
Abstract
The potential of three different logging residues (woody chips, branches and pine needles) as renewable resources to produce environmentally friendly polymeric materials and/or biofuel has been critically evaluated in terms of their structure, chemical composition and thermal properties. Woody chips constitute the most attractive forest residue to be processed into polymeric materials in terms of their highest cellulose content, crystallinity and thermal stability. In contrast, pine needles and branches offer higher heating values and optimum product distribution for solid fuel applications due to their higher lignin content. In general, forest residual biomass has great potential for conversion into new added value products, such as composites or solid biofuel, thus constituting a sustainable waste management procedure from a biorefinery perspective. The correlation between the chemical and structural properties with the thermal/pyrolytic behavior of residual biomass offers valuable insights to assess their sustainable exploitation.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The extraction of biomass from forests to produce paper, furniture and energy generates large amounts of forest residues (branches, tops, bark and un-merchantable wood). These logging residues are often subjected to controlled burning or are left in large piles, causing a negative environmental impact (Siqueira et al. 2010) and risks of pests and fire in warmer countries (Thakur et al. 2011). The use of forest biomass residues to produce environmentally friendly materials constitutes an attractive waste management solution from a biorefinery perspective, contributing to a sustainable future economy. Residual biomass has many significant advantages over fossil resources, such as renewability, sustainability, lower impact on global warming, biodegradability, low cost and non-dangerous emissions during processing, storage and disposal (Mohanty et al. 2000). Nevertheless, interest in using forest residues for polymeric material applications is relatively recent: pine cones were considered for particleboard production (Sahin and Arslan 2011); spruce bark was proposed to design thermoplastic composites (Yemele et al. 2010); pine needles were used to fabricate hydrogels (Chauhan et al. 2007) and to reinforce polymers such as urea–formaldehyde (Thakur et al. 2011), phenol–formaldehyde (Thakur and Singha 2010, 2011) and resorcinol–formaldehyde (Thakur et al. 2010). In general, the main aim of these studies was focused on the determination of the mechanical, thermal, acoustic, flammability and biological properties of the developed products rather than on the characterization of the biomass itself.
Understanding the chemical structure, morphology, composition and thermal properties of the biomass is fundamental to understanding the properties and determining the applicability of the later developed products (Moriana et al. 2014a). However, only a few studies (Dong et al. 2014; Le Normand et al. 2014) have been reported on the physico-chemical characterization of forest residues as a raw material for material applications. On the other hand, greater interest has been devoted to the fundamental understanding of the pyrolytic behavior of forest residual biomass for the design of effective biomass-conversion technologies to produce bioenergy (Amutio et al. 2013; Balogun et al. 2015; Font et al. 2009). In general, pyrolysis plays a key role in determining the reaction kinetics, and it is therefore critical in the thermochemical conversion technologies to the design of reactors and the determination of product distribution, composition and properties (Raveendran et al. 1995). The assessment of the temperature dependence of the biomass by means of thermal studies is also essential to resolve the drawbacks related to their temperature sensitivity in the polymeric material processing technologies (Moriana et al. 2014b).
The aim of this work is to correlate the chemical composition and structure of three logging forest residues (woody chips, pine needles and branches) with their thermal properties, in order to assess their potential to produce polymeric material products as an alternative to the most traditional biofuel production. This study is the first step in a broader scientific project towards the development of an effective forestry waste biomass revalorization procedure into advanced bio-based products, to ensure the optimal exploitation of our natural resources.
Experimental
Materials
The logging residues were kindly provided by SCA R&D Centre (Sundsvall, Sweden) and were collected from a Swedish forest mostly composed of Scots pines (Pinus sylvestris) and Norway spruce (Picea abies). These forest residues from final felling were composed of branches and chips of different sizes together with needles. A first classification of these forest residues was performed according to different physical properties (Table 1). The weight percentage of each type of residue was also assessed to obtain a general distribution of the composition of this Swedish forest biomass waste.
Among these different types of forest residues, medium woody chips (W), large branches (B) and pine needles (PN) were selected for evaluation of their chemical, structural and thermal properties. The selected forest residues were separated and dried at room temperature for 1 week. Later, they were successively milled in different crushers: they were first particulated with a Moretto granulator (Universalmuehle GR 18/10 M1 A, Germany); then, they were milled to powder using a basic analytical IKA A11 mill (Sigma-Aldrich, Germany); and finally, the size was controlled with a Wiley Mill Acm 82302 (Acmas Technocracy Pvt. Ltd, Germany) to a 20 mesh.
Sulfuric acid (96 wt%), ethanol (99 %), sodium hydroxide (NaOH), methyl iodide (CH3I), dimethyl sulfoxide (DMSO), dichloromethane, trifluoroacetic acid (TFA), sodium borohydride (NaBH4), ammonia (NH3), pyridine, acetic anhydride, and ethyl acetate were all analytical grade (Sigma-Aldrich, Germany) and were used without further purification. Ultrapure water (Milli-Q, Millipore, MA) was used in all cases.
Chemical composition analysis
The dry content of the samples was measured using a Mettler Toledo HB43 moisture analyzer (Columbus, OH, USA). The chemical composition of the materials was measured in terms of neutral carbohydrates and Klason lignin following the Tappi test method T222 om-06 (TAPPI 2006). The hydrolysis was carried out using 72 % H2SO4 in an autoclave for 60 min at 120 °C. The released carbohydrates were examined by high-pH anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD) on a ICS3000 system (Dionex, Sunnyvale, CA) using a Carbopac PA1 column (Dionex, Sunnyvale, CA) at 30 °C at a flow rate of 1 mL min−1. Quantification was performed by external calibration using neutral sugar and uronic acid standards (glucose, mannose, xylose, arabinose, galactose, rhamnose, galacturonic and glucuronic acid). The ash content of the samples was calculated after pyrolysis of the dry samples in a furnace at 525 °C for over 6 h, according to the Tappi test method T211 om-02 (TAPPI 2012). The soluble extractive content was evaluated using the same methodology previously proposed for agricultural feedstock (McIntosh and Vancov 2011) and bark (Miranda et al. 2012). According to NREL’s LAP (NREL 2005), 10 g of each dried forest residue was water extracted in a soxhlet for 6 h. A successive ethanol extraction was performed in the same soxhlet instrument for a further 6 h. The obtained extractives from both solvents were dried in an oven at 105 °C and weighed to determine the total amount of soluble extractives. Three replicates of each sample were performed.
Polysaccharide composition by glycosidic linkage analysis
The polysaccharide composition of each forest residue was estimated by glycosidic linkage analysis, following the methodology reported by Pettolino et al. (2012). In brief, 1 mg of each sample was fully methylated with CH3 under alkaline conditions (excess of NaOH) using the conditions reported by Ciucanu and Kerek (1984). The partially methylated polysaccharides were hydrolyzed with 2 M TFA at 121 °C for 3 h and further derivatized into permethylated alditol acetates (PMAAs) by reduction with NaBH4 and acetylation with acetic anhydride and pyridine. The PMAAs were identified and quantified by gas chromatography–mass spectrometry (GC–MS) using a HP-6890 GC coupled to a HP-5973 electron-impact mass spectrometer (EI-MS) (Agilent Technologies) on a SP-2380 capillary column (30 m × 0.25 mm ID, Agilent Technologies) with a temperature ramp of 1 °C min−1 from 160 to 210 °C. The samples were analyzed in triplicate. The fragmentation spectra of the PMAAs by EI-MS could be assigned to the different glycosidic linkages by comparison to reference polysaccharide derivatives. The relative quantification of the different glycosidic linkages was normalized using the monosaccharide composition from HPAEC-PAD. The composition of each polysaccharide could be obtained from the relative abundance of the glycosidic linkages (Pettolino et al. 2012). The hemicellulose content was calculated from the glycosidic linkages assigned to mannans, xylans, and xyloglucan; the pectin content was assigned to the linkages arising from arabinans, galactans and rhamnogalacturonans; and the cellulose content was estimated from the PMAA peak assigned to (1 → 4)-glucopyranosyl units (4-Glcp), corrected according to the expected abundance in galactoglucomannan.
Scanning electron microscopy (SEM)
SEM micrographs from forest residues were obtained using a JEOL JSM-5400 scanning electron microscope (JEOL Ltd., Japan) at 1.0 kV after gold/palladium sputtering on a Cressington 208HR Sputter coater.
X-ray diffraction analysis (XRD)
The X-ray diffractograms of all the samples were determined on a PANalytical X’Pert PRO MRD (X’Pert Pro., Netherlands) equipped with an X’Celerator detector. The analysis was performed with monochromatic Cu Kα radiation (λ = 1.542 Å) at 30 mA and 40 kV in the range 2θ = 10°–60°. The empirical method proposed by Segal et al. (1959) was used to determine the crystallinity index (CrI) (Eq. 1). This approach has been reported to overestimate the CrI values (Peng et al. 2013); however, it is a convenient and quick methodology to compare the relative crystallinity of the cellulosic samples.
Here, I200 is the maximum intensity of the 200 lattice plane, and Iam is the intensity from the amorphous phase around 2θ = 18°.
The apparent crystallite size (D) in the direction perpendicular to the 200 lattice plane was calculated using the Scherrer equation (Eq. 2) (Ahtee et al. 1983):
Here, K is a constant of value 0.94, λ is the X-ray wavelength (0.1542 nm), H is the half height width of the diffraction band in radians and θ is the Bragg angle corresponding to the (200) plane.
The proportion of crystalline interior chains (X) was also calculated (Eq. 3):
Here, D is the apparent crystallite size for the reflection of plane (200), and h = 0.57 nm is the layer thickness of the surface chain (Popescu et al. 2011).
Fourier transform infrared spectrometry (FTIR)
FTIR spectra of the forest residues were recorded on a FTIR spectrometer Spectrum 2000 from Perkin Elmer (Wellesley, MA, USA), equipped with a Golden single-reflection accessory for Attenuated Total Reflection (ATR) measurements. Each spectrum was collected after 16 scans between 4000 and 600 cm−1 at intervals of 1 cm−1. The FTIR spectra were fitted by an automatic base line correction using OMNIC 4.0 software. The measurements were repeated seven times for each sample.
Thermal degradation behavior and kinetics
The thermal degradation behavior, including the thermal stability and kinetics of the forest residues, was evaluated by dynamic thermogravimetric analysis (TGA) using a Mettler-Toledo TGA/SDTA 851 (Columbus, OH). Approximately 6 mg of each sample were heated between 25 and 600 °C at different heating rates in the range of 3–15 °C min under a nitrogen flow of 50 mL min. The thermogravimetric (TG) and derivative thermogravimetric (DTG) curves were obtained using STARe Evaluation Software (Mettler-Toledo, Columbus, OH, USA). The degradation onset (initial decomposition temperature) was obtained by extrapolating the slope of the DTG curve to zero according to the first local maximum in the D2TG curve (Moriana et al. 2014b). The measurements were performed at least four times; the average and the standard deviation from these data were considered representative.
The Friedman (Friedman 1964) differential isoconversional method was proposed to evaluate the thermal degradation kinetics in terms of activation energy (Ea) for each forest residue (Eq. 4). This parameter was defined as the most sensitive to assess the differences between the thermal degraded materials (Moriana et al. 2014c).
where α is the conversion degree defined as α = [(w0 − w)/(w0 − w∞)], where w, w0, and w∞, are the actual, initial and final weights of the sample, respectively; A is a pre-exponential factor; Ea is the apparent activation energy; T is the absolute temperature; and R is the gas constant.
Proximate analysis
The proximate analysis of the forest residues was also performed using Mettler-Toledo TGA/SDTA 851 (Columbus, OH, USA) equipment. In this case, both nitrogen and oxygen atmospheres were used together following the same protocol described by Moriana et al. (2014b). The temperature program consisted of a heating ramp from 25 to 120 °C at 50 °C min, a 3 min isothermal stage at 120 °C, followed by another heating ramp to 950 °C at 100 °C min and finally a cooling process to 450 °C at −100 °C min−1 under a N2 atmosphere. Next, the atmosphere was changed to O2 and a new heating process was started from 450 to 800 °C at 100 °C min. The proximate analysis data (ash, volatile matter and fixed carbon) were obtained by the direct measurement of the weight changes in each experimental curve. The obtained percentages for the ash, volatile matter and fixed carbon contents of each sample were used to determine the net heating value (NHV), as it was indicated in previous studies (Moriana et al. 2014b).
Results
Chemical composition and linkage analysis
Wood and plants are hierarchical biocomposites based on semicrystalline cellulose microfibrils reinforcing amorphous matrices, which are mainly composed of hemicelluloses, pectins, lignin and extractives. The average carbohydrate, lignin, ash, and extractive contents for the forest residues are shown in Table 2. In general, wood chips exhibit a significantly higher carbohydrate content than branches and pine needles. On the other hand, pine needles show a remarkable extractive content together with a higher lignin and ash content. The comprehensive polysaccharide composition was evaluated by combined monosaccharide and glycosidic linkage analysis. The procedure allows the quantification of monosaccharides (both neutral sugar and uronic acids) in the forest residues, and the assignment of the glycosidic linkages of each monosaccharide to an individual polysaccharide population (Pettolino et al. 2012). We are aware that uronic acid containing samples are more resistant to acidic hydrolytic conditions. Other procedures (e.g. methanolysis) have been proposed to determine in a more precise way the content of uronic acids in wood and pulp samples (Willför et al. 2009); however these methods are not capable of hydrolyzing crystalline cellulose completely and therefore are not suitable to assess the carbohydrate composition of our woody samples. The proposed hydrolytic conditions provide a good estimation of the lignin and carbohydrate composition of the woody tissues, balancing the release of monosaccharides (especially those arising from crystalline cellulose) and minimizing the possible degradation of pentoses and the incomplete release of uronic acids. Additionally, methylation analysis provides more insightful information of the polysaccharide fractions present in the woody samples by the relative determination and assignation of the glycosidic linkages to specific polysaccharides. The relative cellulose content ascribed to the linear 4-Glcp linkages (excluding those arising from hemicelluloses such as galactoglucomannan) is highest for wood chips, followed by the branches and pine needles. Pine needles have a large proportion of heterogeneous hemicelluloses and pectins in their structure, which is assigned to the different function of this tissue in the plant. Galactoglucomannan seems to be the main hemicellulose in each forest residue, followed by glucuronoarabinoxylan, which is common for softwoods such as pine and spruce. The (galacto)(gluco)mannan content in woody chips can be estimated as 12–15 wt%, whereas the (glucurono)(arabino)xylan content is 11–12 wt%. Pectic polysaccharides, such as galacturonans, arabinans and arabinogalactans, can be observed in each residue but particularly in branches and pine needles, with an estimated content of around 7 wt% in both residues. The relative polysaccharide composition of each residue will have an important influence on both the thermal properties and on their potential exploitation in bio-based polymeric materials and biofuels.
Scanning electron microscopy
From a structural point of view, lignocellulosic woody materials consist of bundles of elongated and cylindrical honeycomb cells with different sizes, shapes and arrangements depending on the raw material. The walls of conifer cells are called tracheids. Figure 1 shows tangential and radial tracheids of the woody chips. The tracheids have an average width of 26 ± 4 µm, which is typical for Scandinavian softwood tracheid cells (Sjöström 1981). However, the width of the supporting tissue cells for branches and pine needles are smaller (around 20 ± 2 and 18 ± 2 µm, respectively) and more similar in width to hardwood tracheids (Beck-Candanedo et al. 2005). These dimensions give an indication of the size of the cellulosic fibers that could be obtained from the forest residues to be later applied in the development of cellulosic polymeric materials, such as composites.

SEM micrographs of the studied forest residues: a walls of the woody chip tracheids, b transversal image of branches showing the surface and interior morphology, c detail of the surface morphology of branches, d internal aspect of the branches, e pine needle surface morphology, f detail of the surface morphology of pine needles
X-ray diffraction analysis
Naturally occurring cellulose fibers in lignocellulosic tissues normally have a crystalline structure known as cellulose I (Sjöström 1981). Cellulose I has two polymorphs that coexist in the cellulose structure: a two-chain monoclinic structure Iβ and a one-chain triclinic unit cell Iα. The Iα lattice planes for the triclinic structure are (110), (010), and (100), whereas the three lattice planes for the Iβ polymorph are (200), (110) and (110) for the monoclinic structure. The cellulose crystal structure is normally defined by different X-ray diffraction parameters, such as the crystallinity index (CrI), the apparent crystalline size (D) and the proportion of crystallite interior chains (X) (Table 3). Figure 2 shows the X-ray diffraction patterns for the three studied forest residues. All the patterns are typical of semi-crystalline materials, showing an amorphous broad hump and two characteristic crystalline peaks. The first crystalline peak is associated with the overlapping of the (110) and (110) planes, and the second peak is characteristic of the (200) lattice plane, in cellulose I (Park et al. 2010).

Diffraction patterns for the three forest residues
In general, woody chips show the highest diffraction parameters CrI, X and D (Table 3). Andersson et al. (2003) reported similar CrI values for early Norway spruce samples. Woody chips exhibit a higher CrI than the branches, which correlates well with their higher cellulose content. However, the branches and pine needles have similar CrI and X values despite the latter having a lower cellulose content. This fact indicates a higher percentage of crystalline cellulose chains in the pine needles than in the branches. It is known that the crystalline structure of cellulose plays an important role in its thermal properties (Poletto et al. 2011) and determines the mechanical and thermal properties of cellulosic products, such as in reinforced cellulosic composites (Silvério et al. 2013). The crystallite size of the branches and pine needles are similar (approximately 1.7 nm), whereas the woody chips crystallite thickness is higher (approximately 2.7 nm). Crystallite sizes within the range of 2.5–2.8 nm were previously reported for Norway spruce (Andersson et al. 2003), pinus (Tanaka et al. 1981) and other softwoods (Nomura and Yamada 1972). However, lower crystallite dimensions were also found within the range of 1.5–3.0 nm, including other species such as bacteria, algae and coleoptiles of maize (Ohad et al. 1962; Ohad and Danon 1964; Schnepf 1965).
Fourier transform infrared spectrometry
Inter- and intramolecular hydrogen bonds are formed within the Iα and Iβ polymorphs that coexist in cellulose, determining the stability and properties of the crystalline polymorphs and of cellulose itself (Fengel 1978). Fourier transform infrared spectrometry is sensitive to different conformations and local molecular environments of the cellulose molecules, which make it suitable for determining the cellulose crystallinity and hydrogen-bonding patterns, as well as chemical composition. Figure 3 displays the FTIR spectra of each forest residue, where the characteristic bands for hemicellulose/pectins, cellulose and lignin are identified. All three samples present characteristic bands at 3700–2995 cm−1 (OH stretching region); 2915–2900 cm−1 (CH stretching region); 1730 cm−1 (the C=O stretching of methyl esters, carboxylic, uronic ester and acetyl groups); 1640 cm−1 (OH bending of absorbed water); 1509 cm−1 (C=C aromatic skeletal vibration of lignin); 1420–1430 cm−1 (CH2 wagging); 1362–1376 cm−1 (CH bending); 1314–1318 cm−1 (COH bending); 1250 cm−1 (CO stretching of the aryl group in lignin); 1058–1060 and 900 cm−1 (COC, CCO and CCH deformation and stretching from glucopyranose ring); 1130–1000 cm−1 (OH bending); 896–898 cm−1 (CH wagging) and 664 cm−1 (COH bending) (Baeza and Freer 2000).

FTIR spectra of the forest residues
The three forest residues display two overlapped bands at approximately 3331–3347 and 3287–3292 cm−1, related to the intramolecular hydrogen bonding of 3OH···O5 and the intermolecular hydrogen bonding of 6OH···O3. These bands related to the OH stretching modes are more prominent for pine-needles than for woody chips and branches, which could be attributed to a larger number of hydroxyl groups involved in forming hydrogen bonds (Popescu et al. 2011). Woody chips show a maximum peak at 2908 cm−1, which is related to the CH stretching vibrations of cellulose. In contrast, the branches and pine needles show a doublet at approximately 2922 and 2852 cm−1, which is attributed to the antisymmetric and symmetric CH2 stretching vibrations of non-cellulose polysaccharides (such as, hemicelluloses and extractives), respectively (Nelson and O’Connor 1964). The bands at approximately 1730 cm−1 are attributed to the C=O stretching of the methyl esters, acetyl groups and uronic acids in the pectins and hemicelluloses and are of higher intensity for the pine needles and branches than for the woody chips. Again, higher intensities of the bands at approximately 1500 cm−1 that are related to the lignin component are found for pine needles and branches compared to woody chips. The observed differences in the spectra between the residues can be successfully correlated with the chemical analysis, confirming the higher cellulose content in the woody chips and the higher proportion of lignin, pectins and hemicelluloses with respect to cellulose in the pine needles and branches.
The degree of crystallinity of the cellulose can be estimated by infrared spectroscopy using an empirical parameter called crystallinity index (CrI) that was defined as a function of the relative intensity of the bands at 1371 and 2900 cm−1 by Nelson and O’connor (1964). In this study, it was shown how the band at 1371 cm−1 (CH bending) was the most suitable band for indicating crystallinity without being affected by the amount of water adsorbed onto the cellulose or the lattice type. However, it seemed better to use a ratio of this band intensity to that of another band which was unaffected by changes in crystallinity, thus providing a kind of internal standard. The most suitable band found to take this place was the centered at 2900 cm−1 (CH and CH2 stretching). Pine needles show a higher CrI than the branches and woody chips (Table 3), which indicates that the cellulose in pine needles seems to be more crystalline than that in the other forest residues. This has implications for the thermal stability of the cellulose in the forest residues and for the further exploitation possibilities into biomaterials.
Thermal degradation behavior and pyrolysis characteristics
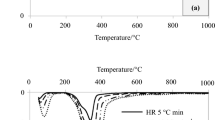
The thermal degradation behavior of the forest residues was analyzed in detail by dynamic thermogravimetry at different heating rates (β). Figure 4a displays the thermogravimetric (TG) and derivative (DTG) curves of the forest residues at a heating rate of 10 °C min, where two main mass loss regions are distinguished at lower and higher temperatures than 150 °C. In the second of these main regions (from 150 to 550 °C), several overlapping peaks can be observed. Similar profiles are found for each material at different heating rates, with a small increase towards higher temperatures with major β values (Fig. 4b). The volatiles released as a function of β, taking into account the different zones within each mass loss region, are classified in Table 4.

Thermogravimetric (TG) and derivative (DTG) curves for the a forest residues at 10 °C min−1 and the b woody chips at different heating rates
The first mass loss region (<150 °C) is attributed to water evaporation (Zone I). The water content of the pine needles and branches is slightly higher than that for the woody chips. This correlates well with a higher pectin content for both pine needles and branches (see chemical composition), since pectins are specially hydrophilic and contribute to a higher hydrated matrix. The second mass loss region (from 150 to 550 °C) shows a complex thermal decomposition with three overlapping peaks in the DTG curve that are related to the main components of the lignocellulosic materials: hemicellulose/pectin, cellulose and lignin (Moriana et al. 2011). The shoulder observed at temperatures below 335 °C is predominantly related to the volatilization of the hemicellulose/pectin (Zone II). The volatiles released in this region show slight differences depending on the forest residue, with higher values for the woody chips, followed by the branches and pine needles. These results are in agreement with the chemical composition analysis for the hemicelluloses/pectins. The second well-defined peak, centered between 346 and 371 °C, is related to the decomposition of cellulose (Zone III). Woody chips exhibit the highest intensity of this main peak attributed to the larger content of cellulose in their composition, as confirmed by the chemical composition analysis. Finally, the tail observed at temperatures exceeding 395 °C is related to the degradation of lignin and/or the final decomposition reactions of the cellulose components to form a pyrolyzed char (Moriana et al. 2014a). The similar amount of released volatiles in this region at temperatures exceeding 395 °C for the branches and pine needles suggests a similar lignin content for both residues and consequently a higher carbohydrate content for branches over pine needles. These results are in good agreement with the chemical composition analysis.
The pyrolysis properties (onset, temperature at maximum rate and endset) and product distribution (char and volatiles) of the forest residues were also assessed and are summarized in Table 4. Among the three studied forest residues, woody chips exhibit the highest production of volatiles together with the highest reactivity and temperature peaks. This result indicates the suitability of this residue to produce gas resources. On the other hand, pine needles and branches have a higher char content due to the higher lignin content (Font et al. 2009), showing their potential for fossil fuel applications. The onset is defined as the initial decomposition temperature of the second main-mass loss region, and it is selected as the most accurate temperature to determine the thermal stability of the samples. The branches and pine needles showed a similar onset, which was lower than the value of the woody chips. The enhanced thermal stability of the woody chips is justified due to its chemical and structural properties, including the higher cellulose and the lower extractive content (Moriana et al. 2014a; Shebani et al. 2008); the higher crystallinity index (Moriana et al. 2014a; Poletto et al. 2014) and larger cellulose crystallite size (Poletto et al. 2014). Therefore, the assessment of the structure and chemical composition of the biomass provides critical information about the thermal stability, which is an important parameter to be controlled in both biomaterials and biofuel applications to determine their processing and service life conditions.
Thermal degradation kinetics
The activation energy (Ea) was experimentally determined via dynamic TGA at different heating rates (β) following a differential isoconversional method, Friedman (Friedman 1964). This method allows the determination of the Ea from the slope of Eq. 4. First, the thermal curves of each forest residue are expressed in terms of α (Fig. 5a), and then, the time derivative of α as a function of temperature is represented for each α, obtaining the Friedman isoconversional plots (Fig. 5b).

a Conversion-degree rate of branches as a function of temperature and b the Friedman isoconversional plot for the woody chips
The plots of the isoconversional method for all the forest residues exhibit a general trend of the Ea values, with changes in the parallelism of the fitted lines at lower (0.1 ≤ α ≤ 0.4) and higher (α ≥ 0.8) α values. This dependence of the Ea on α can be observed more clearly in Fig. 6, where the Ea values obtained by the Friedman method are plotted as a function of α. The Ea calculated and showed in this graph is limited to 0.2 ≤ α ≤ 0.9 due to the lower correlation outside of this range (R2 < 0.89).

Apparent activation energy (Ea) as a function of the conversion degree for the three studied forest residues
The Ea values obtained for the three forest residues are within the range for biomass (110–250 kJ/mol) reported previously by other authors (Amutio et al. 2013; Moriana et al. 2014a, b, c). The woody chips and branches exhibit a first region (0.2 ≤ α ≤ 0.4) with small variations in Ea centered at approximately 212 and 180 kJ/mol, respectively. However, the pine needles display an increasing trend from 215 to 239 kJ/mol in the α range from 0.2 to 0.5. The second region that extends until α = 0.8 shows a stable Ea for all residues, offering the highest Ea for the pine needles (239 kJ/mol), followed by the woody chips (193 kJ/mol) and then the branches (173 kJ/mol). Finally, a third region from α ≥ 0.8 was detected. The Ea for the pine needles and branches decreased, while the Ea for the woody chips increased. These three α regions correspond to Zone II, III and IV, where the devolatilization of the hemicellulose/pectin, cellulose and lignin took place, respectively. In general, the average Ea values at lower conversion in the range 0.2 ≤ α ≤ 0.4 are higher than those reported for the hemicellulose component (105–131 kJ/mol) by Gronli et al. (2002). However, the Ea values for the branches in this region are similar to the previously reported for bast natural fibers (Moriana et al. 2014a), and the Ea values for the woody chips and branches agree with those reported for spruce glucomannan (Moriana et al. 2014b). The Ea values of the region related to the devolatilization of cellulose are in reasonable agreement with literature values for the decomposition of cellulose (Samuelsson et al. 2014). However, significant differences are found for each type of forest residue, indicating that the complexity involved in the lignocellulosic kinetics depends on the characteristics of the biomass resource (Balogun et al. 2015). The highest values obtained for the pine needles (239 kJ/mol) are in agreement with the previously published value of 233 kJ/mol and may be related to the higher crystalline cellulose content. The Ea for the woody chips coincides with those reported for spruce, whereas the lower Ea for the branches could be related to the higher amorphous cellulose content together with the high lignin and extractive content (Font et al. 2009). The differences displayed in the Ea in this α region indicate a characteristic degradation process of the cellulose fibers in each studied forest residue. This behavior suggests that a higher cellulose crystallinity renders higher thermal resistivity once the pyrolysis has started, likely due to the more ordered and packed cellulose crystalline regions. It is known that the higher crystallinity of cellulose fibers may result in higher thermal and mechanical properties for cellulosic products or composites. Therefore, these forest residues may have a high potential as raw materials to produce specific cellulosic fibers.
Proximate analysis
Biomass characterization in terms of proximate analysis and net heating value is a necessary preliminary step for energy generation (Table 5).
The differences in composition of the three residual biomasses affect the volatiles and fixed carbon distribution and consequently the net heating value during pyrolysis. It is known that lignin leads to higher char yields, carbon monoxide and methane; on the other hand, hemicellulose results in higher yields of furans, ketones and aldehydes; and finally, cellulose generates saccharides and aldehydes, as well as carbon dioxide and water (Liu et al. 2011). On the other hand, the ash content has a negative effect on the energetic values (Balogun et al. 2015), and the strong interactions among the components may give a more complex product distribution, enhancing the production of some components and inhibiting that of others during their pyrolysis (Amutio et al. 2013). The significant lignin content of the pine needles and branches give way to a large yield of fixed carbon, resulting in higher heating values than those obtained for the woody chips, despite their high ash content. The obtained values are in agreement with those reported for different forest and agricultural biomass feedstocks (Erol et al. 2010; Balogun et al. 2015), which were in the range of 15.4–20.5 MJ kg−1. The higher fixed carbon content and net heating value for the branches and pine needles point out the suitability of these raw materials to produce solid biofuel.
Conclusions
Forests contain readily available biomass waste that can be used as feedstock to produce fuels and polymeric materials, thus adding extra value to the forest residues and contributing to the technological development of these renewable resources. The correlation of the chemical and structural properties of biomass with their thermal behavior (thermal stability and kinetics, heating value, pyrolytic characteristics and product distribution) provides fundamental information to assess the feasibility of the valorization of three logging residues (woody chips, branches and pine needles) into biofuel or polymeric materials. Woody chips show the highest carbohydrate content but a lower proportion of hemicelluloses/pectins than branches and pine needles. The higher cellulose content of woody chips renders a more crystalline and thermal stable biomass, which has great potential to produce bio-based polymeric materials. However, the cellulose from pine needles is more crystalline than the cellulose in the branches and woody chips, which requires higher activation energy during its pyrolysis and indicates a higher thermal resistivity. Therefore, cellulosic fibres with different structure and morphology could be isolated in principle from the different forest residues and could be used to tailor specific properties in the development of cellulosic polymeric materials, such as cellulose-reinforced composites. On the other hand, the differences in the composition of the residual biomass feedstock affect the volatiles and fixed carbon distribution and consequently the net heating value during pyrolysis. The significant lignin content of the pine needles and branches resulted in the highest yield of fixed carbon, resulting in higher heating values. This behavior suggested that pine needles and branches are biomass wastes with a high potential to be used as solid fuel.
References
Ahtee M, Hattula T, Mangs J, Paakkari T (1983) X-ray diffraction method for determination of crystallinity of wood pulp. Pap Puu 85:475–480
Amutio M, Gartzen L, Alvarez J, Moreira R, Duarte G, Nunes J, Olazar M, Bilbao J (2013) Pyrolyisis kinetics of forestry residues from the Portuguese Central Inland Region. Chem Eng Res Des 91:2682–2690
Andersson S, Serimaa R, Paakkari T, Saranpää P, Pesonen E (2003) Crystallinity of wood and the size of cellulose crystallites in Norway spruce (Picea abies). Wood Sci 49:531–537
Baeza J, Freer J (2000) Chemical characterization of wood and its components. In: Hon D, Shiraishi N (eds) Wood and cellulosic chemistry. Marcel Dekker Inc, NewYork, pp 275–384
Balogun AO, Lasode OA, Li H, McDonald AG (2015) Fourier transform infrared (FTIR) study and thermal decomposition kinetics of Sorghum bicolour Glume and Albizia pedicellaris residues. Waste Biomass Valor 6:109–116
Beck-Candanedo S, Roman M, Gray D (2005) Effect of conditions on the properties behavior of wood cellulose nanocrystals suspensions. Biomacromolecules 6:1048–1054
Chauhan GS, Chauhan K, Chauhan S, Kumar S, Kumari A (2007) Functionalization of pine needles by carboxymethylation and network formation for use as supports in the adsorption of Cr6+. Carbohydr Polym 70(4):415–421
Ciucanu I, Kerek F (1984) A simple and rapid method for permethylation of carbohydrate polymers. Carbohydr Res 131:209–217
Dong C, Parsons D, Davies IJ, Dong C, Parsons D, Davies IJ (2014) Tensile strength of pine needles and their feasibility as reinforcement in composite materials. J Mater Sci 49:8057–8062
Erol M, Haykiri-Acma H, Küçükbayrak S (2010) Calorific value estimation of biomass from their proximate analyses data. Renew Energy 35(1):170–173
Fengel D (1978) On the fibrillar structure of cellulose from wood. Holzforschung 32:37–44
Font R, Conesa JA, Moltó J, Munoz M (2009) Kinetics of pyrolysis and combustion of pine needles and cones. J Anal Appl Pyrol 85:276–286
Friedman HL (1964) Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. J Appl Polym Sci Part C Polym Symp 6:183–195
Gronli MG, Várhegyi G, Di Blasi C (2002) Thermogravimetric analysis and devolatilization kinetics of wood. Ind Eng Chem Res 41:4201–4208
Le Normand M, Moriana R, Ek M (2014) Isolation and characterization of cellulose nanocrystals from spruce bark in a biorefinery perspective. Carbohydr Polym 2014(111):979–987
Liu Q, Zhong Z, Wang S, Luo Z (2011) Interactions of biomass components during pyrolysis: a TG-FTIR study. J Anal Appl Pyrolysis 90:213–218
McIntosh S, Vancov T (2011) Optimisation of dilute alkaline pretreatment for enzymatic saccharification of wheat straw. Biomass Bioenergy 35:3094
Miranda I, Gominho J, Mirra I, Pereira H (2012) Chemical characterization of barks from Picea abies and Pinus sylvestris after fractioning into different particle sizes. Ind Crops Prod 36:395
Mohanty AK, Misra M, Hinrichsen G (2000) Biofibres, biodegradable polymers and biocomposites: an overview. Macromol Mater Eng 276(277):1–24
Moriana R, Vilaplana F, Sigbritt K, Ribes-Greus A (2011) Improved thermo-mechanical properties by the addition of natural fibres in starch-based sustainable biocomposites. Compos Part A Appl Sci Manuf 42:30–40
Moriana R, Vilaplana F, Karlsson S, Ribes A (2014a) Correlation of chemical, structural and thermal properties of natural fibres for their sustainable exploitation. Carbohydr Polym 112:422–431
Moriana R, Zhang Y, Mischnick P, Li J, Ek M (2014b) Thermal degradation behaviour and kinetic analysis of spruce glucomannan: a comparative study with the methylated derivatives. Carbohydr Polym 106:60–70
Moriana R, Strömberg S, Ribes A, Sigbritt K (2014c) Degradation behaviour of natural fibre reinforced starch-based polymer composites under different environments. J Renew Mater 2:145–153
Nelson ML, O’Connor RT (1964) Relation of certain infrared bands to cellulose crystallinity and crystal lattice type. Part II. A new infrared ratio for estimation of crystallinity in celluloses I and 11*. J Appl Polym Sci 8:1325–1341
Nomura T, Yamada T (1972) Structural observation on wood and bamboo by X-ray. Wood Res 52:1–10
NREL (2005) Determination of extractives in biomass. Laboratory analytical procedure. NREL, Golden
Ohad I, Danon D (1964) On the dimensions of cellulose microfibrils. J Cell Biol 22(1):302–305
Ohad I, Danon D, Hestrin S (1962) Synthesis of cellulose by acetobacter xylinum. V. Ultrastructure of polymer. J Cell Biol 12(1):31–46
Park S, Baker JO, Himmel ME, Parilla PA, Johnson DK (2010) Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels 3:1–10
Peng Y, Gardner DJ, Han Y, Kiziltas A, Cai Z, Tshabalala MA (2013) Influence of drying method on the materials propertis of nanocellulose I: thermostability and crystallinity. Cellulose 20:2379–2392
Pettolino FA, Walsh C, Fincher GB, Bacic A (2012) Determining the polysaccharide composition of plant cell walls. Nat Protoc 7(9):1590–1607
Poletto M, Pistor V, Zeni M, Zattera AJ (2011) Crystalline properties and decomposition kinetics of cellulose fibers in wood pulp obtained by two pulping process. Polym Degrad Stab 96:679–685
Poletto M, Júnior OLH, Zattera AJ (2014) Native cellulose: structure, characterization and thermal properties. Materials 7:6105–6119
Popescu MC, Popescu CM, Lisa G, Sakata Y (2011) Evaluation of morphological and chemical aspects of different wood species by spectroscopy and thermal methods. J Mol Struct 988:65–72
Raveendran K, Ganesh A, Khilar KC (1995) Influence of mineral matter on biomass pyrolysis characteristics. Fuel 74(12):1812–1822
Sahin HT, Arslan MB (2011) Weathering performance of particleboards manufactured from blends of forest residues with red pine (pinus brutia) wood. Wood Sci Technol 13(3):337–346
Samuelsson LN, Moriana R, Bables MU, Ek M, Engvall K (2014) Model-free rate expression for thermal decomposition processes: the case of microcrystalline cellulose pyrolysis. Fuel 143:438–447
Schnepf E (1965) Struktur der Zellwände und Cellulosefibrillen bei Glaucocystis. Planta 67:213–224
Segal L, Creely JJ, Martin AE, Conrad CM (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text Res J 29(10):786–794
Shebani AN, Van Reenen AJ, Meincken M (2008) The effect of wood extractives on the thermal stability of different wood species. Thermochim Acta 471(1–2):43–50
Silvério HA, Ilvério HA, Flauzino Neto WP, Dantas NO, Pasquini D (2013) Extraction and characterization of cellulose nanocrystals from corncob for application as reinforcing agent in nanocomposites. Ind Crops Prod 44:427–436
Siqueira G, Abdillahi H, Bras J, Dufresne A (2010) High reinforcing capability cellulose nanocrystals extracted from Syngonanthus nitens (Capim Dourado). Cellulose 17(2):289–298
Sjöström E (1981) Wood chemistry, fundamentals and applications. Academic Press Inc, London
Tanaka F, Koshijima T, Okamura K (1981) Characterization of cellulose in compression and opposite woods of a Pinus densiflora tree grown under the influense of strong wind. Wood Sci Technol 15:265–273
TAPPI (2006) Acid insoluble lignin in wood and pulp T 222 om-06. In: US Technical Association of Pulp and Paper Industry
TAPPI (2012) Ash in wood, pulp, paper and paperboard: combustion at 525 °C T211 om-02. In: US Technical Association of Pulp and Paper Industry
Thakur VK, Singha AS (2010) Mechanical and water absorption properties of natural fibers/polymer biocomposites. Polym Plast Technol Eng 49:694–700
Thakur VK, Singha AS (2011) Physiochemical and mechanical behaviour of cellulosic pine needle-based biocomposites. Int J Polym Anal Charact 16:390–398
Thakur VK, Singha AS, Mehta IK (2010) Renewable resource based green polymer composites: analysis and characterization. Int J Polym Anal Charact 15:137–146
Thakur VK, Singha AS, Thakur MK (2011) Fabrication and physico-chemical properties of high-performance pine needles/green polymer composites. Int J Polym Mater 62:226–230
Willför S, Pranovich A, Tamminen T, Puls J, Laine C, Suurnäkki A, Saake B, Uotila K, Simolin H, Hemming J, Holmbom B (2009) Carbohydrate analysis of plant materials with uronic acid-containing polysaccharides—a comparison between different hydrolysis and subsequent chromatographic analytical techniques. Ind Crops Prod 29:571–580
Yemele MCN, Koubaa A, Cloutier A, Soulounganga P, Wolcott M (2010) Effect of bark fiber content and size on the mechanical properties of bark/HDPE composites. Compos Part A Appl Sci Manuf 41(1):131–137
Acknowledgments
RM would like to acknowledge the Wallenberg and Lars-Erik Thunholm Foundation for the research post-doctoral position.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Moriana, R., Vilaplana, F. & Ek, M. Forest residues as renewable resources for bio-based polymeric materials and bioenergy: chemical composition, structure and thermal properties. Cellulose 22, 3409–3423 (2015). https://doi.org/10.1007/s10570-015-0738-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-015-0738-4