
Abstract
The aim of this work was to develop and characterize microfibrillated cellulose (MFC)/pullulan bionanocomposites. Fourier transform infrared spectroscopy suggested that the affinity between the two polymers resulted in new hydrogen bonding of the nanocomposite materials compared to pristine pullulan. At the same time, an increase in crystallinity was observed proportional to the amount of MFC used, as shown by the X-ray analyses. Accordingly, final films showed improved mechanical properties proportionally to the filler loading, with impressive elastic modulus and tensile strength of ~4.50 GPa and ~60 MPa, respectively, for the sample containing 10 % MFC. However, as demonstrated by the moisture sorption isotherms, the addition of MFC did not help reduce the amount of water adsorbed by the samples. In addition, the oxygen and water vapor permeability data clearly showed that final films still suffered high relative humidity values, whereas their barrier performance toward oxygen was excellent under dry conditions, with O2 permeability coefficients (P′O 2) comparable with those of common high barrier films/coatings. Finally, while the nanocomposites in the form of films had high haze values (from 23 to 40 %), the same nanocomposites in the form of coatings were decidedly more transparent, which suggests that their use as thin layers could be more suitable when the “see-through” capability must be preserved, for example in food packaging applications.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Due to environmental and sustainability issues, recent years have witnessed increased attention in “green” technologies, particularly in the field of materials science, where nanotechnology enables new structures with unprecedented properties and functionalities to be created. One of the most striking outcomes is represented by bionanocomposites, i.e. mixtures of biopolymers with organic and/or inorganic additives having nano-length scale and certain geometries (fibers, flakes, spheres, and particulates) (Siqueira et al. 2010). These nano-fillers are preferentially produced through a “top-down” approach which involves the progressive reduction in size of bulk materials by means of physical/chemical routes (e.g., grinding, milling, and ultrasonication) (Sorrentino et al. 2007).
Cellulose nano-sized derivatives are among the most commonly-employed natural fillers for the current generation of bionanocomposites (Klemm et al. 2011; Faruk et al. 2012). Cellulose is the most abundant biopolymer on earth (Siró and Plackett 2010) and has drawn much attention for its renewable, recyclable, compostable and biodegradable properties. Cellulose is a linear β-glucose homopolymer with subunits called cellobiose, β-1,4 linked glucose (Cabiaca et al. 2011), with several hydroxyl side groups that contribute to its hydrophilic behavior. The cellulose chains are arranged in strands of microfibrils that are partially crystalline, the crystalline parts held together by hydrogen bonding and van der Waals forces immersed in a matrix of hemicellulose and lignin (Berglund 2006). Cellulose fibers can be disassembled into their structural nano-components (Chinga-Carrasco et al. 2011)—cellulose nanoparticles (CNs), such as microfibrillated cellulose (MFC), nanofibrilated cellulose (NFC), and cellulose nanocrystals (CNCs), which differ from each other regarding size, aspect ratio, morphology, crystallinity, crystal structure and properties (Moon et al. 2011).
Microfibrillated cellulose (MFC) in particular has been successfully used to produce bionanocomposite films and coatings to improve barrier properties: for example, against oxygen (Hult et al. 2010; Minelli et al. 2010) and water vapor (Azeredo et al. 2009, 2010; Kaushik et al. 2010); mechanical properties (Peng et al. 2011; Hansen et al. 2012); thermal properties (Tingaut et al. 2009; Jonoobi et al. 2010); and to modulate the release of active compounds (e.g., antimicrobials) (Cozzolino et al. 2013).
Among the wide assortment of biopolymers, pullulan, an exopolysaccharide produced by the fungus-like yeast Aureobasidium pullulans, is increasingly regarded as a suitable candidate for the fabrication of bionanocomposites due to its unique properties (Farris et al. 2014). Pullulan is nontoxic, tasteless, odorless and biodegradable, with high film-forming abilities (Yuen 1974; Trovatti et al. 2012). The α-(1 → 6) linkage between maltotriose residues contributes to the structural flexibility and high solubility of pullulan (Leathers 2003), whereas the presence of hydroxyl groups on the molecular skeleton is responsible of the extensive intermolecular hydrogen bonding and superhydrophilicity, which have been profitably exploited to develop oxygen barrier and anti-fog coatings, respectively (Farris et al. 2012; Introzzi et al. 2012a). As with most biopolymers, pullulan suffers some drawbacks, among which sensitivity to external moisture dramatically affects its performance. Blending pullulan with other biopolymers has been shown as a valid strategy to control moisture sensitivity (Kristo and Biliaderis 2006, 2007; Xiao et al. 2012; Wu et al. 2013). More recently, pullulan has been used in combination with natural montmorillonite (Na+-MMT) to produce bionanocomposite materials with enhanced properties even at high relative humidity values (Introzzi et al. 2012b).
The goal of this work was to develop a green bionanocomposite using MFC as the organic nanosized entity incorporated in the main biopolymer matrix, i.e., pullulan. The sensitivity of the resulting composite toward water in terms of barrier and sorption properties, together with the oxygen barrier, mechanical, and optical properties, was assessed as a function of the nanofiller concentration.
Materials and methods
Materials
MFC was produced at the Paper and Fiber Research Institute (PFI, Trondheim, Norway) by two different types of cellulose: (I) Elemental chlorine-free (ECF) fully bleached sulphate pulp mainly based on juvenile Picea abies and (II) Elemental chlorine-free (ECF) fully bleached sulphate cellulose mainly based on mature Picea abies with up to 5 wt% pine (Pinus sylvestris), according to the same manufacturing procedure described by Turbak and Herrik (Turbak et al. 1983; Herrick et al. 1983). The main physicochemical characteristics of the obtained MFC were described in previous papers (Syverud and Stenius 2009; Iotti et al. 2011). Pullulan powder (PF-20 grade, Mn ~ 200 kDa) was provided by Hayashibara Biochemical Laboratories Inc. (Okayama, Japan). Milli-Q water with a resistivity higher than 18.3 MΩ was used to prepare the starting solution. Reagent-grade lithium chloride anhydrous (LiCl), potassium acetate (CH3COOK), magnesium chloride hexahydrate (MgCl2 6H2O), potassium carbonate (K2CO3), magnesium nitrate hexahydrate [Mg(NO3)2 6H2O], lithium acetate di-hydrate (C2H3LiO2 2H2O), sodium chloride (NaCl), potassium chloride (KCl) and potassium nitrate (KNO3) (Sigma–Aldrich, Milan, Italy) were used to prepare the saturated salt solutions, giving theoretical water activity (a w ) values of 0.11, 0.23, 0.32, 0.44, 0.52, 0.66, 0.75, 0.85 and 0.92 at 25 ± 0.2 °C, respectively.
MFC-pullulan nanocomposites preparation
A fixed amount of pullulan (10 wt%, wet basis) was dissolved in distilled water at 25 °C for 1 h under gentle stirring (500 rpm). Different amounts of MFC were added to the pullulan solution (600 rpm × 6 h) to yield pullulan/MFC ratios of 10:0.1, 10:0.5, and 10:1, which were coded as B1, C5 and D10, respectively. The pure pullulan solution was denoted as A0 and was used as a control. Pullulan–MFC films were obtained by the casting method, i.e., pouring a fixed amount of bionanocomposite water dispersions into acrylic Petri dishes (12.5 × 20.5 cm) and placing them at 30 °C in an oven for 3 days. With the exception of samples used for the sorption experiments, all films were stored in a climatic chamber (23 °C and 52 % relative humidity, RH) for an additional week to ensure the equilibrium moisture content.
The thickness (l) of the dry films was measured using a micrometer (Dialmatic DDI030 M, Bowers Metrology, Bradford, UK) to the nearest 0.001 mm at 10 random locations.
Infrared spectroscopy
Infrared spectra of the films were recorded using a PerkinElmer FT-IR Spectrum 100 Series spectrometer (PerkinElmer, Waltham, MA) equipped with a universal attenuated total reflectance (UATR) accessory featuring a single-reflection sampling plate with a 1.8 mm round germanium surface. The spectra were recorded over a range of 650–4,000 cm−1 with a resolution of 4 cm−1 and averaged over 10 scans. Spectrum 6.0 software was used for data acquisition and analysis.
X-ray diffraction (XRD)
The films were powdered using a IKA MF10 basic mill (IKA-Werke GmbH & Co. KG, Staufen, Germany) equipped with a cutting mill head. Each powder was placed in a zero-background sample mount (silicon crystal specimen holder–Bruker) and analyzed with a Bruker D2 Phaser diffractometer (CuKα radiation, Theta/Theta geometry, spinner, and LynxEye linear PSD detector). The following operating conditions were applied: 30 kV, 10 mA, 2θ range 6–50°, step size 0.02°, time per step 2 s, sample rotation 15 rpm, detector opening of 5° 2θ. X-ray patterns were evaluated using Bruker EVA v14.2 software (Bruker AXS, 2008). MFC profiles were compared with the idealized powder diffraction patterns for cellulose polymorphs calculated by French (2014).
Moisture sorption properties
Water sorption isotherms are useful for predicting water sorption properties of cellulosic materials and provide insight into the interaction between water molecules and cellulosic compounds (Belbekhouche et al. 2011). Moisture sorption data were collected according to the procedures reported in the COST project of European Cooperation in the field of technical and scientific research and the principles described by Bell and Labuza (Bell and Labuza 2000). For this purpose, one square centimeter of each sample was initially kept for 3 weeks in a glass desiccator containing anhydrous calcium sulfate and then moved into sealed glass jars containing saturated salt slurries in the a w range 0.11–0.92 and stored in a refrigerated incubator (model FOC 225E; Velp Scientifica, Usmate, Italy) at 25 ± 0.2 °C for 3 weeks.
The equilibrium moisture content data were then fitted to the Guggenheim-Anderson-DeBoer (GAB) sorption isotherm model, which is widely used to fit the sorption data of cellulosic materials (Mali et al. 2005):
The fitting procedure was performed using nonlinear curve-fitting software (Tablecurve 4.0; Jandel Scientific, San Rafael, CA). In Eq. 1, a w is the water activity generated by each saturated salt solution; M and M 0 are the equilibrium moisture content and the moisture content value in the monolayer, respectively, both expressed as grams per 100 g of dry matter. C and K are constants associated with the enthalpies of sorption of the monolayer and multilayer, respectively. In particular, the constant C should always have a positive value due to the exothermic interaction of water vapor with the primary sorption sites of the hygroscopic matrix, whereas reasonable values of K considerably higher than unity have been never found in the literature. Therefore, the suitability of the mathematical model used in this work was assessed not only by evaluating its ability to fit the experimental data (hence by the R2 and RMSE coefficients) but also by assuming the physicochemical basis of the water sorption phenomenon (Farris et al. 2012). Each isotherm was obtained as an average of three data sets (i.e., three replicates).
Mechanical properties
Elastic modulus (Emod, MPa), elongation at break (ε, %), and tensile strength (TS, GPa) of films were measured according to the ASTM D882-02 by means of a dynamometer (mod. Z005, Zwick Roell, Ulm, Germany) fitted with a 5 kN load cell and connected with two clamps placed at a distance of 125 mm apart. Emod, i.e., the slope of the initial rising part of the stress–strain curve, was determined according to a software-assisted procedure based on a secant method; ε was calculated dividing the extension-at-break of the sample by the initial gauge length and multiplying by 100; TS was calculated using the equation TS = F/A, where TS is the tensile strength in MPa, F is the force in N at maximum load, and A is the initial cross-sectional area (mm2) of the film sample. For each parameter, final results are the mean of twenty replicates.
Oxygen and water vapor barrier properties
For the permeability measurements, a MultiPerm permeability analyzer (ExtraSolution Srl, Capannori, Italy) equipped with one electrochemical sensor (for O2 permeability determination) and one infrared sensor (for water vapor) was used. To minimize the influence of heterogeneities in the thickness of the final films on the barrier properties, the different samples were tested in the form of thin coatings laid on a plastic substrate. For the coating deposition, an aliquot of the water dispersions was placed on the corona-treated side of rectangular (24 × 18 cm2) polyethylene terephthalate (PET) films (12.0 ± 0.5 µm thick) provided by Toray (Saehan, Kyungbuk, South Korea). For the deposition of the coating, an automatic film applicator (ref 1,137, Sheen Instruments, Kingston, UK) at a constant speed of 2.5 mm s−1 was used, according to ASTM D823-07—Practice C. The deposition was performed using a horizontal steel rod with an engraved pattern, which yielded final coatings of comparable nominal thickness of 1 μm after water evaporation. Water evaporation was performed using a constant and perpendicular flux of mild air (25.0 ± 0.3 °C for 2 min) at a distance of 40 cm from the applicator. Coated films were then stored under controlled conditions (23.0 ± 0.5 °C in a desiccator) for 48 h before permeability measurements.
The oxygen barrier properties of film samples were assessed on a 50 cm2 surface according to the standard method ASTM F2622-08, with a carrier flow (N2) of 10 mL min−1, at 23 °C, 0 % relative humidity (RH) and at one atmosphere pressure difference on the two sides of the specimen. The oxygen transmission rate (O 2 TR) data provided by the instrument were converted into permeability coefficients according to the following equation:
In Eq. 2, P′O 2 is the oxygen permeability coefficient (mL μm m−2 24 h−1 atm−1), PO 2, is the permeance (defined as the ratio of the O 2 TR to the difference between the partial pressure of the gas on the two sides of the film, Δp), and t is the total thickness of the material. To quantify the contribution of the bionanocomposite coatings to the total (i.e., substrate plus coating) P′O 2 values, the well-known series resistance formula intended for multilayer systems was used:
where P represents the oxygen permeability coefficient (P′O 2, mL μm m−2 24 h−1 atm−1) of the layer; and l stands for its thickness (μm). With knowledge of l tot, l PET, l coating, P tot, and P PET, the P′O 2 coefficient (P coating) for the nanocomposite coatings can be calculated from Eq. 3.
Water vapor transmission rate (WVTR) was assessed on a 50 cm2 surface sample according to the standard method ASTM F1249-05, with a carrier flow (N2) of 10 mL min−1. Measurements were performed at 23 °C and 65 % relative humidity (RH), which is the humidity gradient between the two semi-chambers between which the sample was mounted. Final WVTR values were expressed as g m−2 24 h−1. Alike O 2 TR, WVTR values were used to calculate the P′WV coefficient of both coated PET films and individual coatings, the latter according to Eq. 3.
For both O 2 TR and P′WV final values resulted from five replicate measurements.
Optical properties
Haze was measured within the wavelength range 780–380 nm, in accordance with ASTM D1003–00, using a UV–Vis high-performance spectrophotometer (Lambda 650, PerkinElmer, Waltham, MA, USA) coupled with a 150 mm integrating sphere, which allows trapping of diffuse transmitted light. Quantification of haze, defined as the percentage of transmitted light deviating by more than an angle of 2.5° from the direction of the incident beam, is important, especially from a commercial point of view, as it is responsible for reduction in the contrast between objects viewed through the specimen. The data shown are the average of nine replicates.
Statistical analysis
Statistical significance of differences between mean values was determined by one-way analysis of variance (ANOVA), using Statgraphic Plus 4.0 software. The mean values, where appropriate, were compared by Student’s t test with a significance level (p) < 0.05. The comparison between the experimental moisture sorption data and those predicted by the GAB model was performed using the root mean square error (RMSE), calculated as follows:
where x exp,i is the observed (measured) value, x pred,i is the value predicted by the model, and n the number of the experimental measurements. RMSE is a widely used parameter to estimate the quality of model fitting as well as to compare the individual model performance. If RMSE tends toward 0 or is very close to the experimental error, it means that model is able to represent the experimental data.
Results and discussion
Infrared spectroscopy
The most relevant peaks of MFC and pullulan films are reported in the supporting information (Figures S1a and S1b, respectively). A first characteristic band at ~3,300 cm−1 for both polymers is assigned to the stretching vibration of –OH groups involved in the formation of hydrogen bonds at the intra- and inter-molecular level (Vieira et al. 2011; Farris et al. 2012). Other common bands concern C–O–C stretching at 1,162 and 1,154 cm−1 for MFC and pullulan, respectively. The peak at 1,110 cm−1 in the spectrum of MFC is assigned to the skeletal vibration involving C–O stretching of the β(1 → 4) glycosidic linkages of the β-d-glucopyranosyl units of cellulose (Marcovich et al. 1996), while the peak at 1,081 cm−1 in the pullulan spectrum is attributed to stretching of the C6–OH bond (Shingel 2002).
FTIR spectra of MFC, pullulan (A0), and of bionanocomposite formulations B1, C5 and D10 are reported in Fig. 1. As shown in Fig. 1a, the addition of the nanofiller (MFC) apparently did not make any significant change in the spectrum of the main polymer (i.e., pullulan). In similar work on MFC–sodium caseinate nanocomposites, the same observation was explained in terms of low filler loading (Pereda et al. 2011). However, a deeper examination reveals a shifting especially in the peaks at 3,300 and 1,078 cm−1 in the bionanocomposite films with respect to the unreinforced ones. The same shifts were also observed in pullulan–silica bionanocomposites (Farris et al. 2012). Shifting of bands specifically assigned to –OH groups are generally associated with the establishment of new hydrogen bonds between phases (MFC and pullulan, in this case) (Tong et al. 2008). Therefore, it is likely interaction between MFC and pullulan in the nanocomposite films was governed by hydrogen bonding.

a Full range (4,000–400 cm−1) FTIR-ATR spectra of MFC (gray line), pullulan (A0, black line), and nanocomposite formulations B1 (red line), C5 (blue line), and D10 (green line); b spectral range 3,750–2,970 cm−1; c spectral range 1,225–975 cm−1 (two zoomed regions are displayed in the insets). (Color figure online)
X-ray diffraction (XRD)
The diffractograms of pristine polymers (cellulose and pullulan) and nanocomposites are displayed in Fig. 2. Pullulan (A0) shows a broad hump ranging from ~7° to ~30° 2θ (maximum at ~18° 2θ), indicative of an amorphous organization of this exopolysaccharide, as found in previous works (Trovatti et al. 2012; Fuentes-Alventosa et al. 2013). The pattern of the cellulose (MFC) employed in this study is characterized by three main peaks at 15.0°, 16.4° and 22.6° 2θ. Two other reflections can be detected at 29.3° (very weak) and 34.5° 2θ. The Miller indices corresponding to these peaks are, respectively: (1–10), (110), (200), (1–22), and (004), as reported by French (2014). This pattern, compared with those calculated for cellulose polymorphs (French 2014), shows the best correspondence with that of cellulose Iβ, the predominant polymorph present in higher plants. The difference concerning the intensities of the two peaks at 15.0° and 16.4° 2θ (in cellulose Iβ the first one is slightly higher than the second) might depend on the stronger contribution of the background at 16.4° 2θ. The absence of the shoulder on the left side of the main peak (between ~20 and ~21° 2θ) suggests that a preferred orientation along the fiber axis has occurred in the powdered sample. This is corroborated by the powder diffraction patterns of cellulose Iβ calculated by French (2014), where the shoulder is present when crystallites are randomly oriented, confirming what was originally found by Segal et al. (1959). Finally, the width of the peaks at half maximum height (2.2° 2θ measured on the main peak) and the hump shape of the background indicate that the MFC contains an amorphous component. On the other hand, bionanocomposite diffraction patterns are characterized by the predominance of the amorphous pullulan profile, progressively modified by the increase of the cellulose content. However, considering the signal to noise ratio and the low quantity of MFC, in particular in the B1 and C5 samples, it is difficult to see the (200) peak in the bionanocomposites. The diffractogram of sample B1 shows a hump, slightly higher in intensity with respect to A0. The effect of cellulose presence in bionanocomposite matrices is more appreciable in sample C5, where a very subtle shoulder came out in correspondence of the MFC main peak (i.e., at ~22.6° 2θ), whereas the same reflection is more evident in D10 diffractogram, according to previous observation by Trovatti et al. (2012). On the whole, the way in which cellulose and pullulan diffraction patterns combine in bionanocomposite materials is analogous to that reported by Lu et al. (2008) for composites prepared with cellulose and polyvinyl alcohol and by Ninan et al. (2013) for pectin/carboxymethyl cellulose/microfibrillated cellulose composites. As reported by the same authors, the fact that neither of the two strong peaks of MFC was found in bionanocomposite materials could be due to the low cellulose content.

X-ray diffraction patterns of MFC (gray line), pullulan (A0, black line), and nanocomposite powders B1 (red line), C5 (blue line), and D10 (green line). A magnification of the 2θ range 20°–25° is shown in the inset. (Color figure online)
Moisture sorption isotherms
The experimental moisture sorption data and the isotherms as predicted by the GAB model (Eq. 1) are displayed in Fig. 3. For all the four formulations, the moisture uptake increased monotonically until a specific a w (~0.7), after which an exponential increase in water uptake was observed. According to Brunauer classification (1945), this shape is typical of type III isotherms for high-crystalline substances. Concerning our systems, it is plausible that at a w values below 0.7–0.8, the amount of moisture gained by the matrix is low because it occurs only at the few free –OH sites on the surface of the films. This would also explain the different shape of pure MFC films, which exhibit a sigmoid or S-shape profile (type II) (Belbekhouche et al. 2011). An increase in a w (>0.8) progressively displaces the MFC-pullulan interactions (hydrogen bonds) by water-polymer (MFC and pullulan) interactions, thus the steep increase in the moisture content of the films.

Experimental (symbols) and predicted (lines) data of moisture sorption isotherms at 25 °C for A0 (open square), B1 (open circle), C5 (open triangle), and D10 (open diamond) samples. The predicted data were obtained by fitting the experimental points with the GAB model (Eq. 1 in the text)
Both the coefficient of determination (R2) and the RMSE values (the former close to the unit, the latter approaching 0) indicated that the fitting procedure of the experimental data with the GAB model was satisfactory (Table 1). However, the statistical analysis also demonstrated that there was no statistical difference between the experimental data at each a w value, with the exception for the moisture content of the sample A0 at a w = 0.53 and the moisture content of the sample D10 at a w = 0.92 (data not shown). This unequivocally indicates that the amount of MFC used to obtain the different nanocomposite films did not impact significantly the adsorption properties of the final materials. Although not significant from a statistical point of view, the modeling parameters arising from the fitting procedure of Eq. 2 to the experimental sorption measurements (see Table 1) suggested the possible scenario at physicochemical level for each matrix. In particular, the moisture content in the monolayer (M 0) predicted by the GAB model was equal to 4.8, 0.65, 1.18, and 1.45 g/100 g solids for samples A0, B1, C5, and D10, respectively, which would suggest that interactions between MFC and pullulan might have reduced the number of active sites on the pullulan backbone available for water molecules. On the other hand, the progressive increase in M 0 within the MFC nanocomposites can be ascribed to an increase in the MFC content, i.e. an increase in hydroxyl groups (beyond those available on the pullulan backbone) able to interact with the surrounding water molecules. The information gathered by the M 0 values is corroborated by the values of the C parameter in the GAB equation (Table 1). In particular, C is a measure of the strength of binding of water to the primary binding sites. The larger C, the more strongly that the water is bound in the monolayer and the larger the difference in enthalpy between the monolayer molecules and multilayer molecules (Quirijns et al. 2005). Therefore, the larger C value for the sample A0 confirms the presence of water beyond the primary binding sites, whereas the highest C value for the sample B1 suggests that the interaction with the adsorbed water molecules is through the primary binding sites of the monolayer.
Mechanical properties
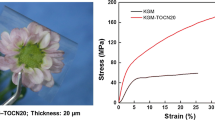
Mechanical properties of pullulan (A0) and bionanocomposite films (B1, C5, and D10) were studied by tensile experiments. Elastic modulus (Emod), elongation at break (ε), and tensile strength (TS), determined from the stress–strain curves (Fig. 4), are shown in Table 2. Elastic modulus of bionanocomposite films was significantly higher for samples C5 and D10, whereas loading below 0.01 % on the total solids apparently did not provide any significant improvement (formulation B1). Interestingly, increasing the amount of MFC loaded into the pullulan matrix did not impair the stretching properties of the final nanocomposites. Although the ε values of bionanocomposites were not high, especially if compared with common plastic films, it must be remembered that no plasticizers were used in the formulation of the final films. The addition of MFC also yielded an increase in the tensile strength of the materials, proportional to the filler loading, up to ~58 MPa for the highest filler loading (10 wt% db, formulation D10). The increase in elastic modulus and tensile strength is also indicative of the uniform dispersion of MFC fibrils in the pullulan matrix (Khalil et al. 2012) and their ability to form a rigid network (Lavoine et al. 2012).

Representative stress–strain curves obtained from the tensile tests on samples A0 (black line), B1 (red line), C5 (blue line), and D10 (green line). A zoom of the first part (ε = 0.3 %) is shown in the inset. (Color figure online)
These results confirm the superior reinforcing effect of cellulose-derived nanoparticles over other nanosized organic fillers. For example, the elastic modulus, tensile strength, and elongation at break measured for starch nanocrystals (SN)/pullulan nanocomposites (SN loading = 10 wt% with respect to the total dry mass, analyses carried out at 53 % RH) were ~300, ~9 MPa, and ~100 % (Kristo and Biliaderis 2007). However, it should be noted that sorbitol (30 wt% with respect to the total dry mass) was used throughout the experiments as a plasticizer. Furthermore, the association between MFC and pullulan led to better performance with respect to other MFC-biopolymer composites. For example, MFC-amylopectin films (MFC loading = 10 wt% with respect to the total dry mass) without plasticizers had an elastic modulus of ~1,800 MPa, tensile strength ~40 MPa, and elongation at break of ~3.0 % (tests carried out at 50 % RH) (López-Rubio et al. 2007). Finally, our results seem to demonstrate the best reinforcing performance of MFC over nanofibrillated celluloce (NFC) nanocomposites (Trovatti et al. 2012). For example, the elastic modulus for NFC-pullulan films at 5 and 10 % NFC loading was 0.028 and 0.032 GPa, respectively, which have to be compared with 3.98 and 4.54 GPa for the same MFC loadings (5 and 10 wt%). This was reflected in the slight inferior elongation at break of MFC films (1.86 and 1.90 % for samples B5 and D10) compared to NFC films (2.0 and 2.4 % for the pullulan films containing 5 and 10 wt% NFC, respectively).
Oxygen and water vapor barrier properties
Results from permeability measurements are reported in Table 3. The oxygen barrier performance of the tested samples was decidedly good under dry conditions, being the P′O 2 values in line with those of commonly used barrier films/coatings of both petrol-based and natural origin (Table 4). The O 2 barrier properties increased by increasing the MFC content. This observation is in line with the XRD data, i.e., besides other factors, the higher the degree of crystal domains, the higher the barrier performance, due to the physical impedance offered by crystal regions to the diffusion of oxygen molecules (Chinga-Carrasco and Syverud 2012; Cozzolino et al. 2012).
As the relative humidity increased to 80 %, overall performance was impaired insofar as the final P′O 2 values of bionanocomposite coatings increased approximately two orders of magnitude. The detrimental effect of humidity on the barrier properties of hydrophilic polymers is well-known (Zhang et al. 2001; Hu et al. 2005). It is ascribed to the plasticizing effect of water molecules adsorbed by the polymer surface and bulk, especially in correspondence with the amorphous regions (Aulin et al. 2010; Kurek et al. 2014). Weakening of the hydrogen bonds at intramolecular and intermolecular level may occur in the long run. Eventually, both phenomena lead to an increase in chain mobility and free volume, which is reflected in a higher diffusion of the permeant (e.g., oxygen) across the polymer matrix. Still, the best performance was recorded for the formulation D10, which can be explained by the lower water sensitivity of crystalline domains compared to the amorphous regions of the polymer matrices (the higher amount of crystallite regions could have induced a decrease in the relaxation rate of the polymer chains) (Cozzolino et al. 2012).
With respect to the P′WV values, it can be clearly seen that both pullulan and bionanocomposite films were moderate barrier against water vapor. This observation confirms what was reported for other biopolymers (Chiellini 2008; Lacroix 2009) and MFC-based films (Hult et al. 2010). Moreover, the fact that there was no statistically significant difference in the P′WV values between samples suggests that the addition of MFC did not bring any specific benefit to the final performance. The hydrophilic nature of these materials (thus the high affinity towards water molecules) is the main reason for this behavior.
Optical properties
Haze, defined as the percentage of transmitted light deviating by more than an angle of 2.5° from the direction of the incident beam, is an important optical property for several applications (Farris et al. 2009). In some cases (e.g., food packaging), when visual inspection through the material must be preserved, it is of the utmost importance to minimize the haze value of the materials to allow an adequate display of the product across the package. Accordingly, high haze values hiding the contents of the package may irreparably affect the final consumers’ choice. For other applications (e.g., solar cells and greenhouses), scattering of the visible light may have a detrimental effect in the yield of the processes involved in the capture of the incident radiation (e.g., energy conversion and photosynthesis). Quantification of haze may thus dictate the market penetration of new materials in specific sectors.
Within this study, haze was first measured on pure pullulan and nanocomposite films. The experimental haze values (h, %) are reported in Fig. 5a (upper data set). To reset any influence arising from differences in thickness of the films, it has been expedient to normalize these values with respect to the highest thickness value (for the one in sample B1, see Table 2). Normalized values are reported in Fig. 5a (lower data set). Increasing the amount of MFC yielded a proportional increase in haze, which is responsible for reducing contrast of objects viewed through it, as clearly shown by the images inset in Fig. 5a. In particular, for the nanocomposite films, the haze variation as a function of the filler loading is governed by a linear relationship (see the h’ trend). The haze values recorded for the final films are too high for most applications. For example, in food packaging, haze values as high as 3 % are generally judged adequate for a proper display of the products (Introzzi et al. 2012b), although some plastics have haze values slightly higher (e.g., low-density polyethylene 48 μm thick ~6 %) (Farris et al. 2009). However, it should be noted that these values rely on the high thickness of the films. Therefore, MFC/pullulan bionanocomposites can more profitably be used in the form of thin films (coatings) rather than bulky sheets, which would allow minimizing light scattering. To support this consideration, we collected haze values of a PET film coated with the different formulations included in this work (thickness of the coatings ~1 μm). As shown in Fig. 5b, the deposition of the coatings modified the haze values of the coated PET substrates to only a limited extent, confirming that coatings may represent a most suitable solution over stand-alone films.

a Haze values for pullulan (A0) and MFC-nanocomposite (B1, C5, and D10) films. Experimental (h, open triangle) and normalized with respect to the thickness (h’, open square) values are reported as a label on top of each data point. Error bars represent the standard deviation around the mean value (n = 9). The see-through capability of each film formulation is shown by the inset images; b haze values of a PET film coated with pure pullulan (A0) and nanocomposite formulations (B1, C5, and D10). Note that the haze value of bare PET was measured to be 2.78 % ± 0.14
The increase in haze observed for film and coating samples can be explained in terms of surface roughness (Tilley 2011) rather than presence of scattering centers (Introzzi et al. 2012b). As observed in previous works, the addition of MFC to polymer systems (e.g., films and coatings) leads to an increase in surface roughness as measured by laser profilometry or atomic force microscopy (Syverud et al. 2011; Chinga-Carrasco and Syverud 2012; Cozzolino et al. 2013). In particular, the increase in roughness was in the micron-scale and was attributed to the inherent fibrillated nature of MFC. Conversely, the presence of macro-sized domains has not been described, as opposed to nanocomposite systems based on inorganic fillers (e.g., clays).
Conclusions
Novel “green” nanocomposites based on pullulan and microfibrillated cellulose were prepared and characterized. Infrared spectroscopy analysis suggested that the two polymers presumably interacted by cooperative hydrogen bonding, while the incorporation of the highly crystalline MFC was responsible for the excellent mechanical properties of final films as well as the high oxygen barrier properties under dry conditions. However, the nanocomposite films still showed sensitivity to high relative humidity values, which has to be linked to the highly hydrophilic nature of both MFC and pullulan. The high degree of haze measured for the nanocomposite materials clearly indicated that they are not suitable in the form of films, especially when clear display of objects through the films is required. The use of MFC/pullulan bionanocomposites in the form of thin coatings (e.g., on plastic films) is thus suggested toprevent this drawback.
References
Aulin C, Gällstedt M, Lindström T (2010) Oxygen and oil barrier properties of microfibrillated cellulose films and coatings. Cellulose 17:559–574
Azeredo HM, Mattoso LH, Wood D, Williams TG, Avena-Bustillos RJ, McHugh TH (2009) Nanocomposite edible films from mango puree reinforced with cellulose nanofibers. J Food Sci 74:31–35
Azeredo HM, Mattoso LH, Avena-Bustillos RJ, Filho GC, Munford ML, Wood D, McHugh TH (2010) Nanocellulose reinforced chitosan composite films as affected by nanofiller loading and plasticizer content. J Food Sci 75:1–7
Belbekhouche S, Bras J, Siqueira G, Chappey C, Lebrun L, Khelifi B, Marais S, Dufresne A (2011) Water sorption behavior and gas barrier properties of cellulose whiskers and microfibrils films. Carbohyd Polym 83:1740–1748
Bell LN, Labuza TP (2000) Practical aspects of moisture sorption isotherm measurement and use, 2nd edn. Egan Press, Egan, MN
Berglund L (2006) New concepts in natural fibres composites. 27th Risø international symposium on material science. Risø National Laboratory, Roskilde, Denmark
Cabiaca A, Guillon E, Chambon F, Pinel C, Rataboul F, Essayem N (2011) Cellulose reactivity and glycosidic bond cleavage in aqueous phase by catalytic and non catalytic transformations. Appl Catal a-gen 402:1–10
Chiellini E (ed) (2008) Environmentally compatible food packaging. CRC Press, Boca Raton
Chinga-Carrasco G, Syverud K (2012) On the structure and oxygen transmission rate of biodegradable cellulose nanobarriers. Nanoscale Res Lett 7:192
Chinga-Carrasco G, Miettinen A, Luengo Hendriks CL, Gamstedt EK, Kataja M (2011) Structural characterisation of kraft pulp fibres and their nanofibrillated materials for biodegradable composite applications. In: Nanocomposites and polymers with analytical methods: book 3, ISBN 979-953-307-136-6, pp 243–260
Cozzolino CA, Blomfeldt TOJ, Nilsson F, Piga A, Piergiovanni L, Farris S (2012) Dye release behavior from polyvinyl alcohol films in a hydro-alcoholic medium: influence of physicochemical heterogeneity. Colloids Surf A Physicochem Eng Asp 403:45–53
Cozzolino CA, Nilsson F, Iotti M, Sacchi B, Piga A, Farris S (2013) Exploiting the nano-sized features of microfibrillated cellulose (MFC) for the development of controlled release packaging. Colloid Surf B 110:208–216
Farris S, Introzzi L, Piergiovanni L (2009) Evaluation of a bio-coating as a solution to improve barrier, friction and optical properties of plastic films. Packag Technol Sci 22:69–83
Farris S, Introzzi L, Fuentes-Alventosa JM, Santo N, Rocca R, Piergiovanni L (2012) Self-assembled pullulan-silica oxygen barrier hybrid coatings for food packaging applications. J Agric Food Chem 60(3):782–790
Farris S, Uysal Unalan I, Introzzi L, Fuentes-Alventosa JM, Cozzolino CA (2014) Pullulan-based films and coatings for food packaging: present applications, emerging opportunities, and future challenges. J App Polym Sci 131:40539–40551
Faruk O, Bledzki AK, Fink HP, Sain M (2012) Biocomposites reinforced with natural fibers: 2000–2010. Prog Polym Sci 37:1552–1596
French AD (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21:885–896
Fuentes-Alventosa JM, Introzzi L, Santo N, Cerri G, Brundu A, Farris S (2013) Self-assembled nanostructured biohybrid coatings by an integrated ‘sol-gel: intercalation’ approach. RSC Adv 3:25086–25096
Hansen NML, Blomfeldt TOJ, Hedenqvist MS, Plackett DV (2012) Properties of plasticized composite films prepared from nanofibrillated cellulose and birch wood xylan. Cellulose 19:2015–2031
Herrick FW, Casebier RL, Hamilton JK, Sandberg KR (1983) Microfibrillated cellulose: morphology and accessibility. J Appl Polym Sci Appl Polym Symp 37:797–813
Hinterstoisser B, Salmén L (2000) Application of dynamic 2D FTIR to cellulose. Vib Spectrosc 22:111–118
Hu YS, Mehta S, Schiraldi DA, Hiltner A, Baer E (2005) Effect of water sorption on oxygen-barrier properties of aromatic polyamides. J Polym Sci Pol Phys 43:1365–1381
Hult EL, Iversen T, Sugiyama J (2003) Characterization of the supermolecular structure of cellulose in wood pulp fibres. Cellulose 10:103–110
Hult EL, Iotti M, Lenes M (2010) Efficient approach to high barrier packaging using microfibrillar cellulose and shellac. Cellulose 17:575–586
Introzzi L, Fuentes-Alventosa JM, Cozzolino CA, Trabattoni S, Tavazzi S, Bianchi CL, Schiraldi A, Piergiovanni L, Farris S (2012a) ‘Wetting enhancer’ pullulan coating for anti-fog packaging applications. ACS Appl Mater Interfaces 4:3692–3700
Introzzi L, Blomfeldt TOJ, Trabattoni S, Tavazzi S, Santo N, Schiraldi A, Piergiovanni L, Farris S (2012b) Ultrasound-assisted pullulan/montmorillonite bionanocomposite coating with high oxygen barrier properties. Langmuir 28:11206–11210
Iotti M, Gregersen ØW, Moe S, Lenes M (2011) Rheological studies of microfibrillar cellulose water dispersions. J Polym Environ 19:137–145
Jonoobi M, Harun J, Mathew AP, Oksman K (2010) Mechanical properties of cellulose nanofiber (CNF) reinforced polylactic acid (PLA) prepared by twin screw extrusion. Compos Sci Technol 70:1742–1747
Kaushik A, Singh M, Verma G (2010) Green nanocomposites based on thermoplastic starch and steam exploded cellulose nanofibrils from wheat straw. Carbohyd Polym 82:337–345
Khalil HPSA, Bhat AH, Yusra IAF (2012) Green composites from sustainable cellulose nanofibrils: a review. Carbohyd Polym 87:963–979
Klemm D, Kramer F, Moritz S, Lindström T, Ankerfors M, Gray D, Dorris A (2011) Nanocelluloses: a new family of nature-based materials. Angew Chem Int Ed 50:5438–5466
Kristo E, Biliaderis CG (2006) Water sorption and thermo-mechanical properties of water/sorbitol-plasticized composite biopolymer films: caseinate–pullulan bilayers and blends. Food Hydrocolloid 20:1057–1071
Kristo E, Biliaderis CG (2007) Physical properties of starch nanocrystal-reinforced pullulan films. Carbohyd Polym 68:146–158
Kurek M, Guinault A, Voilley A, Galić K, Debeaufort F (2014) Effect of relative humidity on carvacrol release and permeation properties of chitosan based films and coatings. Food Chem 144:9–17
Lacroix M (2009) Mechanical and permeability properties of edible films and coatings for food and pharmaceutical applications. In: Embuscado ME, Huber KC (eds) Edible films and coatings for food applications. Springer, Berlin, pp 347–366
Lavoine N, Desloges I, Dufresne A, Bras J (2012) Microfibrillated cellulose: its barrier properties and applications in cellulosic materials: a review. Carbohyd Polym 90:735–764
Leathers TD (2003) Biotechnological production and applications of pullulan. Appl Microbiol Biotechnol 62:468–473
López-Rubio A, Lagaron JM, Ankerfors M, Lindström T, Nordqvist D, Mattozzi A, Hedenqvist MS (2007) Enhanced film forming and film properties of amylopectin using micro-fibrillated cellulose. Carbohydr Polym 68:718–727
Lu J, Wang T, Drzal LT (2008) Preparation and properties of microfibrillated cellulose polyvinyl alcohol composite materials. Compos A 39:738–746
Mali S, Sakanaka LS, Yamashita F, Grossmann MVE (2005) Water sorption and mechanical properties of cassava starch films and their relation to plasticizing effect. Carbohydr Polym 60:283–289
Marcovich NE, Reboredo MM, Aranguren MI (1996) FTIR spectroscopy applied to woodflour. Compos Interfaces 4:119–132
Minelli M, Baschetti MG, Doghieri F, Ankerfors M, Lindström T, Siró I, Plackett D (2010) Investigation of mass transport properties of microfibrillated cellulose (MFC) films. J Membrane Sci 358:67–75
Moon RJ, Martini A, Nairn J, Simonsen J, Youngblood J (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3941–3994
Ninan N, Muthiah M, Park I-K, Elain A, Thomas S, Grohens Y (2013) Pectin/carboxymethyl cellulose/microfibrillated cellulose composite scaffolds for tissue engineering. Carbohyd Polym 98:877–885
Peng XW, Ren JL, Zhong LX, Sun RC (2011) Nanocomposite films based on xylan-rich hemicelluloses and cellulose nanofibers with enhanced mechanical properties. Biomacromolecules 12:3321–3329
Pereda M, Amica G, Rácz I, Marcovich NE (2011) Structure and properties of nanocomposite films based on sodium caseinate and nanocellulose fibers. J Food Eng 103:76–83
Quirijns EJ, van Boxtel AJB, van Loon WKP, van Straten G (2005) Sorption isotherms, GAB parameters and isosteric heat of sorption. J Sci Food Agric 85:1805–1814
Segal L, Creely JJ, Martin AE, Conrad CM (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text Res J 29:786–794
Shingel KI (2002) Determination of structural peculiarities of dextran, pullulan and γ-irradiated pullulan by Fourier-transform IR spectroscopy. Carbohyd Res 337:1445–1451
Siqueira G, Bras J, Dufresne A (2010) Cellulosic bionanocomposites: a review of preparation, properties and applications. Polymers 2:728–765
Siró I, Plackett D (2010) Microfibrillated cellulose and new nanocomposite materials: a review. Cellulose 17:459–494
Sorrentino A, Gorrasi G, Vittoria V (2007) Potential perspectives of bio-nanocomposites for food packaging applications. Trends Food Sci Technol 18:84–95
Sothornvit R, Rhim JW, Hong SI (2009) Effect of nano-clay type on the physical and antimicrobial properties of whey protein isolate/clay composite films. J Food Eng 91:468–473
Syverud K, Stenius P (2009) Strength and barrier properties of MFC films. Cellulose 16:75–85
Syverud K, Chinga-Carrasco G, Toledo J, Toledo PG (2011) A comparative study of Eucalyptus and Pinus radiata pulp fibres as raw materials for production of cellulose nanofibrils. Carbohyd Polym 84:1033–1038
Tilley RJD (2011) Colour and the optical properties of materials, 2nd edn. Wiley, Hoboken, p 42
Tingaut P, Zimmermann T, Lopez-Suevos F (2009) Synthesis and characterization of bionanocomposites with tunable properties from poly(lactic acid) and acetylated microfibrillated cellulose. Biomacromolecules 11:454–464
Tong Q, Xiao Q, Lim LT (2008) Preparation and properties of pullulan–alginate–carboxymethylcellulose blend films. Food Res Int 41:1007–1014
Trovatti E, Fernandes SCM, Rubatat L, da Silva Perez D, Freire CSR, Silvestre AJD, Neto CP (2012) Pullulan–nanofibrillated cellulose composite films with improved thermal and mechanical properties. Compos Sci Technol 72:1556–1561
Turbak AF, Snyder FW, Sandberg KR (1983) Microfibrillated cellulose, a new cellulose product: properties, uses, and commercial potential. J Appl Polym Sci: Appl Polym Symp 37:815–827
Vieira AP, Santana SAA, Bezerra CWB, Silva HAS, Chaves JAP, de Melo JCP, da Silva Filhod EC, Airoldi C (2011) Epicarp and Mesocarp of Babassu (Orbignya speciosa): characterization and application in copper phtalocyanine dye removal. J Braz Chem Soc 22(1):21–29
Wu J, Zhong F, Li Y, Shoemaker CF, Xia W (2013) Preparation and characterization of pullulanechitosan and pullulanecarboxymethyl chitosan blended films. Food Hydrocolloid 30:82–91
Xiao Q, Lim LT, Tong Q (2012) Properties of pullulan-based blend films as affected by alginate content and relative humidity. Carbohyd Polym 87:227–234
Yuen S (1974) Pullulan and its applications. Process Biochem 9:7–9
Zhang Z, Britt IJ, Tung MA (2001) Permeation of oxygen and water vapor through EVOH films as influenced by relative humidity. J Appl Polym Sci 82:1866–1872
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cozzolino, C.A., Cerri, G., Brundu, A. et al. Microfibrillated cellulose (MFC): pullulan bionanocomposite films. Cellulose 21, 4323–4335 (2014). https://doi.org/10.1007/s10570-014-0433-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-014-0433-x