
Abstract
Cellulose crystallinity assessment is important for optimizing the yield of cellulose products, such as bioethanol. X-ray diffraction is often used for this purpose for its perceived robustness and availability. In this work, the five most common analysis methods (the Segal peak height method and those based on peak fitting and/or amorphous standards) are critically reviewed and compared to two-dimensional Rietveld refinement. A larger (\(n=16\)) and more varied collection of samples than previous studies have presented is used. In particular, samples (\(n=6\)) with low crystallinity and small crystallite sizes are included. A good linear correlation (\(r^{2} \ge 0.90\)) between the five most common methods suggests that they agree on large-scale crystallinity differences between samples. For small crystallinity differences, however, correlation was not seen for samples that were from distinct sample sets. The least-squares fitting using an amorphous standard shows the smallest crystallite size dependence and this method combined with perpendicular transmission geometry also yielded values closest to independently obtained cellulose crystallinity values. On the other hand, these values are too low according to the Rietveld refinement. All analysis methods have weaknesses that should be considered when assessing differences in sample crystallinity.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose makes up the largest biomass portion of all organic matter. In wood, cellulose comprises up to 50 % of the dry mass. Wood and paper-making industries naturally have strong interest in cellulose products. More recently, byproducts from these industries have also been suggested as a renewable energy source that does not compete with food production (Himmel et al. 2007). Developing enzyme mixtures that are optimized for cellulose hydrolysis requires knowledge of the cellulose crystallinity since different enzymes are used for crystalline and amorphous cellulose (Thygesen et al. 2005).
Crystallinity of cellulose also affects the mechanical properties, such as strength and stiffness, of both natural and man-made cellulosic products. The strength of a biocomposite material can be increased by the inclusion of highly crystalline cellulose (Siró and Plackett 2010).
X-ray diffraction (XRD) has also been used to study cellulosic materials—for over 80 years (Sisson 1933)—and it is still a prominent method of determining crystallinity of these materials due to its perceived robustness, non-destructive nature and accessibility (Zavadskii 2004; Driemeier and Calligaris 2010; Kim et al. 2013; Lindner et al. 2015). In addition to XRD, crystallinity in cellulose samples can be determined with many other methods, such as Raman spectroscopy (Schenzel et al. 2005; Agarwal et al. 2013; Kim et al. 2013), infrared spectroscopy (Kljun et al. 2011; Chen et al. 2013; Kim et al. 2013), differential scanning calorimetry (Gupta et al. 2013; Kim and Kee 2014), sum frequency generation vibration spectroscopy (Barnette et al. 2012; Kim et al. 2013), and solid state nuclear magnetic resonance (NMR) (Davies et al. 2002; Liitiä et al. 2003; Park et al. 2009; Kim et al. 2013).
In contrast to NMR, XRD cannot yield the cellulose crystallinity directly, but rather the mass fraction of crystalline cellulose among the entire sample. The latter is referred henceforth as sample crystallinity. In this article cellulose crystallinity refers to the mass fraction of crystalline cellulose among the total cellulose content. It follows that the values for sample crystallinity and cellulose crystallinity are directly comparable only if the sample is pure cellulose. Otherwise, the cellulose content of the sample should be determined using independent methods if cellulose crystallinity should be obtained from XRD measurements. Furthermore, sample crystallinity may include crystalline contribution from other crystalline material besides cellulose. In this case the crystalline contributions need to be separated before cellulose crystallinity can be evaluated. Cellulose exists in several polymorphs (French 2014) but this study focuses on cellulose I, which is the prominent polymorph in unprocessed wood and other higher plants.
In XRD crystallinity studies, many authors do not attempt to obtain an absolute value for cellulose crystallinity but rather discuss only a crystallinity index or refer to relative crystallinity values. In some cases, the absolute sample crystallinity may be a more useful metric. Absolute crystallinity is obtained for isotropic samples by calculating the area under the intensity curve for the crystalline contribution relative to the combined areas of crystalline and amorphous contributions. However, there are various methods of performing this calculation and different models for amorphous material have been used. For samples with preferred orientation, the used measurement geometry also affects the obtained crystallinity values. As there is no standard method to determine sample crystallinity from XRD data, comparing results from different literature sources is challenging.
A literature survey of 244 articles published between 2010 and 2014 (inclusive) that discussed cellulose crystallinity determination with XRD was conducted. The most common method was the Segal peak height method (Segal et al. 1959), which was used in 64 % of these articles. The second most common method was peak fitting (25 %, sometimes referred to as peak deconvolution), which was performed either with an amorphous standard or using a mathematical model for the amorphous contribution. The third most common method, amorphous subtraction, was used in 2.0 % of the articles. These three methods were also found to be the most common by Park et al. (2010) for the crystallinity analysis of commercial cellulose.
Recently there has been a vivid discussion on comparisons between the XRD crystallinity analysis methods (Thygesen et al. 2005; Park et al. 2010; Bansal et al. 2010; Terinte et al. 2011; Barnette et al. 2012). Most of these articles discuss the Segal method, an amorphous subtraction method and a peak fitting method and find differences between the methods. Park et al. (2010) concluded that the Segal method gave values that were too high and recommended the use of other methods. Bansal et al. (2010) also showed that the Segal method performed poorly with samples with known crystallinity, showing a mean error of over 20 %-point for crystallinity values. Terinte et al. (2011) found that values obtained by a peak fitting method by different experts were consistent.
This article includes the Segal method (method 1), the amorphous subtraction method (method 4) and three different peak fitting method implementations. Peak fitting methods vary in the choice of the amorphous model, which is here modeled with a wide Gaussian peak (method 2), with a combination of a linear fit and a wide Gaussian peak (method 3) or with an amorphous standard (method 5). Another peak fitting method, which originates from crystallography, is Rietveld refinement (Rietveld 1969; De Figueiredo and Ferreira 2014), which focuses on fitting the crystalline contribution accurately and includes all crystalline diffraction peaks. Rietveld refinement has been recently applied for the analysis of plant cellulose samples by Oliveira and Driemeier (2013). Although this method is not as common as the other methods considered here, it is very promising for the accurate analysis of two-dimensional (2D) scattering data. Thus, a 2D Rietveld method is included here as a comparison method.
The purpose of this article is to compare the chosen sample crystallinity determination methods and to see under which conditions—if any—comparisons could be made. The recent literature (Bansal et al. 2010; Park et al. 2010; Terinte et al. 2011) on this topic has focused on highly crystalline and pure cellulose samples. The samples compared here vary in degree of crystallinity, average crystallite size, degree of preferred orientation, and cellulose content. In particular, a collection of samples with small crystallite sizes and lower crystallinities were chosen for this study. These samples are more challenging to analyze than the samples in the previously cited crystallinity analysis comparison articles due to extensive peak overlap.
Although the Segal method is the most commonly used, criticism towards it is on the rise (Park et al. 2010; Terinte et al. 2011; French and Santiago Cintrón 2013; Nam et al. 2016). A secondary aim of this study is to further quantify this critique, in particular with respect to the effect of the crystallite size and the unrealistic cellulose crystallinity values obtained with the Segal method.
Materials and methods
Samples
Three forms of commercial microcrystalline cellulose (MCC) were selected to represent standard cellulose samples. MCC1 is known as Avicel PH-102, MCC2 as Vivapur 105 and MCC3, which was measured earlier (Tolonen et al. 2011), is from Merck (No. 1.02330.0500). Commercial (Milouban) cotton linter pulp (CLP) was also used. These cellulose samples were pressed in the shape of a disc into metal holders. Sample thicknesses were 0.95 (CLP), 1.4 (MCC1) and 1.1 mm (MCC2). Wood with a high average microfibril angle was represented by a juniper sample (Hänninen et al. 2012) of 1.4 mm thickness.
Additionally, XRD data was obtained from recent publications. Samples of low- and medium-density balsa (86 and 159 g/cm\(^{3}\), respectively) (Borrega et al. 2015), spruce-pine sulphite pulp and nata de coco (Parviainen et al. 2014), birch pulp (Testova et al. 2014), bamboo (Dixon et al. 2015), and MCC from birch sulphite, poplar kraft and cotton linters (Leppänen et al. 2009) were analyzed. The published properties of these samples are summarized in Online Resource 1. The bamboo samples represent values calculated as averages from three replicates.

Crystallinity determination with the five chosen methods for a the MCC2 (Avicel PH-102) sample and b the juniper sample, both measured with perpendicular transmission (two-dimensional scattering pattern shown in top left of each subfigure). The asterisks denote the positions of the fitted Gaussian crystalline peaks.
Experimental set-up
MCC1, MCC2, CLP and the juniper sample were measured using both perpendicular transmission (PT) geometry and symmetrical transmission (ST) geometry. Set-up 1 is based on a rotating anode source (Kontro et al. 2014) and was used for the PT measurements using a mar345 image plate detector. Set-up 2 is a four-circle goniometer (Andersson et al. 2003) that was used for all ST measurements. For the ST measurements the samples were rotated to reduce preferred orientation effects. All measurements were done using copper K\(\alpha\) energies (wavelength \(\lambda =0.154\) nm) and for compatibility with the Segal method scattering angles (\(2\theta\)) are discussed.
Data analysis
The XRD data was corrected for read-out noise (set-up 1) and normalized with the transmission calculated from the primary beam before air scattering was subtracted. After this, polarization correction was applied (taking into account the monochromator angle of 28.44\(^{\circ }\) for set-up 1). Geometrical corrections were applied for set-up 1. After this angle-dependent absorption (irradiated volume) correction was applied. For set-up 1 the diffraction data was averaged radially before data analysis in MATLAB. From the samples with published data, original corrected intensities were used if they were available.
A total of five different analysis methods were used to determine sample crystallinity for each of the 23 measurements included in this study. All five analysis methods are visualized in Fig. 1 for an MCC standard sample (high crystallinity) and a wood sample (low crystallinity). For comparison, 2D Rietveld refinement was included for the samples with 2D data available.
Method 1: Segal peak height
In the Segal peak height method (Segal et al. 1959) a maximum intensity value \(I_{200}\) is found between the scattering angles of \(2\theta =22^{\circ }\) and \(23^{\circ }\). The region between the cellulose I\(\beta\) 200 diffraction peak and the 110 and \(1\overline{1}0\) peaks is assumed to have very little crystalline contribution and is approximated as comprising of only an amorphous contribution. The minimum value \(I_{min}\) is taken using a minimum in the data, typically between \(2\theta =18^{\circ }\) and \(19^{\circ }\). The sample crystallinity (usually referred to as the crystallinity index) is then calculated as
Method 2: Gaussian peak fitting without a linear background (Gaussian peaks)
In method 2 a relatively small \(2\theta\) range between \(2\theta _{1}=13^{\circ }\) and \(2\theta _{2}=25^{\circ }\) is used and four cellulose diffraction peaks, corresponding to reflections 110, \(1\overline{1}0\), 102 and 200 are fitted with Gaussian peaks. A fifth Gaussian is fitted as the amorphous contribution. Peak positions for cellulose reflections are limited here to within 0.3\(^{\circ }\) of the literature values (Nishiyama et al. 2002) in the least square fit except for the 200-diffraction peak, which is fitted to the right of the observed 200-peak maximum. The amorphous peak maximum is limited between 18\(^{\circ }\) and 22\(^{\circ }\). The area of the crystalline peaks (\(A_{cr}\)) is used to calculate crystallinity as
where \(A_{sample}\) is the area under the sample intensity curve.
Method 3: Gaussian peak fitting with a linear background (Gaussian+ linear)
Method 3 includes a larger scattering angle region (from \(2\theta _{1}=13^{\circ }\) to \(2\theta _{2}=50^{\circ }\)) than method 2 and correspondingly more reflections (18 reflections of cellulose I\(\beta\) (Nishiyama et al. 2002)). In this method the amorphous model is also more sophisticated since it is represented by a superposition of a linear fit and a wide Gaussian peak, with a peak maximum between 18\(^{\circ }\) and 22\(^{\circ }\) and peak full width at half maximum between 10\(^{\circ }\) and 22.5\(^{\circ }\). The linear fit is assumed to be part of the amorphous model since the scattering intensities are already corrected before the crystallinity analysis.
The peak positions in this model are allowed to vary by 0.3 degrees, whereas peak widths and peak heights are taken essentially as free fitting parameters, with the starting values taken from a 36-chain crystallite model (Ding and Himmel 2006). The 200-diffraction peak position is fitted to the right of the observed 200-peak maximum instead of the exact literature position. The crystallinity is calculated with Eq. (2).
Method 4: Amorphous subtraction
In the Amorphous subtraction method an amorphous standard is chosen that should fit the amorphous contribution from the sample. The shape of the model is taken from a measured amorphous sample and may thus be more complicated and asymmetric than the ones of methods 2 and 3.
Before analysis the experimental data is smoothed with a Gaussian filter. The amorphous curve is then fitted to the data using a constant scaling factor so that it touches the experimental data in at least one point but does not surpass it. The area under the amorphous curve (\(A_{am}\)) is then taken as the amorphous contribution and crystallinity is then calculated as
The scattering angle range used to calculate the area is chosen to include a large wide-angle X-ray scattering region. Here the values of \(2\theta _{1}=13.5^{\circ }\) and \(2\theta _{2}=49.5^{\circ }\) are used for the Amorphous subtraction method.
Method 5: Gaussian peak fitting with an amorphous standard (Amorphous fitting)
Similarly to method 4, the Amorphous fitting method uses also an experimental amorphous standard obtained from a chosen amorphous sample. The crystalline model is the same as in method 3 and the crystallinity is calculated using Eq. (3) with \(2\theta _{1}=13^{\circ }\) and \(2\theta _{2}=50^{\circ }\). A linear superposition of the crystalline and amorphous models is used in the least squares fit. In contrast to method 4, method 5 features fitting which allows the amorphous model to surpass the measurement intensities slightly at some scattering angles if this improves the fit. This can happen due to differences in the actual shape of the amorphous contribution and the selected amorphous standard.
Comparison method: Two-dimensional Rietveld refinement
Rietveld refinement (RR) represents a more sophisticated method of fitting crystalline cellulose peaks to the experimental data. RR was conducted using the Cellulose Rietveld analysis for fine structure (CRAFS) software (Oliveira and Driemeier 2013; Driemeier 2014) using corrected two-dimensional scattering data. The standard CRAFS background model was replaced with the linear+Gaussian amorphous model of method 3. Otherwise the fitting algorithm and the fitting model was the same as explained in Oliveira and Driemeier (2013). Because the samples represent cellulose from different sources, all the parameters for unit cell, crystallite size and diffraction peak shape were refined. The starting values and upper and lower boundaries for all these parameters were from Oliveira and Driemeier (2013) except for the parameters that account for differences in the 110 and 1\(\overline{1}\)0 peak intensities.Footnote 1 The amount of preferred orientation in the samples varied from weak (powder-like samples) to very strong (wood and bamboo) and an orientation distribution was fitted to all the samples using a single Gaussian peak and a positive smoothly-varying background described with Legendre polynomials. Refined models for a microcrystalline cellulose standard and for two highly oriented samples are shown in Fig. 2. The 2D RR sample crystallinity was calculated using Eq. (3).

Rietveld refinement done with the CRAFS software (Oliveira and Driemeier 2013) shows how the experimental data (top row) is fitted with the Rietveld model (middle row). The residual (bottom row) is relatively small for the highly oriented Moso bamboo sample (left column), medium-density balsa (middle column) and the microcystalline cellulose standard Avicel PH-102 (right column). All intensities are given as relative to the maximum intensity of the corresponding experimental scattering data.

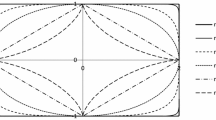
Fully crystalline cellulose I\(\beta\) models (top) constructed from the unit cell parameters of Nishiyama et al. (2002). Arrows on bottom right indicate directions perpendicular to the lattice planes (hkl). Models with equal number of glucose chains (\(n=6\ldots 13\)) in the [110] and [\(1\overline{1}0\)] directions were created and the calculated scattering intensities are shown
Fully crystalline models: the crystallite size effect
Fully crystalline cellulose models were constructed from the unit cell parameters of Nishiyama et al. (2002) for the purposes of seeing if the size of the crystallites affects the crystallinity values obtained with the chosen methods. These idealized crystallite models contain no surface, or other, disorder. Each model represents an ideal cellulose crystallite with both the cellulose and the sample crystallinity of 100 %. Any variation from this value in sample crystallinities reported in the Results section is due to the systematical error in the fitting method. Scattering intensities were calculated using the Debye formula (Debye and Bueche 1949) for the models shown in Fig. 3. The length of each model was 20 glucose residues. The size of the models were chosen to represent typical cellulose crystallite sizes (3–7 nm). The size was calculated in the [110] and [\(1\overline{1}0\)] directions from atomic coordinates.
Results
Crystallinity values
Ideally, the crystallinity value should not depend on the crystallite size. However, looking at the values of the fully crystalline models (Fig. 4), the values of the Segal peak height method show a positive linear correlation (\(r^{2}=0.92\)) with the crystallite size, as does the Amorphous subtraction method (\(r^{2}=0.92\)). The largest variation as a function of the crystallite size was seen in the Gaussian+ linear method, whereas the Amorphous fitting showed the least variation as a function of crystallite size (Table 1). The linear component of the Gaussian+ linear model increases for the larger crystallite sizes resulting in larger amorphous contributions. All methods yield crystallinity values significantlyFootnote 2 below the ideal value of 100 %. The Segal method values at the largest crystallite sizes are closest to the correct values whereas the average crystallinity value for the other methods ranges from 77 to 84 %.

Effect of the crystallite size on the sample crystallinity value for artificial, fully crystalline cellulose data. A third order fit is plotted for each data set for visualization purposes. Crystallite size is given along the [110] and [\(1\overline{1}0\)] directions (Fig. 3).
For the real samples, a complete list of sample crystallinity values obtained with the considered analysis methods are shown in Table 2. The average sample crystallinity values for the Segal method are higher than for the other methods (66 % higher than Gaussian peaks, 63 % higher than Gaussian + linear, 52 % higher than Amorphous fitting and 40 % higher than Amorphous subtraction).
The values of sample crystallinities obtained can also be compared to NMR crystallinity results if the cellulose content is available. For the samples where this information was available, cellulose crystallinity was calculated as C/cc, where cc is the cellulose content and C is the sample crystallinity. The values in Table 3 show that the Segal method produces unrealistically high values, over 100 % for samples with low cc. Results from methods 4 and 5, based on an amorphous standard, correspond best with the NMR results.
The unprocessed plant and wood material have strong preferred orientation. The effects of the orientation can be assessed by measuring the same sample using multiple measurement geometries. For the medium-density balsa sample that was measured with three measurement geometries, only the symmetrical reflection geometry produces systematically cellulose crystallinity values of over 100 %. This can be explained by the optimal scattering orientation of the cellulose I\(\beta\) 200 reflection for wood samples, which causes overestimation of its contribution in the scattering pattern (Paakkari et al. 1988) and leads to too high cellulose crystallinity values. Thus for samples with wood-like texture, PT and ST geometries yield more realistic cellulose crystallinity values. Samples (n=5) that were measured with both of these geometries showed on average higher sample crystallinity values with PT than with ST (Table 2; 8, 14, 9, 14 % and 12 % higher, with methods 1 through 5, respectively).
Correlation between the methods
If all the crystallinity analysis methods correlate with the actual sample crystallinity, there should be a linear correlation between the values of different methods. The linearity of other methods relative to the Amorphous fitting method is shown in Fig 5a. The strongest linear correlation (\(r^{2}=0.98\)) is seen with the Amorphous subtraction method and the weakest with the Gaussian+ linear method (\(r^{2}=0.90\)). The two Gaussian peak fitting methods show a similar linear trend. The Gaussian+ linear model shows large scatter at higher crystallinity values.

Sample crystallinity values of methods 1–4 relative to those of method 5, the Amorphous fitting method. Solid line indicates one-to-one correspondence. Possible linear correlation of the methods is assessed with the \(r^{2}\) value. Methods without such value show no statistically significant linear correlation. a All samples in one group. b Samples divided into two groups. c Individual bamboo samples (n=9)
To see if the correlations hold at smaller crystallinity differences the data was divided into two data sets (Table 2), those with low crystallinity (n = 8) and those with high crystallinity (n = 15). For the Amorphous fitting method low crystallinity samples vary in sample crystallinity from 20.7 to 28.1 % and the high crystallinity ones from 42.3 to 61.9 %. The linearity between the methods diminishes or disappears compared to Fig 5a as can be seen in Fig. 5b. Only the Amorphous subtraction method shows a linear correlation with the Amorphous fitting method.
The samples compared in Fig. 5b are not from a single sample set of similar samples. An analysis of a set of bamboo samples is shown in Fig. 5c. These nine bamboo samples were measured in the same conditions, with the same measurement geometry and data-corrected in the same way. A good linear correlation was seen with the Segal method (\(r^{2}=0.91\)) and the Amorphous subtraction method (\(r^{2}=0.97\)), compared to the Amorphous fitting method. The other methods did not show significant linearity. The sample crystallinity values for the bamboo samples were between 20 and 30 %, according to the Amorphous fitting method.
Comparison to Rietveld refinement
In order to further evaluate the chosen crystallinity fitting methods, a 2D RR was carried out on the samples with 2D data (Fig. 2). RR yielded higher sample crystallinity values than the methods 2–5 (especially for the low crystallinity samples with a strong preferred orientation) and lower values than those of the Segal method (Table 2).
Discussion
A good linear correlation (\(r^{2}\ge 0.90\)) was found between all crystallinity fitting methods. This suggests that the choice of the analysis method will usually not affect the relative differences between samples (i.e. relative crystallinities), as long as the relative differences are large. If the relative differences are small, however, the methods will not show the same differences in relative crystallinity. This negative result stands for dissimilar samples, measured with different measurement geometries.
As shown in the result section, differences in sample crystallinity values obtained with the Segal method can also be due to differences in crystallite sizes. A positive correlation between the crystallite size and the Segal crystallinity value has also been shown by Nam et al. (2016).
The Segal method also produced too high sample crystallinity values (Table 3). It did, however, have a linear correlation with the values obtained from the amorphous standard methods when a single sample set (n = 9, Fig. 5c) was considered. When a sample set (n = 8, Fig. 5b) consisted of different types of samples, the linearity was no longer present. This is consistent with the fact that the Segal method is an empirical method which was not meant to be used to compare different types of samples but rather quantify changes within a single sample set.
The Gaussian fitting methods 2 and 3 give the lowest crystallinity values, possibly due to over-fitting of the amorphous components. These methods may yield unrealistic amorphous contributions if fitting limits are too loose. On the other hand if the limits are too strict they may lead to wrong crystallinity values. For example, if the lower limit for the width of the amorphous Gaussian peak is too low, there is a risk of fitting crystalline contribution with this peak and thus over-fitting the amorphous contribution, especially for the Gaussian+ linear method. Publishing the enforced fitting limits along with the obtained crystallinity values will make these results more comparable with other research. The 2D Rietveld method was used with the same amorphous model as the Gaussian+ linear model, but yielded higher crystallinity values. This further suggests that the simpler Gaussian+ linear method might overestimate the amorphous contribution.
Methods 4 and 5, Amorphous subtraction and Amorphous fitting, are closely related to each other. Amorphous subtraction is more sensitive to the exact shape of the amorphous standard than the Amorphous fitting method. In the Amorphous subtraction method the amorphous model cannot surpass the sample intensity even if the shape of the model is wrong in some part of the selected scattering angle range. Since the Amorphous subtraction method does not model the crystalline contribution it is also difficult to quantify how well the chosen amorphous standard fits the data.
Method 5, the Amorphous fitting, is not as vulnerable to crystallite size effects as other methods. However, direct comparisons between crystallinity values obtained by it for different data can be difficult due to factors such as the choice of the scattering angle region, the choice of the amorphous model and the different corrections and background subtractions. Since the Amorphous fitting method gave values below 80 % even for the computational models that were 100 % crystalline, it is not a good method for determining whether a sample is fully crystalline or not. Furthermore, the crystalline model of methods 3 and 5 includes only the 18 most significant peaks. This can cause a systematic underestimation of the crystalline component. However, for samples with 60 % cellulose crystallinity, the values obtained by Amorphous fitting were similar to those obtained by NMR.
One of the biggest challenges in using the Amorphous fitting method and the Amorphous subtraction method is to find an appropriate amorphous model. Ideally the amorphous component should be measured separately and then used in the fitting process. As the choice of an amorphous model affects the absolute values of sample crystallinity values obtained, amorphous standards should be freely available.

Scattering intensities from materials considered for an amorphous model, vertically shifted for clarity.

The crystallinity determined using different amorphous backgrounds as a function of corresponding crystallinity values using the sulphate lignin background. All results are from the Amorphous fitting method
In this paper, different standards were considered for the amorphous component of the Amorphous fitting method (Fig. 6). A two-sided t-test showed no differences (for significance level \(\alpha = 0.05\)) in the means of obtained crystallinities for the amorphous standards. The exception was the crystallinity obtained with the ball-milled cellulose (Avicel), which yielded statistically significantly (\(\alpha =0.01\)) lower crystallinity means than all the other curves. Also an excellent linearity \(r^{2}\ge 0.94\) was found for all the other amorphous standards except for the ball-milled cellulose (\(r^{2}=0.82\), Fig. 7), where the variation from linearity was the highest for the low-crystallinity samples. The sulphate lignin data has been used extensively for wood and wood-like samples (Andersson et al. 2003; Leppänen et al. 2011; Borrega et al. 2015; Dixon et al. 2015) and was chosen here as well for the low crystallinity samples, which had high non-cellulosic content. In this approach, the sulphate lignin is used as a model for all amorphous material in the sample: for example lignin, xylan and amorphous cellulose. For samples of high cellulose content and samples of highly processed cellulosic materials, the ball-milled cellulose model was chosen because these samples contain little or no lignin.
In Rietveld refinement, the crystalline contribution contains more fitting parameters (18) than the amorphous contribution (5). The crystalline contribution may then be over-fitted and the sample crystallinity values overestimated. On the other hand, since the RR is done in 2D, it takes into consideration the preferred orientation. Assuming that the amorphous contribution is isotropic and the crystalline cellulose has a strong preferred orientation, a more accurate upper limit for the amorphous contribution can be obtained from the 2D diffraction pattern than from the averaged one-dimensional data. Both of these factors explain why the RR yields higher sample crystallinity values than methods 2 to 5. De Figueiredo and Ferreira (2014) have used a one-dimensional RR with a corundum calibration standard to assess the crystallinity of Avicel PH-102 (MCC2). Their symmetrical transmission geometry measurement resulted in a crystallinity value of 51 % (compare with Table 2).
Careful crystallinity analysis should also account for other factors that may have a measurable effect on obtained crystallinity values. These include the contribution from non-cellulosic crystalline material, water background, effects from sample texture and measurement geometry. Different devices and geometries can result in peak shapes that are different from the Gaussian shape used here. Several different peak shapes have been suggested (Wada et al. 1997) and each user should check with a calibration sample which peak shape fits best to the data from their device. Other factors such as inelastic scattering and paracrystallinity can be included in a more sophisticated model if the data quality is high. The lack of features in challenging cellulose samples measured with tabletop devices calls for a simplified model, such as the two-phase model used in this article.
Information on sample paracrystallinity can be obtained with NMR by separating the signal into multiple components (Larsson et al. 1997). NMR yields information on the physical and chemical environment of individual atoms whereas XRD is sensitive to long-range order. Due to these underlying differences between the measurement modalities, NMR-crystallinity should not be expected to be identical with XRD-crystallinity. However, both methods can be interpreted with a simplified two-phase model in which a material consists of only purely crystalline and amorphous components. In this model the paracrystalline contribution is included in the NMR-crystallinity (Tolonen et al. 2011). This streamlined model is used in this article when NMR- and XRD-crystallinities are compared.
This study assumed that contribution from water background is negligible. As moisture content was not measured separately for all samples used in the analysis, no direct correction could be made. For the case of wood samples, zero moisture content is a reasonable approximation for low humidity (equilibrium moisture content (EMC) 2.4 % at 298 K and 10 % humidity), but not for high humidity conditions (EMC 10.8 % at 50 % humidity) (Simpson 1998). For bamboo samples similar to the ones used in this study (measured at relative humidities between 35.8 and 39.3 %) a mass drop of (\(4.6\pm 0.2\)) % was experienced when the samples were heated in oven at 50 \(^{\circ }\)C for 98 h. These values suggest that in the general case the water background is not negligible and careful analysis should consider also the water background. If the measurement cannot be performed under low humidity conditions and absolute crystallinity values are of interest, water background should be subtracted from the measured intensities. In any case, all samples should be measured under similar humidity conditions.
Finally, for non-powder samples, different measurement geometries result in different sample crystallinity values due to texture effects. Using the peak weight parameters from Paakkari et al. (1988) and the relative peak heights for cellulose I\(\beta\) from French (2014), the difference in total intensity of the major diffraction peaks (110, \(1\overline{1}0\), 102, 200 and 004) between symmetrical reflection and symmetrical transmission is approximately 40 %. Values obtained with perpendicular transmission were found to fall between the values obtained with the two other geometries. The texture effects can be reduced to some extent by using multiple measurement geometries (Paakkari et al. 1988) or by choosing the most appropriate measurement geometry. However, neither of these approaches work for 2D diffraction, where the measurement geometry is effectively limited to perpendicular transmission. For samples with strong preferred orientation, 2D diffraction is therefore more suitable for determining differences in sample crystallinity values rather than for assessing absolute crystallinity values. In this case only samples with similar preferred orientation should be compared as preferred orientation affects the crystallinity values.
Conclusions
In order to avoid crystallite size effects it is better to use area-based fitting methods than peak height based methods. The Amorphous fitting method showed the least variation with respect to the crystallite size for fully crystalline cellulose models and thus it should be used when comparing samples of different crystallite sizes. That method also showed the best correspondence with the available NMR crystallinity results. The values obtained from the Segal peak height method should be considered relative values and comparisons of values obtained from different studies should be avoided.
An ideal, fully quantitative and optimized assessment of cellulose crystallinity should include the contribution of all diffraction peaks. For samples with preferred orientation, this requires the use of at least two measurement geometries and is more reliably performed using two-dimensional scattering data. Although the choice of refined parameters and their fitting limits affects the obtained crystallinity values, the 2D Rietveld method is a promising method for evaluating sample crystallinity.
Relative differences in crystallinity within a sample set can be distinguished with many different crystallinity analysis methods. Comparison between results from different research groups is more challenging and the availability of good, open-access amorphous standards would be beneficial to the field. We include the amorphous sulphate lignin model in Online Resource 2 for this purpose. Comparing the crystallinity of different samples by their XRD-crystallinity values is problematic unless identical measurement and analysis protocols have been used.
Notes
The nata de coco sample could not be fitted without increasing the upper boundaries of the \(L_{\delta }\) and \(p_{\delta }\) parameters of Oliveira and Driemeier (2013). These parameters model the differences in the crystallite size and diffraction peak shape corresponding to the 110 and 1\(\overline{1}\)0 peaks.
Statistical significance based on a one-sided t-test with a significance level of 0.01.
References
Agarwal UP, Reiner RR, Ralph SA (2013) Estimation of cellulose crystallinity of lignocelluloses using near-IR FT-Raman spectroscopy and comparison of the Raman and Segal-WAXS methods. J Agric Food Chem 61:103–113. doi:10.1021/jf304465k
Andersson S, Serimaa R, Paakkari T, Saranpää P, Pesonen E (2003) Crystallinity of wood and the size of cellulose crystallites in Norway spruce (Picea abies). J Wood Sci 49:531–537. doi:10.1007/s10086-003-0518-x
Bansal P, Hall M, Realff MJ, Lee JH, Bommarius AS (2010) Multivariate statistical analysis of X-ray data from cellulose: a new method to determine degree of crystallinity and predict hydrolysis rates. Bioresour Technol 101:4461–4471. doi:10.1016/j.biortech.2010.01.068
Barnette AL, Lee C, Bradley LC, Schreiner EP, Park YB, Shin H, Cosgrove DJ, Park S, Kim SH (2012) Quantification of crystalline cellulose in lignocellulosic biomass using sum frequency generation (SFG) vibration spectroscopy and comparison with other analytical methods. Carbohydr Polym 89:802–809. doi:10.1016/j.carbpol.2012.04.014
Borrega M, Ahvenainen P, Serimaa R, Gibson L (2015) Composition and structure of balsa (Ochroma pyramidale) wood. Wood Sci Technol 49:403–420. doi:10.1007/s00226-015-0700-5
Chen C, Luo J, Qin W, Tong Z (2013) Elemental analysis, chemical composition, cellulose crystallinity, and FT-IR spectra of Toona sinensis wood. Monatshefte für Chem Chem Mon 145:175–185. doi:10.1007/s00706-013-1077-5
Davies LM, Harris PJ, Newman RH (2002) Molecular ordering of cellulose after extraction of polysaccharides from primary cell walls of Arabidopsis thaliana: a solid-state CP/MAS (13)C NMR study. Carbohydr Res 337:587–593. doi:10.1016/S0008-6215(02)00038-1
De Figueiredo LP, Ferreira FF (2014) The rietveld method as a tool to quantify the amorphous amount of microcrystalline cellulose. J Pharm Sci. doi:10.1002/jps.23909
Debye P, Bueche AM (1949) Scattering by an inhomogeneous solid. J Appl Phys 20:518–525. doi:10.1063/1.1698419
Ding SY, Himmel ME (2006) The maize primary cell wall microfibril: a new model derived from direct visualization. J Agric Food Chem 54:597–606. doi:10.1021/jf051851z
Dixon P, Ahvenainen P, Aijazi A, Chen S, Lin S, Augusciak P, Borrega M, Svedström K, Gibson L (2015) Comparison of the structure and flexural properties of Moso, Guadua and Tre Gai bamboo. Constr Build Mater 90:11–17. doi:10.1016/j.conbuildmat.2015.04.042
Driemeier C (2014) Two-dimensional rietveld analysis of celluloses from higher plants. Cellulose 21:1065–1073. doi:10.1007/s10570-013-9995-2
Driemeier C, Calligaris GA (2010) Theoretical and experimental developments for accurate determination of crystallinity of cellulose I materials. J Appl Crystallogr 44:184–192. doi:10.1107/S0021889810043955
French AD (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21:885–896. doi:10.1007/s10570-013-0030-4
French AD, Santiago Cintrón M (2013) Cellulose polymorphy, crystallite size, and the Segal Crystallinity Index. Cellulose 20:583–588. doi:10.1007/s10570-012-9833-y
Gupta B, Agarwal R, Sarwar Alam M (2013) Preparation and characterization of polyvinyl alcohol-polyethylene oxide-carboxymethyl cellulose blend membranes. J Appl Polym Sci 127:1301–1308. doi:10.1002/app.37665
Hänninen T, Tukiainen P, Svedström K, Serimaa R, Saranpää P, Kontturi E, Hughes M, Vuorinen T (2012) Ultrastructural evaluation of compression wood-like properties of common juniper (Juniperus communis L.). Holzforschung 66:389–395. doi:10.1515/hf.2011.166
Himmel ME, Ding SY, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315:804–807. doi:10.1126/science.1137016
Jeoh T, Ishizawa CI, Davis MF, Himmel ME, Adney WS, Johnson DK (2007) Cellulase digestibility of pretreated biomass is limited by cellulose accessibility. Biotechnol Bioeng 98:112–122. doi:10.1002/bit.21408
Kim SH, Lee CM, Kafle K (2013) Characterization of crystalline cellulose in biomass: Basic principles, applications, and limitations of XRD, NMR, IR, Raman, and SFG. Korean J Chem Eng 30:2127–2141. doi:10.1007/s11814-013-0162-0
Kim SS, Kee CD (2014) Electro-active polymer actuator based on PVDF with bacterial cellulose nano-whiskers (BCNW) via electrospinning method. Int J Precis Eng Manuf 15:315–321. doi:10.1007/s12541-014-0340-y
Kljun A, Benians TAS, Goubet F, Meulewaeter F, Knox JP, Blackburn RS (2011) Comparative analysis of crystallinity changes in cellulose I polymers using ATR-FTIR, X-ray diffraction, and carbohydrate-binding module probes. Biomacromolecules 12:4121–4126. doi:10.1021/bm201176m
Kontro I, Wiedmer SK, Hynönen U, Penttilä PA, Palva A, Serimaa R (2014) The structure of Lactobacillus brevis surface layer reassembled on liposomes differs from native structure as revealed by SAXS. Biochim Biophys Acta 1838:2099–2104. doi:10.1016/j.bbamem.2014.04.022
Larsson PT, Wickholm K, Iversen T (1997) A CP/MAS 13C NMR investigation of molecular ordering in celluloses. Carbohydr Res 302:19–25. doi:10.1016/S0008-6215(97)00130-4
Leppänen K, Andersson S, Torkkeli M, Knaapila M, Kotelnikova N, Serimaa R (2009) Structure of cellulose and microcrystalline cellulose from various wood species, cotton and flax studied by X-ray scattering. Cellulose 16:999–1015. doi:10.1007/s10570-009-9298-9
Leppänen K, Bjurhager I, Peura M, Kallonen A, Suuronen JP, Penttilä PA, Love J, Fagerstedt K, Serimaa R (2011) X-ray scattering and microtomography study on the structural changes of never-dried silver birch, European aspen and hybrid aspen during drying. Holzforschung 65:865–873. doi:10.1515/HF.2011.108
Liitiä T, Maunu SL, Hortling B, Tamminen T, Pekkala O, Varhimo A, Liiti T, Varhimo A (2003) Cellulose crystallinity and ordering of hemicelluloses in pine and birch pulps as revealed by solid-state NMR spectroscopic methods. Cellulose 10:307–316. doi:10.1023/A:1027302526861
Lindner B, Petridis L, Langan P, Smith JC (2015) Determination of cellulose crystallinity from powder diffraction diagrams. Biopolymers 103:67–73. doi:10.1002/bip.22555
Nam S, French AD, Condon BD, Concha M (2016) Segal crystallinity index revisited by the simulation of X-ray diffraction patterns of cotton cellulose I\(\beta\) and cellulose II. Carbohydr Polym 135:1–9. doi:10.1016/j.carbpol.2015.08.035
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose I\(\beta\) from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124:9074–9082. doi:10.1021/ja0257319
Oliveira RP, Driemeier C (2013) CRAFS: a model to analyze two-dimensional X-ray diffraction patterns of plant cellulose. J Appl Crystallogr 46:1196–1210. doi:10.1107/S0021889813014805
Paakkari T, Blomberg M, Serimaa R, Järvinen M (1988) A texture correction for quantitative X-ray powder diffraction analysis of cellulose. J Appl Crystallogr 21:393–397. doi:10.1107/S0021889888003371
Park S, Johnson DK, Ishizawa CI, Parilla PA, Davis MF (2009) Measuring the crystallinity index of cellulose by solid state 13C nuclear magnetic resonance. Cellulose 16:641–647. doi:10.1007/s10570-009-9321-1
Park S, Baker JO, Himmel ME, Parilla PA, Johnson DK (2010) Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels 3:10. doi:10.1186/1754-6834-3-10
Parviainen H, Parviainen A, Virtanen T, Kilpeläinen I, Ahvenainen P, Serimaa R, Grönqvist S, Maloney T, Maunu SL (2014) Dissolution enthalpies of cellulose in ionic liquids. Carbohydr Polym 113:67–76. doi:10.1016/j.carbpol.2014.07.001
Rietveld HM (1969) A profile refinement method for nuclear and magnetic structures. J Appl Crystallogr 2:65–71. doi:10.1107/S0021889869006558
Schenzel K, Fischer S, Brendler E (2005) New method for determining the degree of cellulose I crystallinity by means of FT Raman spectroscopy. Cellulose 12:223–231. doi:10.1007/s10570-004-3885-6
Segal L, Creely J, Martin A, Conrad C (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the X-Ray diffractometer. Text Res J 29:786–794. doi:10.1177/004051755902901003
Simpson WT (1998) Equilibrium moisture content of wood in outdoor locations in the United States and worldwide. Technical report, U.S. Department of Agriculture, Forest Service, Forest Products Laboratory
Siró I, Plackett D (2010) Microfibrillated cellulose and new nanocomposite materials: a review. Cellulose 17:459–494. doi:10.1007/s10570-010-9405-y
Sisson WA (1933) X-ray analysis of fibers. Part I, literature survey. Text Res J 3:295–307
Terinte N, Ibbett R, Schuster KC (2011) Overview on native cellulose and microcrystalline cellulose I structure studied by x-ray diffraction (WAXD): comparison between measurement techniques. Lenzing Ber 89:118–131
Testova L, Borrega M, Tolonen LK, Penttilä PA, Serimaa R, Larsson PT, Sixta H (2014) Dissolving-grade birch pulps produced under various prehydrolysis intensities: quality, structure and applications. Cellulose 21:2007–2021. doi:10.1007/s10570-014-0182-x
Thygesen A, Oddershede J, Lilholt H, Thomsen AB, Ståhl K (2005) On the determination of crystallinity and cellulose content in plant fibres. Cellulose 12:563–576. doi:10.1007/s10570-005-9001-8
Tolonen LK, Zuckerstätter G, Penttilä PA, Milacher W, Habicht W, Serimaa R, Kruse A, Sixta H (2011) Structural changes in microcrystalline cellulose in subcritical water treatment. Biomacromolecules 12:2544–2551. doi:10.1021/bm200351y
Wada M, Okano T, Sugiyama J (1997) Synchrotron-radiated X-ray and neutron diffraction study of native cellulose. Cellulose 4:221–232
Zavadskii AE (2004) X-ray diffraction method of determining the degree of crystallinity of cellulose materials of different anisotropy. Fibre Chem 36:425–430. doi:10.1007/s10692-005-0031-7
Acknowledgments
P.A. has received funding from the Finnish National Doctoral Program in Nanoscience. We would like to thank Dr. Seppo Andersson and Dr. Paavo Penttilä for providing some of the used X-ray diffraction data, and Dr. Carlos Eduardo Driemeier for giving access to the CRAFS package source code. We would also like to thank professor Ritva Serimaa for fruitful discussions on cellulose crystallinity and for motivating us to carry out this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ahvenainen, P., Kontro, I. & Svedström, K. Comparison of sample crystallinity determination methods by X-ray diffraction for challenging cellulose I materials. Cellulose 23, 1073–1086 (2016). https://doi.org/10.1007/s10570-016-0881-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-016-0881-6