toxicol 41:319–326
Drozdov AD (2009) Mullins’ effect in semicrystalline polymers. Int J Solids Struct 46:3336–3345
Gong L, Kyriakides S, Jang WY (2005) Compressive response of open-cell foams. Part I: morphology and elastic properties. Int J Solids Struct 42:1355–1379
Gonzalez JS, Alvarez VA (2014) Mechanical properties of polyvinylakohol/hydroxyapatite cryogel as potential artificial cartilage. J Mech Behav Biomed 34:47–56
Gottrup F, Agren MS, Karlsmark T (2000) Models for use in wound healing research: a survey focusing on in vitro and in vivo adult soft tissue. Wound Repair Regen 8:83–96
Gousse C, Gandini A (1997) Acetalization of polyvinyl alcohol with furfural. Eur Polym J 33(5):667–671
Harrass K, Krueger R, Moeller M, Albrecht K, Groll J (2013) Mechanically strong hydrogels with reversible behaviour under cyclic compression with mPa loading. Soft Matter 9:2869–2877
Imai K, Shiomi T, Tezuka Y, Miya M (1984) Acetalization of poly(vinyl alcohol) by kornblum reaction. J Polym Sci Pol Chem 22(3):841–842
Isogai A, Saito T, Fukuzumi H (2011) TEMPO-oxidized cellulose nanofibers. Nanoscale 3:71–85
Iwasaki K, Maeda S, Oomori Y, Kawakami H (1985) Odorless phenolic foam. Japanese Patent No. JPS60149638 (A)
Karimi A, Navidbakhsh M (2014) Mechanical properties of PVA material for tissue engineering applications. Mater Technol Adv Perform Mater 29(2):90–100
Karimi A, Navidbakhsh M, Razaghi R (2014) An experimental-finite element analysis on the kinetic energy absorption capacity of polyvinyl alcohol sponge. Mater Sci Eng, C 39:253–258
Kumar A, Negi YS, Bhardwaj NK, Choudhary V (2013) Synthesis and characterization of cellulose nanocrystals/PVA based bionanocomposite. Adv Mater Lett 4(8):626–631
Kumar A, Negi YS, Choudhary V, Bhardwaj NK (2014) Microstructural and mechanical properties of porous biocomposite scaffolds based on polyvinyl alcohol, nano-hydroxyapatite and cellulose nanocrystals. Cellulose 21:3409–3426
Lacroix M, Khan R, Senna M, Sharmin N, Salmieri S, Safrany A (2014) Radiation grafting on natural films. Radiat Phys Chem 94:88–92
Lani NS, Ngadi N, Johari A, Jusoh M (2014) Isolation, characterisation, and application of nanocellulose from oil palm empty fruit bunch fiber as nanocomposites. J Nanomater. doi:10.1155/2014/702538
Lee SY, Mohan DJ, Kang IA, Doh GH, Lee S, Han SO (2009) Nanocellulose reinforced PVA composite films: effects of acid treatment and filler loading. Fiber Polym 10(1):77–82
Li X, Li Y, Zhang S, Ye Z (2012) Preparation and characterization of new foam adsorbents of poly(vinyl alcohol)/chitosan composites and their removal for dye and heavy metal from aqueous solution. Chem Eng J 183:88–97
Li W, Zhao X, Huang Z, Liu S (2013) Nanocellulose fibrils isolated from BHKP using ultrasonication and their reinforcing properties in transparent poly(vinyl alcohol) films. J Polym Res 20:210–216
Liu D, Sun X, Tian H, Maiti S, Ma Z (2013) Effects of cellulose nanofibrils on the structure and properties on PVA nanocomposites. Cellulose 20:2981–2989
Liu D, Ma Z, Wang Z, Tian H, Gu M (2014) Biodegradable poly(vinyl alcohol) foams supported by cellulose nanofibrils: processing, structure, and properties. Langmuir 30:9544–9550
Mariano M, Kissi NE, Dufresne A (2014) Cellulose nanocrystals and related nanocomposites: review of some properties and challenges. J Polym Sci Pol Phys 52:791–806
Nishimura H, Sato SM, Sato S (1972) Method of producing polyvinyl acetal porous articles and the shaped porous articles made therefrom. U.S. Patent No. 3663470
Pampolini G, Piero GD (2009) Strain localization in polyurethane foams: experiments and theoretical model. In: Pfeiffer F, Wriggers P (eds) Mechanics of microstructured solids: cellular materials, fibre reinforced solids and soft tissues. Springer, Berlin
Pan Y, Wang W, Peng C, Shi K, Luo Y, Ji X (2014) Novel hydrophobic polyvinyl alcohol-formaldehyde foams for organic solvents absorption and effective separation. RSC Adv 4:660–669
Peppas NA, Tennenhouse D (2004) Semicrystalline poly(vinyl alcohol) films and their blends with poly(acrylic acid) and poly(ethylene glycol) for drug delivery applications. J Drug Deliv Sci Technol 14(4):291–297
Peresin MS, Habibi Y, Vesterinen AH, Rojas OJ, Pawlak JJ, Seppälä JV (2010) Effect of moisture on electrospun nanofiber composites of poly(vinyl alcohol) and cellulose nanocrystals. Biomacromolecules 11:2471–2477
Răpă M, Grosu E, Stoica P, Andreica M, Hetvary M (2014) Polyvinyl alcohol and starch blends: properties and biodegradation behavior. J Environ Res Prot 11(1):34–42
Rosenblatt S (1996) Injection molded PVA sponge, U.S. Patent No. 5554658
Saito T, Kimura S, Nishiyama Y, Isogai A (2007) Cellulose nanofibers prepared by TEMPO-mediated oxidation of native cellulose. Biomacromolecules 8:2485–2491
Siró I, Plackett D (2010) Microfibrillated cellulose and new nanocomposite materials: a review. Cellulose 17:459–494
Souza SF, Leão AL, Cai JH, Wu C, Sain M, Cherian BM (2010) Nanocellulose from curava fibers and their nanocomposite. Mol Cryst Liq Cryst A 522:342–352. doi:10.1080/15421401003722955
Srithep Y, Turng LS, Sabo R, Clemons C (2012) Nanofibrillated cellulose (NFC) reinforced polyvinyl alcohol (PVOH) nanocomposites: properties, solubility of carbon dioxide, and foaming. Cellulose 19:1209–1223
Sueoka A, Okamoto T, Ohmori A, Kawai S, Ueda M (1981) Polyvinyl alcohol semi-permeable membrane and method for producing same. U.S. Patent No. 4279752
Tashiro K, Kobayashi M (1985) Calculation of crystallite modulus of native cellulose. Polym Bull 14:213–218
Toncheva VD, Ivanova SD, Velichkova RS (1992) Modified poly(vinyl acetals). Eur Polym J 28(2):191–198
Toncheva VD, Ivanova SD, Velichkova RS (1994) Poly(vinyl acetal)s from poly(vinyl alcohol) and 4-dimethylaminobenzaldehyde. Eur Polym J 30(6):741–747
Virtanen S, Vartianen J, Setälä H, Tammelin T, Vuoti S (2014a) Modified nanofibrillated cellulose-polyvinyl alcohol films with improved mechanical performance. RSC Adv 4:11343–11350
Virtanen S, Vuoti S, Heikkinen H, Lahtinen P (2014b) High strength modified nanofibrillated cellulose-polyvinyl alcohol films. Cellulose 21:3561–3571
Wajira SR, David SJ (2006) Gelatinization and solubility of corn starch during heating in excess water: new insights. J Agric Food Chem 54(10):3712–3716
Wang C, Chiu K (2003) Foam filter and the manufacturing method thereof. U.S. Patent No. 20030140794 A1
Wang X, Chung YS, Lyoo WS, Min BG (2006) Preparation and properties of chitosan/poly(vinyl alcohol) blend foams for copper adsorption. Polym Int 55:1230–1235
Wilson CL (1952) Method of making expanded polyvinyl alcohol-formaldehyde reaction product and product resulting therefrom. U.S. Patent No. 2609347 A
Wongsuban B, Muhammad K, Ghazali Z, Hashim K, Hassan MA (2003) The effect of electron beam irradiation on preparation of sago starch/polyvinyl alcohol foams. Nucl Instrum Methods Phys Res B 211:244–250
Yoshizawa I (1990) Production of polyvinyl alcohol foam. Japanese Patent No. JPH02107648 (A)
Zhao CX, Jin DD, Zhou GX, Li JC (2014) Preparation method of polyvinyl alcohol modified waterborne polyurethane. China Patent No. CN103589135A
Zhou YM, Fu SY, Zheng LM, Zhan HY (2012) Effect of nanocellulose isolation techniques on the formation of reinforced poly(vinyl alcohol) nanocomposite films. Exp Polym Lett 6(10):794–804
Zhou X, Sain MM, Oksman K (2015) Semi-rigid biopolyurethane foams based on palm-oil polyol and reinforced with cellulose nanocrystals. Compos Part A. doi:10.1016/j.compositesa.2015.06.008
Acknowledgments
We thank Dr. Alejandro Leiro for technical help with SEM instruments, and Dr. Farid Touaiti for help with AFM operation and integration. Postdoctoral scholarship was received from the Kempe Foundations, Sweden and financial support from Tekes, FiDiPro Project. This work was also part of the activities of Wallenberg Wood Science Center (WWSC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Song, T., Tanpichai, S. & Oksman, K. Cross-linked polyvinyl alcohol (PVA) foams reinforced with cellulose nanocrystals (CNCs). Cellulose 23, 1925–1938 (2016). https://doi.org/10.1007/s10570-016-0925-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-016-0925-y