
Abstract
The effect of surface hydrophobicity and side-chain variation on xyloglucan adsorption onto cellulose microfibrils (CMF) is investigated via molecular dynamics simulations. A molecular model of CMF with (100), (010), (1–10), (110) and (200) crystal faces was built. We considered xylogluco-oligosaccharides (XGO) with three repeating units, namely (XXXG)3, (XXLG)3, and (XXFG)3 (where each (1,4)-β-d-glucosyl residue in the backbone is given a one-letter code according to its substituents: G = β-d-Glc; X = α-d-Xyl-(1,6)-β-d-Glc; L = β-d-Gal-(1,2)-α-d-Xyl-(1,6)-β-d-Glc; F = α-l-Fuc-(1,2)-β-d-Gal-(1,2)-α-d-Xyl-(1,6)-β-d-Glc). Our work shows that (XXXG)3 binds more favorably to the CMF (100) and (200) hydrophobic surfaces than to the (110), (010) and (1–10) hydrophilic surfaces. The origin of this behavior is attributed to the topography of hydrophobic CMF surface, which stabilizes (XXXG)3 in flat conformation. In contrast, on the rough hydrophilic CMF surface (XXXG)3 adopts a less favorable random-coil conformation to facilitate more hydrogen bonds with the surface. Extending the xyloglucan side chains from (XXXG)3 to (XXLG)3 hinders their stacking on the CMF hydrophobic surface. For (XXFG)3, the interaction with the hydrophobic surface is as strong as (XXXG)3. All three XGOs have similar binding to the hydrophilic surface. Steered molecular dynamics simulation was performed on an adhesive model where (XXXG)3 was sandwiched between two CMF hydrophobic surfaces. Our analysis suggests that this sandwich structure might help provide mechanical strength for plant cell walls. Our study relates to a recently revised model of primary cell walls in which extensibility is largely determined by xyloglucan located in limited regions of tight contact between CMFs.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The plant cell wall is a complex biomaterial in which cellulose microfibrils (CMF) are embedded in a matrix of polysaccharides, proteins, and polyphenolics (Bacic et al. 1988; Cosgrove 2005; McNeil and Darvill 1984). One such polysaccharide is xyloglucan, which has a cellulose-like β-(1,4)-d-glucose backbone with branches of xylose, galactose, and occasionally fucose groups (Fry 1989; Hayashi 1989; Hisamatsu et al. 1992). Xyloglucan was proposed to function as a load-bearing tether between CMF in the primary cell wall based on the observation of wall loosening induced by xyloglucan breakdown (Hayashi 1989). Three distinct xyloglucan domains were identified in primary cell walls based on extractability (Pauly et al. 1999): a xyloglucanase-accessible domain, a domain that binds to cellulose surface, and a trapped domain that is only released after complete wall breakdown with cellulase. Recent results are at odds with the idea that CMFs are linked together by xyloglucan tethers and instead indicate a key role for relatively inaccessible xyloglucan trapped between CMFs (Park and Cosgrove 2012).
One fundamental research question is the binding mechanism of CMF and xyloglucan in the cell wall, particularly in the trapped domain. Using two- and three-dimensional magic-angle-spinning solid state NMR, Dick-Pérez et al. (2011) suggested that a limited fraction of xyloglucans of the promary cell wall of Arabidopsis thaliana might have intimate interaction with interior cellulose based on the presence of cross-peaks between interior cellulose and the xyloglucan backbone glucose and side chain residues in the three-dimensional spectra. These xyloglucans have dynamic properties intermediate between rigid cellulose and the more mobile pectins. To date, no molecular modeling work has been performed on the possible configurations of this trapped domain. This work investigates xyloglucans trapped between CMFs and the mechanical significance of this type of interaction.
Jean et al. (2009) built biomimetic thin films consisting of regular stacking of single layers of cellulose nanocrystals separated by a single layer of xyloglucan thin layers, benefiting from the strong cellulose–xyloglucan interaction. This suggests that in vitro xyloglucan can form a single layer between cellulose nanocrystals. However, the detailed molecular-level understanding of the cellulose–xyloglucan interaction in this layer model is not clear.
Many of the functional properties of CMF and xyloglucan depend on the surface characteristics of cellulose (Besombes and Mazeau 2005; Brown Jr 1990; Brumer et al. 2004; Fink et al. 2011; Mazeau 2011; Pérez and Mazeau 2005) and the structural characteristics of xyloglucan (Fry 1989; Hayashi and Takeda 1994; Peña et al. 2004; Vincken et al. 1995). For cellulose, adsorption and adhesion depend on surface properties such as hydrophobicity and surface morphology (Besombes and Mazeau 2005; Mazeau 2011; Mazeau and Vergelati 2002; Mazeau and Wyszomirski 2012; Pérez and Mazeau 2005). Transmission electron microscopy revealed a rectangular cross-section for large crystalline microfibrils such as those of Valonia, Micrasterias or tunicin (Van Daele et al. 1992; Hanley et al. 1997; Helbert et al. 1998; Helbert and Nishiyama 1998; Sugiyama et al. 1991) with four surfaces defined by the hydrophilic (110) and (1–10) planes for the I-β allomorph (Sugiyama et al. 1991). Several authors observed that the corners of the crystals are frequently blunt. The blunt corners could be a thermodynamic equilibrium shape that is lower in surface energy based on the Gibbs-Wulff theorem. Therefore surfaces corresponding to other planes are also present. The minor hydrophobic (100) surface for the I-β phase may be particularly important as the site of binding by matrix polymers as well as type-A cellulose binding modules. With the view that xyloglucan crosslinks between CMFs in plant cell walls provide structural integrity (Hayashi et al. 1987), it is important to understand at the molecular-level the binding between CMF and xyloglucan in the cell wall construction. This knowledge could be useful for potential industrial applications such as biomass conversion (Himmel et al. 1999; Sun and Cheng 2002; Wegner and Jones 2009) or use of modified cellulose or xyloglucan in the evolving field of cellulose-based nanocomposites (Brumer et al. 2004; Dufresne 2008; Hubbe et al. 2008; Mishra and Malhotra 2009; Moon et al. 2011; Wegner and Jones 2009).
Molecular models have been used to characterize the interaction between cellulose and xyloglucans. Early theoretical studies (Levy et al. 1991, 1997) of xyloglucan conformation employed high temperature Monte Carlo procedures and constrained dynamics. Results suggest that a planar glucan backbone is essential for optimal cellulose-binding and also the fucosyl residue enables increased backbone planarity and steric accessibility. However, the study is limited to an isolated xyloglucan molecule.
The landmark work of Hanus and Mazeau (2006) suggested that in vacuo, the xyloglucan fragments (namely XXXG, XXLG, and XXFG, where each (1,4)-β-linked d-glucosyl residue in the backbone is given a one-letter code according to its substituents: G = β-d-Glc; X = α-d-Xyl-(1,6)-β-d-Glc; L = β-d-Gal-(1,2)-α-d-Xyl-(1,6)-β-d-Glc; F = α-l-Fuc-(1,2)-β-d-Gal-(1,2)-α-d-Xyl-(1,6)-β-d-Glc) bind equally to hydrophobic and hydrophilic cellulose surfaces. One caveat for this study is that the lack of water molecules could exaggerate the contribution of hydrogen bonding to the binding. Zhang et al. (2011) later examined larger xyloglucan models (namely GXXXGXXXG, GXXLGXXXG, and GXXFGXXXG) and their interactions with cellulose in the presence of explicit water. However, this modeling work mainly focused on the adsorption of xyloglucans onto the hydrophilic cellulose I-β (1s10) surface. Hydrophobic surfaces should provide a particularly strong potential binding site in aqueous solutions. How surface hydrophobicity of CMF affects xyloglucan adsorption in an aqueous environment is still unclear.
Isothermal titration calorimetry (ITC) and adsorption isotherm studies of xyloglucan–cellulose interaction (Lopez et al. 2010) suggested a minimum of 12 glucosyl residues on the backbone is required to observe significant interactions between CMF and xyloglucan. Moreover, this study suggested bacterial microcrystalline cellulose (BMCC) presents two types of interaction sites for tamarind xylogluco-oligosaccharides (XGO). Type 1 binding sites have a much higher binding affinity constant (3.4 × 10−2 mL · μg −1xyloglucan ) than type 2 binding sites (0.2 × 10−2 mL · μg −1xyloglucan ). However, type 1 binding sites are rapidly saturated, accompanied by a low xyloglucan maximum adsorption capacity. The mechanism of the two XGO binding sites in BMCC and the possibility of the favorable binding sites being the minor hydrophobic (100) surfaces are not clear.
Another important question concerns the specific roles of the backbone and various side chain residues in the xyloglucan–cellulose interaction. Both the complexity of the system and a lack of experimental methods for atomic-level structural analysis contribute to our insufficient knowledge about how side chain substitutions affect xyloglucan–CMF interactions. The subject remains as a debate despite significant efforts in experimental studies both in vivo (Burgert 2006; Cavalier and Lerouxel 2008; Desveaux et al. 1998; Madson et al. 2003; Peña et al. 2004; Ryden and Sugimoto-Shirasu 2003; Vanzin and Madson 2002) and in vitro (Hayashi and Takeda 1994; Lima et al. 2004; de Lima and Buckeridge 2001; Lopez et al. 2010; Vincken et al. 1995). In this regard, in silico simulations provide a powerful tool, because the complexity can be controlled systematically to determine the influence of xyloglucan side chains on their adsorption onto cellulose.
The present paper investigates the effect of surface hydrophobicity and side chain substitutions on xyloglucan adsorption onto CMF in water. The investigation focuses on the characterization of the interactions of the hydrophobic and hydrophilic surfaces with XGOs containing various side chain substitutions (namely (XXXG)3, (XXLG)3, and (XXFG)3). To link atomic-level phenomena to macro-scale cell wall mechanical behavior, steered molecular dynamics (SMD) simulation was employed to estimate the rupture force for an adhesive model where two cellulose microfibrils are held together by a (XXXG)3 between their hydrophobic (100) surfaces.
Methods
All simulations were performed with the CHARMM package (Brooks et al. 2009). The CHARMM all-36 parameter force field for carbohydrates was employed (Guvench et al. 2009). XGOs, composed of the three basic repeat units, namely (XXXG)3, (XXLG)3 and (XXFG)3 as shown in Fig. 1, were built and positioned at a distance of 9–10 Å from the cellulose (110), (1–10), (010), (100), (200) surfaces with their glycosidic backbone parallel to the cellulose chains as shown in Fig. 2. Initial side-chain orientations of xyloglucan were obtained through rotating the XGOs so that both their backbone and side-chains were near the CMF surfaces. The advantage of simultaneous adsorption of multiple XGOs onto CMF is that the analysis can be performed on one simulation, and one has the possibility of observing xyloglucan aggregation on the CMF surface, which could be biologically relevant.

Schematic representation of xylogluco-oligosaccharide models used in this study: (XXXG)3, (XXLG)3 and (XXFG)3

Initial and final (25 ns) structures of (XXXG)3 adsorbed onto cellulose I-β microfibril (110), (100), (010), (200), and (1–10) surfaces. The cellulose microfibril has 25 glucan chains, each 20 glucose residues long. Same color code as Fig. 1 is used for (XXXG)3. (Color figure online)
Based on the coordinates of cellulose I-β crystal structure (Nishiyama et al. 2002), a small cellulose microfibril with different exposed crystallographic faces [(110), (1–10), (010), (100) and (200)] was fabricated using the crystal-building and segment-select facilities in CHARMM. The cellulose microfibril contains 25 glucan chains, each 20 glucose residues long. Fernandes et al. (2011) suggested a 24-chain cellulose microfibril model with both hydrophobic and hydrophilic surfaces exposed based on fitting to the spectroscopy and scattering data. This is at odd with the common assumption of 36 chains based on the cellulose synthase rosette structure. We chose the 25 glucan chains to build the CMF model to be consistent with the 24-chain model of cellulose microfibril and to allow the interactions for different CMF surfaces.
The system was placed in the center of a periodic box with explicit H2O molecules. Water molecules were represented using the modified TIP3P force field (Jorgensen et al. 1983; Durell et al. 1994). The initial coordinates of cellulose used to build the model of fiber and for the water model are included in the Supplementary data (Coor01_cell and Coor02_water). A minimum 10 Å solvation shell was created for each system.
Long-range electrostatics were handled using the particle-mesh Ewald (PME) algorithm (Darden et al. 1993; Essmann et al. 1995). Non-bonded interactions were truncated at 10 Å. All chemical bonds involving H atoms were kept at fixed lengths using the SHAKE algorithm (Van Gunsteren and Berendsen 1977). One hundred steps of steepest descent minimization followed by 100 steps of conjugate gradient minimization were first applied to the system to relieve any large strains associated with the initial conditions. The system was then further prepared by a stepwise heating of 10 ps each at 100, 200, and 300 K, followed by a 20 ps equilibration at 300 K. The 25 ns production trajectory was collected in the NVE ensemble. A Verlet integrator with a step size of 2 fs was used to calculate atomic motions. Configurations were saved every 0.1 ps for subsequent analysis.
In addition, an adhesive model in which a (XXXG)3 molecule was sandwiched between two CMF (100) surfaces was built. The initial configuration was equilibrated in an aqueous environment for 10 ns. After equilibration, a non-equilibrium steered molecular dynamics (SMD) simulation was carried out using the NAMD package (Nelson and Humphrey 1996), with the same force field as described above. One CMF was fixed by external constraints and the center of mass of the other CMF was constrained to move at a constant speed of 0.1 nm ns−1. The pulling velocity is a tradeoff between computing time and the need to stay close to the equilibrium. Although this speed is still much faster than that used in AFM experiments, it represents the computational limit in the simulation regime. The pulling force was applied to all atoms in the pulled CMF in a direction perpendicular to the cellulose (100) face. The MD simulation continued until the pulled CMF detached from the other CMF. Forces and coordinates of the gliding spring and the pulled atoms were stored every 1 ps. The force acting on the spring is calculated as
where k is the spring constant (100 pN Å−1), r and r 0 are the instantaneous position and the initial position of the SMD atoms and n is the direction of pulling.
Results and discussion
Interactions between xyloglucans and cellulose I-β (100) and (1–10) surfaces
To test the accuracy of the CHARMM carbohydrate force field, we extracted three surface slab models as shown in Fig. 3 from a randomly selected configuration within the equilibration trajectory of the MD simulation where multiple chains of (XXFG)3 were adsorbed onto the CMF surfaces in an aqueous environment. We benchmarked the interaction energy for the surface models calculated using the CHARMM force field against dispersion-corrected density functional theory (DFT) using M05-2X methodology (Zhao and Truhlar 2006) with the 6-311G(d,p) basis set (Krishnan and Binkley 1980), comparing the binding energies, i.e.,
for these three surfaces: (1–10), (100) and (010), where Ecellulose–xyloglucan is the total potential energy for the xyloglucan and cellulose assembly for the model, Exyloglucan and Ecellulose are the potential energies for xyloglucan and cellulose for the sample model. The DFT-based energies are −43, −26 and −21 (kcal mol −1XXFG ) for the (1–10), (100) and (010) configurations shown in Fig. 3, while the corresponding CHARMM results are −36, −25 and −21 (kcal mol −1XXFG ). The results are in good agreement except for the hydrophobic surface, whose binding is underestimated by 16 % in CHARMM. Nevertheless, the relative stabilities of the three interfaces are properly described by the empirical force field with reasonable quantitative accuracy. Note that the DFT energies are likely to have inaccuracies on the order of 1 kcal mol−1 (Zhao and Truhlar 2007).

Surface slab models for (XXFG)3 adsorbed onto cellulose I-β microfibril (1–10), (100) and (010) surfaces (Same color code as Fig. 1 is used for (XXFG)3). (Color figure online)
For MD calculations, simulations of 25 ns were performed using the classical CHARMM carbohydrate potentials. The interaction energy profiles for the 25 ns are plotted in Fig. S1 and show equilibrium or near-equilibrium conditions around 10 ns for (XXXG)3, 10 ns for (XXLG)3, and 16 ns for (XXFG)3.
After reaching equilibration, the instantaneous interactions between XGOs and CMF surfaces for independent configurations were estimated using Eq. (2). The average binding energy was obtained as the average of the interaction energies over the independent configurations for the last 5 ns of the 25 ns simulation. The average interaction energy is divided by the number of repeating XXXG units in each fragment to facilitate the comparison. To determine if the average interactions for different xyloglucan fragments are significantly different from each other, we calculated the 95 % confidence interval.
This binding energy method has been widely used for adsorption studies (Shen et al. 2008; Kang et al. 2009). The binding energy analysis does not include other energy components, such as CMF–water, xyloglucan–water, and xyloglucan–xyloglucan interaction energy as well as the water entropy. Therefore it is less rigorous than the binding free energy method, but computationally it is more efficient and less expensive. Table 1 shows the average interaction energies per XGO repeat unit on the CMF (100) and (1–10) surfaces. To obtain further insight into the nature of the interactions, the interaction energy was decomposed into van der Waals (vdw) and electrostatic (elec) contributions.
For (XXXG)3, which has only xylose substitutions, the average binding energy on the (100) surface is −67 kcal mol −1XXXG . In contrast, on the cellulose (1–10) surface, the binding is significantly weaker (−32 kcal mol −1XXXG ). To understand the origin of the binding difference, the interaction energies were calculated for the xylose and backbone glucose residues in (XXXG)3 (Table S1). The xylose–cellulose interactions on both surfaces are similar (−18 kcal mol−1 xylose−1 on the (100) surface and −14 kcal mol−1 xylose−1 on the (1–10) surface). The decreased binding energy on the (1–10) surface is mainly due to the decreased interaction between the surface and the xyloglucan backbone glucose residues (−18 kcal mol−1 glucose−1 on the (1–10) surface and −49 kcal mol−1 glucose−1 on the (100) surface). On the (100) surface, the interaction between the xyloglucan backbone glucose residues and cellulose surface has stronger vdw component (−20 kcal mol−1 glucose−1) as well as higher electrostatic contribution (−29 kcal mol−1 glucose−1). Visual inspection of the trajectory suggests a flat ribbon conformation of (XXXG)3 on the cellulose (100) surface. In contrast, on the cellulose hydrophilic (1–10) surface, (XXXG)3 shows a combination of kink and straight conformations (Fig. 5). Note that our binding energy analysis does not include the water-mediated interaction between (XXXG)3 and cellulose surfaces, which might be another component of the interaction.
For (XXLG)3, the average interaction energy with the cellulose surfaces is −39 kcal mol −1XXXG on (100) and −35 kcal mol −1XXXG on (1–10). Statistically, the difference between these two means is not significant at the 5 % significance level. However the interaction on the (100) surface has higher vdw component than the one on the (1–10) surface.
Compared with (XXXG)3, the interaction energy of (XXLG)3 with the (1–10) surface is not significantly different from the one of (XXXG)3 with the same surface. However, the interaction of (XXLG)3 has more electrostatic contributions than (XXXG)3. Looking at the individual sugar interaction, the addition of the galactose residue does not contribute much in terms of direct interaction with the surface because the average galactose–cellulose interaction energy is −1 kcal mol−1 galactose−1. However, the addition of the galactose residue in (XXLG)3 increases the electrostatic interaction and decreases the vdw interaction between the backbone glucose residue and the (1–10) surface. A look at the trajectory suggests the galactose residue forms hydrogen bonds with the backbone glucose residues and modifies the glucose conformation with respect to the (1–10) surface. There are more hydrogen bond interactions between the backbone glucose residue with the surface than the C–H stacking interactions in (XXLG)3. On the (100) surface, the average XXLG–cellulose interaction is lower than the average XXXG–cellulose interaction with both decreased electrostatic and vdw components mainly due to the loss of the flat ribbon conformation in (XXLG)3.
For (XXFG)3 on the (1–10) surface, the average interaction energy is −29 (kcal mol −1XXFG ), which is a little lower than that of (XXXG)3 and (XXLG)3 on the same surface. The vdw component is similar to that of (XXLG)3, while the electrostatic component of the interaction is lower than (XXLG)3 on that surface. Detailed sugar residue interaction analysis suggests that in (XXFG)3, the backbone glucose residues interact a little more weakly with the cellulose (1–10) surface compared with (XXLG)3. The side-chain galactose and fucose residues does not interact directly with the surface.
However, on the (100) surface, the addition of fucose increases the average interaction energy to −63 (kcal mol −1XXFG ), which is close to the interaction energy of −67 (kcal mol −1XXXG ) for (XXXG)3 on the same surface. The interaction has a vdw component [−24 (kcal mol −1XXFG )] and an electrostatic component [−38 (kcal mol −1XXFG )]. Compared with (XXLG)3, the increased binding energy is mainly due to the stronger electrostatic component. Similar to (XXXG)3, (XXFG)3 has favorable binding to the cellulose hydrophobic (100) surface.
The interaction energy reported by Hanus and Mazeau (2006) calculated in an in vacuo model was significantly higher, approximately −200 kcal mol−1 of XGO unit (e.g. XXXG, XXLG or XXFG). More hydrogen bonds form between xyloglucan and cellulose in vacuo than in an aqueous environment, since hydrogen bonds to water molecules reduce the direct interaction between xyloglucan and cellulose.
Zhang et al. (2011) reported the interaction energy of XGO consisting of two repeating units (e.g. GXXXGXXXG) with cellulose I-β (1–10) surface in water being around −126 kcal mol−1 using the GLYCAM force field (Kirschner 2008), correspond to −63 (kcal mol −1XXXG ) on the (1–10) surface, which is much larger than our result (−32 kcal mol−1). Zhang’s work modeled a GXXXGXXXG molecule on an infinite cellulose (1–10) surface with periodic boundary conditions to two dimensions, whereas we considered a (XXXG)3 molecule on a finite 25-chain cellulose microfibril model in which the (1–10) surface has very limited spatial extent. A close study of the conformation of GXXXGXXXG in Zhang’s work on the (1–10) surface suggests that the addition of one more glucose residue at one end and the short length of the xyloglucan fragment resulted in a straighter conformation which has more efficient binding interaction with the infinite surface. In our model, the CMF (1–10) surface has a width around 3 nm (a more realistic model), which greatly limited the interactions. In addition, (XXXG)3 is longer and has a bent conformation on the (1–10) surface. All these factors might contribute to the less-efficient binding interaction in our model compared with the one found in the previous study. A different force field parameterization might also contribute to the differences. However, we believe our model is more biologically relevant than the one in Zhang et al. (2011).
To further understand adsorption dynamics, we analyzed the distributions of the binding energy per XGO repeat unit for the last 1 ns of the MD run, as shown in Fig. 4. All the distributions are very broad in the range of 0–100 kcal mol−1 and approximately Gaussian. Since the total system energy involves partial cancellations of various distinct energetic contributions (i.e. various bond deformations plus multiple non-bonded interactions), the variance in binding energies is much larger than the thermal energy scale. Nevertheless, trends in the magnitude of the attractive electrostatic-plus-vdw components will reflect trends in the overall favorability of binding, since the countervailing terms in the total energy (e.g. the energetic costs of molecular deformations to assume a more favorable registry at the interface) are largely driven by these attractive components. Most of the variance arises from variations in the electrostatic component (e.g. hydrogen bonding), not the vdw component, probably due to the strong directionality of the hydrogen bond component and the competition from ambient water for hydrogen bond sites. The average electrostatic contribution to the binding varies from −10 to −40 kcal mol−1, depending on the specific XGO (XXXG, XXLG or XXFG); with hydrogen bonding being the major contributor to the electrostatic part, this corresponds to approximately 2–8 hydrogen bonds per XGO unit with the cellulose surface. All XGOs studied here have four glucose backbone residues and three xylose side-chain residues, so this corresponds to less than one hydrogen bond per residue. All the distributions have similar variance, between 40 and 60 kcal mol−1, corresponding to fluctuation of ±5 hydrogen bonds per XGO unit. Most of this large variation is associated with competition from hydrogen bonding to ambient water.

The binding energy distribution for the (XXXG)3, (XXLG)3 and (XXFG)3 adsorbed onto the cellulose I-β microfibril (100) (filled circles) and (1–10) (open circles) surfaces (the energy measured per xyloglucan unit (i.e. XXXG, XXLG or XXFG)). The ln(P) is defined to be the natural logarithm of the probability. By taking the natural logarithm, the probability distribution was fitted to a parabola curve for a Gaussian distribution (the blue line for the fitted curve on (100) surface, and the green line for the one on (1–10) surface). (Color figure online)
In summary, on the (1–10) surface, the interaction energies are statistically not different for (XXXG)3, (XXLG)3 and (XXFG)3 (e.g. (XXXG)3 ≈ (XXFG)3 ≈ (XXLG)3). However, on the (100) surface, the binding energy is ordered as (XXXG)3 ≈ (XXFG)3 > (XXLG)3, where the galactose residue appears to interfere with the interaction between the xyloglucan backbone and the surface. One might expect that increasing the side-chain length of xyloglucan to produce a monotonic trend, either increasing or decreasing, to CMF binding. Our results suggest there is no such a trend. Further analysis of the interaction from individual sugar residue shows that, on average, the galactose and the fucose residues do not contribute directly to the binding interactions, but they can modify the interaction between the backbone glucose residue and the cellulose surface through the interaction with the backbone residues. The cellulose surface topography, instead, appears to have a larger effect on the binding. For example, the binding energy of (XXXG)3 and (XXFG)3 to the (100) surface is significantly higher than the other ones.
Conformations of adsorbed xyloglucans
The glycosidic backbone of the xyloglucans was initially aligned parallel to the cellulosic chains. During equilibration, the xyloglucan backbone partially deviates from the CMF axis through bending or tilting, except for the case of (XXXG)3 adsorbed on the (100) hydrophobic surface, which retains its initial ribbon-like backbone conformation throughout the 25 ns trajectory. The glucose backbone of (XXXG)3 aligns between two glucan chains on this (100) surface, similar to the stacking conformation found between different sheets inside a CMF. The xylose side-chain residues adopt similar stacking. The XXXG can apparently adapt well to the flat ribbon-like structure of the CMF (100) surface. Figure 5 provides snapshots of typical XXXG conformations on the CMF (100) and (1–10) surfaces.

Backbone conformations at 25 ns for (XXXG)3, (XXLG)3 and (XXFG)3 absorbed onto cellulose microfibril I-β hydrophobic (100) and hydrophilic (1–10) surfaces. Same color code as Fig. 1 is used. (Color figure online)
We characterize the rigidity of the xyloglucan backbone by monitoring the angle θ between the geometric centers of three successive glucose residues on the backbone, as indicated by the blue lines in Fig. 6. This angle is similar to the bond bending variable in a coarse-grained model. For (XXXG)3 on the hydrophobic (100) surface, the distribution is narrow and close to 180° (as shown in Fig. 6) indicating a rigid, linear chain. Surface hydrophobicity promotes the rigidity of bound (XXXG)3 in a flat ribbon-like conformation. However, this rigidity is lost when (XXXG)3 is placed on the hydrophilic (1–10) surface, as demonstrated by the much broader distribution. The distribution has two centers around 165° and 140°, indicating two populations of backbone conformations: straight and kinked.

The backbone angle distribution for (XXXG)3, (XXLG)3 and (XXFG)3 adsorbed onto cellulose I-β microfibril (100) and (1–10) surfaces. Same definition for ln(P) as Fig. 4 is used. The angle θ is defined by the geometric centers of three successive backbone glucosyl residues as indicated by the blue lines. Filled circles for xyloglucan on (100) surface, and open circles for xyloglucan on (1–10) surface. (Color figure online)
The density profile for the backbone bending angle of (XXLG)3 on the (100) and (1–10) surfaces has a distribution centered around 165°, suggesting a close-to-straight backbone conformation. Compared with (XXXG)3 on the (1–10) surface, the broadness of the distribution is similar, indicating similar rigidity of the backbone. But in (XXLG)3, a kinked conformation forms on the (100) surface of cellulose microfibril, as indicated by a second population around 140°. For (XXFG)3, the main distribution of the backbone on the (100) and (1–10) surfaces is in the range of 160–170° (Fig. 6), indicating their backbones are also close to a straight conformation. The result suggests that in general there are two populations of conformation for xyloglucan backbone on the surface of cellulose microfibril: kinked and straight.
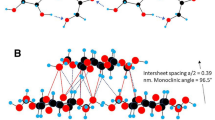
To better understand the factors that determine the conformations of adsorbed xyloglucan, we examined the intra-molecular hydrogen bonding patterns. On the hydrophobic surface, the lightly substituted (XXXG)3 is flat, with all residues stacking to the surface. This flat ribbon conformation is made possible by the intra-molecular hydrogen bonding between the xylose C2 hydroxyl group and the C3 hydroxyl group of the backbone glucose residue in the reducing end direction, as indicated by the black arrow in Fig. S2. The distribution of the O2(xylose)–O3(glucose) distance in (XXXG)3 on the (100) surface (Fig. S3) confirms a high density at the hydrogen bond distance of 0.3 nm. Visual inspection of the trajectory also confirms the dominant populations of these hydrogen bonds in (XXXG)3. The surface topography of the hydrophobic surface of the CMF further stabilizes this conformation because of structural similarity. Note that the surface chains of real CMFs have a certain amount of disorder, so the stacking of the (XXXG)3 to the hydrophobic surface in real systems may be less ideal than that seen in for our well-ordered fibril surfaces.
On the rough hydrophilic (1–10) surface, a flat conformation of (XXXG)3 is not stable because the twisted backbone conformation better facilitates the formation of hydrogen bonds both between (XXXG)3 and CMF and also between (XXXG)3 and ambient water. The density profile of the O2(xylose)–O3(glucose) distance in (XXXG)3 on the (1–10) surface suggests a more dynamic interaction between the xylose C2 hydroxyl group and the C3 hydroxyl group of the backbone glucose residue in this twisted conformation.
The substitution of a galactosyl residue at the xylosyl residue C2 position inhibits the hydrogen bonding formation between the xylose C2 hydroxyl group and the backbone glucose C3 hydroxyl group, instead allowing hydrogen bonds between galactose and adjacent xylose residues as well as direct hydrogen bonds between galactose residues and the backbone glucose residue (Fig. S2). The overall conformation of the (XXLG)3 backbone is twisted and bent (Fig. 5) on both the hydrophobic and hydrophilic surfaces of the CMF and some (XXLG)3 segments are expelled from the surface (consistent with the similar binding energy distributions of (XXLG)3 on both surfaces). For (XXFG)3, the increase in the size of the side chain greatly increases the number of possible hydrogen bonding geometries (Fig. S2), causing the backbone to favor a twisted and bent conformation (Fig. 5).
The initial distance between the xyloglucan chain and the surface is approximately 9 Å in each case, which is about three water layers. At equilibrium the distance decreases to 5–6 Å for bound xyloglucans, with a single incomplete water layer at the interface, as shown in Fig. 7 for (XXXG)3 bound to the (100) and (110) surfaces. The exclusion of water is more complete for the hydrophobic (100) surface due to the flat stacking of the glucose rings. The hydrophobic interaction between XXXG and the CMF surface might be the driving force for the preferential binding of (XXXG)3 to the hydrophobic CMF surface.

Snapshots of interfacial water layer (in blue) between (XXXG)3 (shown in color) and cellulose I-β microfibril (100) and (110) surfaces (in grey) at 25 ns. More water molecules are exclude at the (100) surface due to the flat stacking of sugar rings. (Color figure online)
To quantify the distances of different xyloglucan backbones and side-chains from the surface of the CMF, we analyzed the distance distributions between the geometric centers of the sugar residues of interest with respect to the glycosyl residues of the CMF surface. Figure 8 shows the resulting radial distribution functions of each xyloglucan sugar residue in the three systems studied. For (XXXG)3 on the hydrophobic surface (Fig 8), the first peak appears at 0.58 nm for the side chain xylose residues and 0.64 nm for the backbone glucose residues. This is only slightly larger than the distance observed in common direct sugar stacking (0.4–0.5 nm), indicating that both backbone and side chain sugar residues of (XXXG)3 are in close vicinity to the CMF surface; they interact strongly across the whole simulation time, with few interstitial water molecules between them. On the hydrophilic surface (Fig. 8), the peaks for both the backbone and side chain residues are much weaker than on the hydrophobic surface, reflecting a less ordered (XXXG)3 structure with fewer bound sugar residues for (XXXG)3. This is consistent with our previous discussion that the topography of the hydrophilic CMF surface does not support a flat ribbon-like conformation of (XXXG)3.

The radial distribution function between the glucose residue on the (100) surface of cellulose microfibril and the sugar residues in (XXXG)3, (XXLG)3 and (XXFG)3 (the backbone glucose residue in black, the xylose residue in red, the galactose residue in blue, and the fucose residue in orange). The corresponding distribution on the (1–10) surface of cellulose microfibril is shifted down vertically for clarity. (Color figure online)
For (XXLG)3 on the hydrophobic CMF surface (Fig 8), several peaks exist for the backbone glucose residues, ranging from 0.50 to 0.75 nm, consistent with a more dynamic backbone conformation compared to (XXXG)3 on the same surface. (XXLG)3 side-chain xylose and galactose residues are farther from the CMF surface, between 0.60 and 0.80 nm. The pair distribution profile for (XXLG)3 on the hydrophilic surface (Fig. 8) is similar to (XXXG)3 on the same surface. This is consistent with weaker binding of (XXLG)3 to both hydrophobic and hydrophilic surfaces. For (XXFG)3 on the hydrophobic (100) surface, both backbone glucose residue and side-chain fucose residues are approximately 0.60–0.70 nm from the surface (Fig. 8), reflecting the contribution of surface interaction with both backbone and side-chain fucosyl residues. Residue pair correlations for (XXFG)3 on the hydrophilic (1–10) surface show favorable surface interactions with side-chain xylosyl and fucosyl residues, while the backbone glucosyl and side-chain galactosyl residues are farther away.
In summary, the structural and binding properties of adsorbed xyloglucans are greatly affected by both side chain substitution and the topography of the CMF surfaces. (XXXG)3 has a specific binding preference to hydrophobic CMF surfaces, while binding to hydrophilic CMF surfaces is relatively weak and nonspecific.
Rupture force evaluated by SMD simulation for an adhesive model
Based on the effectiveness of different substrate-specific endoglucanases for causing creep and increasing cell wall compliances, Park and Cosgrove (2012) proposed that site of wall loosening by these enzymes is limited to small region of tight CMF contact mediated by trapped xyloglucans. To test this idea, we sandwiched an (XXXG)3 molecule—the strongest interacting xyloglucan studied here—between two hydrophobic CMF surfaces (Fig. 9) and used steered molecular dynamics to characterize the mechanical strength of this interface. After equilibration in water, one CMF was fixed, and the other one was pulled away with a constant speed of 0.1 nm ns−1 within the SMD module of NAMD. The SMD model applies a fictitious spring force to the pulled CMF, similar to that which would be applied by an atomic force microscope cantilever. The force-extension profile (Fig. 10) can be extracted as the simulation proceeds.

The adhesive model consists of one (XXXG)3 oligosaccharide (red) sandwiched between two cellulose I-β microfibril (100) surfaces. The blue cellulose microfibril is fixed. The green cellulose microfibril is constrained to move at a constant velocity along the direction perpendicular to the (100) surface. (Color figure online)

The force experienced by the cellulose microfibril as pulled away from its original position in the adhesive model (refer to Fig. 9). The pulling velocity is 0.1 nm ns−1
Initially, the force grows rapidly and almost linearly with the separation, as expected for a Hooke’s law spring. The force and displacement display fluctuations with a characteristic energy on the order of kT and a characteristic distance on the order of half an Angstrom, consistent with an origin in thermal fluctuations of non-bonded CMF–xyloglucan interactions. The interface ruptures when the CMF is displaced approximately 0.3 nm from its initial equilibrium position, after which the force trends downwards. Fluctuations in this downward ramp are less pronounced than in the quasi-Hooke’s law region, since the interaction after rupture is dominated by smoother long-ranged forces. The (XXXG)3 remains bound to the fixed CMF. The rupture force Fmax is approximately 1,000 pN. Taking a cross-sectional area Across of approximately 6 nm2 (1 nm × 6 nm), the failure stress of this hydrophobic link is roughly 1,000 pN 6 nm−2 ≈ 0.2 GPa. This value is on the same order of magnitude as wall stresses (0.01–0.1 GPa). The stress or tension within the wall is approximately estimated, i.e.,
where S is the equatorial stress, P is the turgor pressure, r is the radius of the sphere and t is the thickness of the wall (Carpita 1985). For spherical cells, the equatorial stress is opposite to the pressure applied over the area of an equatorial plane through the sphere. The stress is distributed only over the area of the thin wall in this plane, or approximately thickness times circumference. Taking the radius of the cell of approximately 10 μm and the thickness of the wall of 0.1 μm (Carpita 1985), the stress within the wall is roughly 0.01–0.1 GPa with the turgor pressure being 0.3–1 MPa in typical plant cell walls (Cosgrove 1993). In real walls, CMFs are likely to be glued together in larger configurations than what was simulated here, with the results that thermal fluctuations will have a diminished influence on force-extension behavior. Multiple adhesion points concentrated in the larger configuration might provide more rigidity to the cell wall network.
The stress from our simulation is an upper bound on the true quasi-static value, due to the finite (and rapid) pulling speed in the simulation. Fig. S4 shows the force curves for several pulling speeds and demonstrates that the estimated rupture force decreases with decreasing pulling speed. Although even the slowest speed (0.1 nm ns−1) is still seemingly large, further slowing SMD simulations face challenges of computational resources. Besides, we believe the 0.1 nm ns−1 is slow enough to produce the rupture force in the right order of magnitude. In a real AFM experiment, slower pulling will allow a greater role for fluctuations in overcoming the interfacial adhesion. Also, while all interactions are totally lost at the point of rupture in the SMD simulation, some interactions remain in place in the experiment. Thus it is not uncommon to notice that the rupture force from SMD is high when compared to the value measured by AFM (Morfill et al. 2008; Guzman et al. 2008; Izrailev et al. 1997).
Taking these into consideration, we believe that the extensional strength of this cellulose–XXXG junction could bear a significant portion of the overall wall stress, but that other modes of mechanical resiliency, including the shear response of this junction and other possible modes of CMF–CMF interaction, should also be considered.
Conclusion
We have, by means of atomistic MD simulations, investigated the adsorption of three XGOs, (XXXG)3, (XXLG)3, and (XXFG)3, on the cellulose I-β hydrophobic and hydrophilic surfaces in an aqueous environment, with a particular focus on the effects of surface hydrophobicity and side chain variation on adsorption. For (XXXG)3 and (XXFG)3, the hydrophobic surface is favored, especially for (XXXG)3, which adopts a conformation on this surface which is close to that of the CMF strands themselves. (XXLG)3 binds more weakly to both hydrophobic and hydrophilic surfaces. The topography of the hydrophobic surface of CMF stabilizes the flat backbone conformation of (XXXG)3, apparently driven by a strong hydrophobic interaction. In contrast, the rough hydrophilic surface has weaker interactions with all of the xyloglucan systems studied. One has to keep in mind that for MD simulations there can always be some issues with incomplete sampling, especially at this size of a system (105 atoms). The conformational sampling in this study might not be perfect with possibilities of other binding modes being not sampled. In addition, the binding energy analysis has no entropy term, therefore further refinement of the method and experimental validation of the model generated in this study is needed to strengthen the conclusion. Using non-equilibrium steered MD simulations, we further estimate an upper bound on the extensional stress that a hydrophobic link can sustain when a (XXXG)3 chain is sandwiched between two hydrophobic CMF surfaces. The result suggests that this configuration may contribute significantly to CMF–CMF adhesion and thus cell wall mechanical response.
References
Bacic A, Harris P, Stone B, Preiss J (1988) Structure and function of plant cell walls. Biochem Plant 14:297–371
Besombes S, Mazeau K (2005) The cellulose/lignin assembly assessed by molecular modeling. Part 1: adsorption of a threo guaiacyl β-O-4 dimer onto a Iβ cellulose whisker. Plant Physiol Biochem 43:299–308
Brooks B, Brooks C, Mackerell AD, MacKerell A et al (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614
Brown Jr RM (1990) Microbial cellulose modified during synthesis. U.S. Patent No. 4,942,128
Brumer H, Zhou Q, Baumann M et al (2004) Activation of crystalline cellulose surfaces through the chemoenzymatic modification of xyloglucan. J Am Chem Soc 126:5715–5721
Burgert I (2006) Exploring the micromechanical design of plant cell walls. Am J Bot 93:1391–1401
Carpita N (1985) Tensile strength of cell walls of living cells. J Plant Physiol 79:485–488
Cavalier D, Lerouxel O (2008) Disrupting two Arabidopsis thaliana xylosyltransferase genes results in plants deficient in xyloglucan, a major primary cell wall component. Plant Cell Online 20:1519–1537
Cosgrove D (1993) Wall extensibility: its nature, measurement and relationship to plant cell growth. New Phytol 124:1–23
Cosgrove D (2005) Growth of the plant cell wall. Nat Rev Mol Cell Bio 6:850–861
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an Nlog (N) method for Ewald sums in large systems. J Chem Phys 98:10089
de Lima D, Buckeridge M (2001) Interaction between cellulose and storage xyloglucans: the influence of the degree of galactosylation. Carbohydr Polym 46:157–163
Desveaux D, Faik A, Maclachlan G (1998) Fucosyltransferase and the biosynthesis of storage and structural xyloglucan in developing nasturtium fruits. Plant Physiol 118:885–894
Dick-Pérez M, Zhang Y, Hayes J, Salazar A, Zabotina OA, Hong M (2011) Structure and interactions of plant cell-wall polysaccharides by two- and three-dimensional magic-angle-spinning solid-state NMR. Biochemistry 50:989–1000
Dufresne A (2008) Polysaccharide nano crystal reinforced nanocomposites. Can J Chem 86:484–494
Durell SR, Brooks BR, Ben-Naim A (1994) Solvent-induced forces between two hydrophilic groups. J Phys Chem 98:2198–2202
Essmann U, Perera L, Berkowitz ML et al (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593
Fernandes AN, Thomas LH, Altaner CM, Callow P, Forsyth VT, Apperley DC, Kennedy CJ, Jarvis MC (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc Natl Acad Sci USA 108:1195–1203
Fink H, Ahrenstedt L, Bodin A (2011) Bacterial cellulose modified with xyloglucan bearing the adhesion peptide RGD promotes endothelial cell adhesion and metabolism—a promising modification for vascular grafts. J Tissue Eng Regen Med 5:454–463
Fry S (1989) The structure and functions of xyloglucan. J Exp Bot 40:1–11
Guvench O, Hatcher ER, Venable RM et al (2009) CHARMM additive all-atom force field for acyclic polyalcohols, acyclic carbohydrates and inositol. J Chem Theory Comp 5:2353–2370
Guzman D, Roland J, Keer H et al (2008) Using steered molecular dynamics simulations and single-molecule force spectroscopy to guide the rational design of biomimetic modular polymeric materials. Polymer 49:3892–3901
Hanley S, Revol J, Godbout L et al (1997) Atomic force microscopy and transmission electron microscopy of cellulose from Micrasterias denticulata; evidence for a chiral helical microfbril twist. Cellulose 4:209–220
Hanus J, Mazeau K (2006) The xyloglucan–cellulose assembly at the atomic scale. Biopolymers 82:59–73
Hayashi T (1989) Xyloglucans in the primary cell wall. Annu Rev Plant Biol 40:139–168
Hayashi T, Takeda T (1994) Effects of the degree of polymerization on the binding of xyloglucans to cellulose. Plant Cell Physiol 35:893–899
Hayashi T, Marsden M, Delmer D (1987) Pea xyloglucan and cellulose VI. Xyloglucan-cellulose interactions in vitro and in vivo. Plant Physiol 83:384–389
Helbert W, Nishiyama Y (1998) Molecular imaging of Halocynthia papillosa cellulose. J Struct Biol 124:42–50
Helbert W, Sugiyama J, Kimura S, Itoh T (1998) High-resolution electron microscopy on ultrathin sections of cellulose microfibrils generated by glomerulocytes in Polyzoa vesiculiphora. Protoplasma 203:84–90
Himmel M, Ruth M, Wyman C (1999) Cellulase for commodity products from cellulosic biomass. Curr Opin Biotechnol 10:358–364
Hisamatsu M, York W, Darvill A, Albersheim P (1992) Characterization of seven xyloglucan oligosaccharides containing from seventeen to twenty glycosyl residues. Carbohyd Res 227:45–71
Hubbe M, Rojas O, Lucia L, Sain M (2008) Cellulosic nanocomposites: a review. Bioresources 3:929–980
Izrailev S, Stepaniants S, Balsera M et al (1997) Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys J 72:1568–1581
Jean B, Heux L, Dubreuil F, Chambat G, Cousin F (2009) Non-electrostatic building of biomimetic cellulose-xyloglucan multilayers. Langmuir 25:3920–3923
Jorgensen WL, Chandrasekhar JM, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Kang Y, Liu YC, Wang Q, Shen JW, Wu T, Guan W (2009) On the spontaneous encapsulation of proteins in carbon nanotubes. Biomaterials 30:2807–2815
Kirschner K (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem 29:622–655
Krishnan R, Binkley J (1980) Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys 72:650
Levy S, York WS, Stuike-Pril R, Meyer B, Staehelin A (1991) Simulations of the static and dynamic molecular conformations of xyloglucan. The role of the fucosylated sidechain in surface-specific sidechain folding. Plant J 1:195–215
Levy S, Maclachlan G, Staehelin A (1997) Xyloglucan sidechains modulate binding to cellulose during in vitro binding assays as predicted by conformational dynamics simulations. Plant J 11:373–386
Lima D, Loh W, Buckeridge M (2004) Xyloglucan–cellulose interaction depends on the sidechains and molecular weight of xyloglucan. Plant Physiol Biochem 42:389–394
Lopez M, Bizot H, Chambat G (2010) Enthalpic studies of xyloglucan-cellulose interactions. Biomacromolecules 11:1417–1428
Madson M, Dunand C, Li X (2003) The MUR3 gene of Arabidopsis encodes a xyloglucan galactosyltransferase that is evolutionarily related to animal exostosins. Plant Cell Online 7:1662–1670
Mazeau K (2011) On the external morphology of native cellulose microfibrils. Carbohydr Polym 84:524–532
Mazeau K, Vergelati C (2002) Atomistic modeling of the adsorption of benzophenone onto cellulosic surfaces. Langmuir 18:1919–1927
Mazeau K, Wyszomirski M (2012) Modelling of Congo red adsorption on the hydrophobic surface of cellulose using molecular dynamics. Cellulose 19:1495–1506
McNeil M, Darvill A (1984) Structure and function of the primary cell walls of plants. Annu Rev Biochem 53:625–663
Mishra A, Malhotra AV (2009) Tamarind xyloglucan: a polysaccharide with versatile application potential. J Mater Chem 19:8528–8536
Moon R, Martini A, Nairn J (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3941–3994
Morfill J, Neumann J, Blank K et al (2008) Force-based analysis of multidimensional energy landscapes: application of dynamic force spectroscopy and steered molecular dynamics simulations to an antibody fragment–peptide complex. J Mol Biol 381:1253–1266
Nelson M, Humphrey W (1996) NAMD: a parallel, object-oriented molecular dynamics program. J High Perform Comput Appl 10:251–268
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124:9074–9082
Park Y, Cosgrove D (2012) A revised architecture of primary cell walls based on biomechanical changes induced by substrate-specific endoglucanases. Plant Physiol 158:1933–1943
Pauly M, Andersen L, Kauppinen S (1999) A xyloglucan-specific endo-β-1, 4-glucanase from Aspergillus aculeatus: expression cloning in yeast, purification and characterization of the recombinant enzyme. Glycobiology 9:93–100
Peña M, Ryden P, Madson M (2004) The galactose residues of xyloglucan are essential to maintain mechanical strength of the primary cell walls in Arabidopsis during growth. Plant Physiol 134:443–451
Pérez S, Mazeau K (2005) Conformations, structures, and morphologies of celluloses. In: Dumitriu S (ed) Polysaccharides: structural diversity and functional versatility, 2nd edn. Dekker, New York, pp 41–68
Ryden P, Sugimoto-Shirasu K (2003) Tensile properties of Arabidopsis cell walls depend on both a xyloglucan cross-linked microfibrillar network and rhamnogalacturonan II-borate complexes. Plant Physiol 132:1033–1040
Shen JW, Wu T, Wang Q, Pan HH (2008) Induced stepwise conformational change of human serum albumin on carbon nanotube surfaces. Biomaterials 29:513–532
Sugiyama J, Vuong R, Chanzy H (1991) Electron diffraction study on the two crystalline phases occurring in native cellulose from an algal cell wall. Macromolecules 24:4168–4175
Sun Y, Cheng J (2002) Hydrolysis of lignocellulosic materials for ethanol production: a review. Bioresour Technol 83:1–11
Van Daele Y, Revol J, Gaill F, Goffinet G (1992) Characterization and supramolecular architecture of the cellulose-protein fibrils in the tunic of the sea peach (Halocynthia papillosa, Ascidiacea, Urochordata). Biol Cell 76:87–96
Van Gunsteren W, Berendsen H (1977) Algorithms for macromolecular dynamics and constraint dynamics. Mol Phys 34:1311–1327
Vanzin G, Madson M (2002) The mur2 mutant of Arabidopsis thaliana lacks fucosylated xyloglucan because of a lesion in fucosyltransferase AtFUT1. Proc Natl Acad Sci USA 99:3340–3345
Vincken JP, de Keizer A, de Keizer A et al (1995) Fractionation of xyloglucan fragments and their interaction with cellulose. Plant Physiol 108:1579–1585
Wegner T, Jones E (2009) A fundamental review of the relationships between nanotechnology and lignocellulosic biomass. Nanosci Technol Renew Biomater 1:1–41
Zhang Q, Brumer H, Ågren H, Tu Y (2011) The adsorption of xyloglucan on cellulose: effects of explicit water and side chain variation. Carbohydr Res 346:2595–2602
Zhao Y, Truhlar D (2006) A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J Chem Phys 125:194101
Zhao Y, Truhlar D (2007) Density functionals for noncovalent interaction energies of biological importance. J Chem Theory Comput 3:289–300
Acknowledgments
This work is supported as part of The Center for Lignocellulose Structure and Formation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001090. We acknowledge the Penn State University RCC Center for supercomputer time.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zhao, Z., Crespi, V.H., Kubicki, J.D. et al. Molecular dynamics simulation study of xyloglucan adsorption on cellulose surfaces: effects of surface hydrophobicity and side-chain variation. Cellulose 21, 1025–1039 (2014). https://doi.org/10.1007/s10570-013-0041-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-013-0041-1