
Abstract
A family of polysaccharide based scaffold materials, bacterial cellulose/chitosan (BC/CTS) porous scaffolds with various weight ratios (from 20/80 to 60/40 w/w%) were prepared by freezing (−30 and −80 °C) and lyophilization of a mixture of microfibrillated BC suspension and chitosan solution. The microfibrillated BC (MFC) was subjected to 2,2,6,6-tetramethylpyperidine-1-oxyl radical (TEMPO)-mediated oxidation to introduce surface carboxyl groups before mixing. The integration of MFC within chitosan matrix was performed by 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC)-mediated cross-linking. The covalent amide bond formation was determined by ATR-FTIR. Because of this covalent coupling, the scaffolds retain their original shapes during autoclave sterilization. The composite scaffolds are three-dimensional open pore microstructure with pore size ranging from 120 to 280 μm. The freezing temperature and mean pore size take less effect on scaffold mechanical properties. The compressive modulus and strength increased with increase in MFC content. The results show that the scaffolds of higher MFC content contribute to overall better mechanical properties.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Rapid progresses of microbially derived bacterial cellulose as biomedical materials in recent studies are wound-healing applications (Czaja et al. 2006) and artificial blood vessels (Klemm et al. 2001) mainly because of its high purity, native mechanical property, mouldability, and biocompatibility (Helenius et al. 2006; Jonas and Farah 1998; Yamanaka et al. 1989). BC as scaffolds for tissue engineering applications has become latest emerging field in this decade (Svensson et al. 2005). Tissue engineering, a technique to create artificial constructs (scaffolds) to direct tissue generation from culture cells, is now being considered as a potential alternative to organ or tissue transplantation. It is generally based on using three dimensional biodegradable polymer scaffolds with appropriate porosity for mechanical support and tissue guidance. In some cases, the scaffolds serve as carrier for growth factors to accelerate healing process when placed in vivo (Ikada 2006).
Any kind of living tissues is composed of cells and extracellular matrix (ECM). For example: in plant, ECM is primarily composed of cellulose; in arthropods and fungi, ECM is largely composed of chitin; in vertebrates, ECM is made of a complex mixture of collagen fibers, proteoglycans and glycoproteins (plus minerals in the case of bone). Proteoglycan is composed of unbranched heteropolysaccharides chains, glycosaminoglycan (GAGs), which is associated with a small portion of protein core. The GAGs chains consist of repeated disaccharide units with the general structure: [uronic acid-amino sugars] n . Several sugars are incorporated in proteoglycans (Di Martino et al. 2005; Suh and Matthew 2000). The most abundant one is N-acetylglucosamine (GlcNAc), which has similar chemical structure to that of chitin, the second most abundant natural biopolymer commonly found in shells of marine crustaceans and cell walls of fungi (Roberts 1992). Chitosan, a deacetylated derivative of chitin, is readily soluble in dilute acids. The resulting cationic nature, high charge density chitosan solution favors formation of insoluble ionic complexes with anionic GAGs, alginates and other negatively charged molecules or polymers. Chitosan has been used as scaffolding materials in articular cartilage tissue engineering due to its structural similarity with various GAGs that found in articular cartilage (Di Martino et al. 2005; Suh and Matthew 2000).
Because GAGs are considered to play an important role in stimulating the chondrogenesis, such as modulating chondrocyte morphology, differentiation, and function (Di Martino et al. 2005; Kosher et al. 1973), using chitosan as GAGs analogue in components of scaffold preparation appears to be potentially applicable for repair of articular cartilage (Suh and Matthew 2000) together with its non-toxicity, excellent biocompatibility and biodegradability, and excellent formation capability (Madhihally and Matthew 1999). Lahiji et al. (2000) reported that chitosan supports the expression of ECM proteins in human osteoblasts and chondrocytes. Moreover, the scaffolds designed for tissue engineering purpose should be three-dimensional, highly- porous-interconnected pore structure to support cell attachment and proliferation; sufficient structural integrity matching the mechanical properties of native tissue. Notably, scaffold microstructure (porosity, mean pore size, pore shape, interconnectivity, specific surface area)(Nehrer et al. 1997; O’Brien et al. 2005; Zeltinger et al. 2001) and mechanical properties (Freyman et al. 2001; Schulz-Torres et al. 2000) have been shown to influence cell behaviors such as adhesion, growth and differentiation.
On the basis of the facts mentioned above, development of BC/CTS three-dimensional porous scaffolds that can serve as biomimetic ECM is the goal of this study. However, never-dried native BC is composed of several lamella layers of asymmetric network structure with a small void volume (~10 to 50 μm) (Nge and Sugiyama 2007). In order to use BC as a tissue scaffold, a more uniform porous structure with larger pore size is needed to develop. Therefore, we intend to fabricate a family of polysaccharide-based BC/CTS porous scaffold by two-step chemical modifications. The experimental design includes: (a) disintegration of never-dried BC pellicles followed by TEMPO-mediated oxidation (Nge and Sugiyama 2007; Saito and Isogai 2004) to introduce surface carboxyl groups; (b) EDC-mediated cross-linking (Araki et al. 2002; Harley et al. 2007; Mao et al. 2003; Olde Damink et al. 1996; Pfeiffer et al. 2008) to integrate BC nanofibrils within chitosan matrix; (c) freezing and lyophilization to produce three-dimensional porous scaffolds.
The water-soluble carbodiimide, 1-ethyl-3-(3-dimethyl aminopropyl)carbodiimide (EDC) is known to mediate ester or amide bond formation between hydroxyl or amine and carboxyl groups, and has been used widely to cross-link various proteins and polysaccharides (Araki et al. 2002; Harley et al. 2007; Mao et al. 2003; Olde Damink et al. 1996; Pfeiffer et al. 2008). Cross-linking reaction between active chemical groups enhances structural stability and strength. In this way, the structural integrity of the heterogeneous system—BC nanofibrils in chitosan solution—can be achieved. Here, we report the updated information of bacterial cellulose-based BC/CTS porous scaffolds with open pore microstructure in order to know the basic understanding of structure-property relationship as a function of fiber content for further evaluation of this kind of scaffolds applicable for cartilage tissue engineering.
Experimental
Materials
Acetobacter Aceti (AJ 12368) and Hestrin-Schramm (HS) medium (Hestrin and Schramm 1954) were used for BC pellicle production. Chitosan 100 (MW 150,000 with degree of deacetylation about 80%, data provided from supplier), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 2,2,6,6-tetramethylpyperidine-1-oxyl radical (TEMPO), sodium bromide (NaBr), and 12% sodium hypochlorite (NaClO) solution (Wako Pure Chemicals, Japan) were used without further purification.
TEMPO-oxidized bacterial cellulose suspension
BC pellicle production was described in detail in our previous report (Nge and Sugiyama 2007). The never-dried BC pellicles after purification were fibrillated by using homogenizer (Physcotron NS 50, Microtech Nichion) followed by TEMPO-mediated oxidation to introduce carboxyl functional groups (Nge and Sugiyama 2007; Saito and Isogai 2004). Briefly, the microfibrillated cellulose (MFC) 3 g was suspended in water (1 L) containing TEMPO (0.075 g) and NaBr (0.375 g). The TEMPO-oxidation was started by addition of NaClO (4.84 mmol/g cellulose) while stirring. The reaction was carried out at room temperature and the reaction pH was maintained at 10.5 by addition of 0.5 M NaOH until no more consumption of alkali was observed. The oxidation was quenched by adding ethanol (5 mL) and the pH was adjusted to 7 with 0.1 M HCl. After several centrifugal washing, the TEMPO-oxidized BC suspension was stored at 4 °C until use. The carboxylate content of the BC suspension measured by conductimetric titration (Saito and Isogai 2004) was 0.74 mmol/g cellulose.
EDC-mediated cross-linking
Chitosan solution (1%, w/v) was prepared by dissolving chitosan powder in 0.1 M HCl and filtering through eight layers of nylon cloth (mesh size −50 μm). A covalent coupling between surface carboxylate groups of TEMPO-oxidized BC suspension and amino groups of chitosan was carried out through EDC-mediated amide binding. Molar ratio of chitosan, EDC, and NHS was 1:1:1. Various weight ratios of TEMPO-oxidized BC suspension and chitosan solution (BC/CTS—from 20/80 to 60/40 w/w%) were designated for scaffold preparation (Table 1). Note that moles of CTS-NH2, EDC, and NHS are excess amount compared to moles of carboxylate groups available on BC microfibrils surface in all compositions.
The coupling reaction was performed at room temperature according to the method D of Araki et al. (Araki et al. 2002) with some modifications. Briefly, NHS was added into the TEMPO-oxidized cellulose suspension with stirring followed by slow addition of chitosan solution. The pH of the mixture was adjusted to 5.5–6.0 with 0.5 M NaOH and the mixture was continuously stirring for 30 min. The desired amount of EDC aqueous solution was then added drop-wise and the mixture was stirred overnight. The reaction mixture were subjected to dialysis (Seamless Cellulose Tubing with MWCO-14,000 and pore size- 50 Å from Viskase Co., Inc., Japan) to remove excess EDC, NHS, and possible byproduct for 3 days. The dialysis medium was exchanged with fresh water several times. The dialysis tubes were then immersed in aqueous poly(ethylene glycol) (MW-20,000) solution for 3–4 days to obtain concentrated BC/CTS slurry (6–7 mg/mL).
Bacterial cellulose/chitosan (BC/CTS) scaffold preparation
The BC/CTS scaffolds were fabricated by freezing and lyophilization (Madhihally and Matthew 1999). The BC/CTS slurries with various weight ratios were degassed under vacuum for 2 h to remove air bubbles created by the mixing process. The slurries were loaded into 48-well tissue culture plates (1 mL/well) or 60-mm polystyrene tissue culture dishes (10 mL/dish) (Iwaki- Asahi Techno Glass, Japan). All the culture plates and dishes were transferred into freezers at a preset temperature of −30 °C (FDU-2100 freeze dryer attached with DRC-1000 pre-freezing chamber, EYELA) and −80 °C (deep freezer) to solidify the solvent and induce solid–liquid phase separation. The solidified mixture was maintained at each freezing temperature for 24 h and subsequently sublimated under a vacuum (ca. 15 Pa) at 0 °C for 3 days.
Microstructural characterization
ATR-FTIR spectroscopy
The covalent coupling reaction of BC/CTS scaffolds with various weight ratios were determined by fourier transformed infrared spectroscopy (Spectrum 2000 FT-IR, PerkinElmer) equipped with an attenuated total reflection (single reflection diamond crystal ATR top plate) accessory. The spectra were collected over the range of 4,000–700 cm−1 with an accumulation of 32 scans and a resolution of 4 cm−1.
Field emission scanning electron microscopy (FE-SEM)
Freeze-dried cylindrical scaffolds were carefully sectioned at horizontal plane with a razor blade, mounted on the copper stubs with conductive adhesive tape, and sputter-coated with platinum (JEOL, JFC 1600 Auto fine coater). Field emission scanning electron microscopy (FE-SEM, JOEL, JSM-6700F), operating at accelerating voltage of 1.5 kV, was used to observe qualitative pore microstructure of the scaffolds with various weight ratios. Estimated pore sizes were determined by analysis of FE-SEM images using Adobe Photoshop (version 5.0.2).
Porosity
The porosities were calculated from the mass and the volume of each cylindrical scaffold. The weight was measured by an electronic balance. The volume (V) was determined by measuring scaffold’s dimensions with a digital caliper. The measurements were conducted on three to four replicates, with three times per each dimension. The density of solid nonporous polymers were determined by compacting freeze-dried fibrous MFC and CTS/HCl powder samples, respectively, at 2 ton to form solid discs of known dimensions using the KBr-disc apparatus. The weights as well as the volumes of the discs were determined after vacuum-dried at 40 °C for 1 day. The solid densities of BC-MFC and CTS/HCl are 1.14 and 1.11 g/cm3, respectively. Then we can calculate the polymer volume (V BC and V CTS) of the scaffolds with respective weight ratio. Taking into account that V = V polymer + V pore, a rough estimation of the samples porosity ϕ was then calculated for each sample using the equation;
Alternatively, the porosity of the BC/CTS scaffolds was also measured by liquid displacement (Hsu et al. 1997). Hsu et al. used water as the displacement liquid. However, absolute ethanol was used for our BC/CTS scaffolds because it is a nonsolvent for BC and CTS and is able to permeate easily through the scaffolds without swelling or shrinking. Briefly, a scaffold sample of weight W was immersed in a graduated cylinder containing a known volume (V 1) of ethanol. The sample was kept in the ethanol for approximately 5 min. In this time, an evacuation-repressurization cycle was repeated to force the ethanol through the pores of the scaffold. The total volume of ethanol and the ethanol-impregnated scaffold was recorded as V 2. The volume difference (V 2 − V 1) was the volume of the polymer skeleton of the scaffold. The ethanol-impregnated scaffold was removed from the cylinder and the residual ethanol volume was recorded as V 3. The void volume of the scaffold (V 1 − V 3) occupied by ethanol was then obtained. Thus, the total volume of the scaffold was V = (V 2 − V 1) + (V 1 − V 3).
The porosity of the scaffold (ϕ) was obtained by:
The value was reported as mean ± SD (n = 3).
In vitro degradation
The biodegradation study of the scaffolds was carried out by incubating the scaffolds in lysozyme containing phosphate buffer saline (PBS) solution (pH 7.4) at 37 °C. Egg-white lysozyme (Sigma–Aldrich) was used without further purification. The various weight ratios of BC/CTS scaffolds with known weight were immersed in 10 mg/L lysozyme solution. The PBS solution was renewed every week. At predetermined time interval, the scaffolds were taken out from the lysozyme solution, thoroughly rinsed with distilled water, freeze-dried, and weighed. The extent of the degradation was expressed as the percent weight loss of the scaffolds after treatment from three replicates as follow:
where W, W 0, and W t denote the percent weight loss after lysozyme treatment, initial weight, and weight at time t, respectively. The values were expressed as the means with standard deviation (n = 3).
Mechanical testing
Mechanical tests were conducted on a Thermomechanical Analyzer (TMA/SS6100, S II Nanotechnology Inc., Japan) under compression mode. Freeze-dried cylindrical scaffolds measuring 9.3 mm in diameter and ranging between 9.3 and 10.3 mm in height were tested under compression in the direction parallel to the cylinder axis from 1 mN to 980 mN at a rate of 98 mN/min. The compression force was applied using a probe (3 mm diameter) acting on the central area of the test specimen, which was setting between two quartz crystal parallel plates. The compression tests were performed on dry (at ambient temperature) and hydrated (in PBS, at 37 °C) BC/CTS scaffolds. Scaffolds were hydrated in PBS (pH = 7.4, at 37 °C) for 3 h prior to testing and were kept immersed in PBS throughout the test. Compressive stress-strain curves were plotted and compressive elastic modulus (E*) and compressive strength (σ*) were determined for all scaffolds fabricated at both freezing temperatures. The initial cross sectional area was used for the calculation whenever it was necessary. Mechanical compression data are described as an average of three to four test specimens with standard deviation.
Results and discussion
Because of the asymmetric network nature with a variable small void volume of native BC although it possesses native mechanical properties, BC scaffold with more-uniform porous structure and larger-pore size is intended to develop in favor of tissue engineering application in this study.
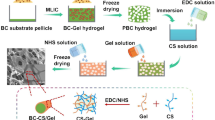
First, never-dried BC pellicles were fibrillated and surface modified by TEMPO-mediated oxidation. Then, BC/CTS scaffolds of various weight ratios were fabricated by freezing (at −30 and −80 °C) and lyophilization after EDC-mediated amide bond formation between surface carboxyl functional groups of BC nanofibrils and amino groups of chitosan (Fig. 1). It was found that all the BC/CTS scaffolds maintained the original cylindrical shape after autoclave (at 121 °C and 15 psi for 15 min), whereas chitosan (non-cross-linked) and pure BC (TEMPO-oxidized MFC) scaffolds showed deformation of structural integrity (Fig. 2). This observation indicates the occurrence of amide bond between BC and CTS where BC nanofibrils are held within chitosan matrix. In this study, hydrochloric acid (0.1 M HCl) was used for chitosan dissolution instead of acetic acid (HAc) to avoid extra source of carboxyl groups other than those from BC. Chitosan scaffold (CTS/HCl) prepared from HCl dissolution was weak in its structural integrity than that (CTS/HAc) from HAc dissolution. The CTS/HCl scaffold became shrunken and completely dissolved in water while the CTS/HAc scaffold maintained its shape until after autoclave sterilization. As a result, the BC/CTS(HCl) scaffolds without EDC/NHS showed deformation and dissociation of BC nanofibrils after autoclave sterilization as shown in Fig. 2b. Therefore, the structure-property relationship of polysaccharide-based scaffolds emphasized in present study is scaffolds prepared from CTS/HCl with EDC/NHS cross-linking (Fig. 2c).

a Schematic representation of bacterial cellulose/chitosan scaffold preparation. b Never-dried BC pellicle and FE-SEM image of BC nanobrillar network (left) and photograph of BC/CTS scaffolds (right)

Structural integrity of a CTS scaffolds, b BC/CTS scaffolds without EDC/NHS, c with EDC/NHS cross-linking (scaffolds are prepared at −30 °C). CTS/HCl and CTS/HAc denote the chitosan scaffold prepared from dissolution of chitosan in hydrochloric acid and acetic acid. Both are non-cross-linked scaffolds. (1) freeze-dried chitosan scaffolds, BC/CTS scaffolds with various MFC content, and BC scaffold, (2) soaking in water before autoclave, (3) after autoclave (at 121 °C and 15psi for 15 min)
EDC-mediated amide bond formation
ATR-FTIR spectra of freeze-dried scaffolds (BC/CTS −30 °C) with various weight ratios are shown in Fig. 3a. This technique is a powerful tool for detecting amide bonds on solid surfaces as well as in aqueous systems (Li et al. 2006; Parikh and Chorover 2006; Vigano et al. 2000). The characteristic absorption bands of chitosan and TEMPO-oxidized BC are shown at the top and the bottom. The peaks at 1,652 and 1,588 cm−1 correspond to the amide absorption band (amide I) and the NH2 scissoring mode of the primary amine of chitosan powder spectrum (top), whereas the carboxylate absorption band of TEMPO-oxidized BC nanofibrils was observed at about 1,600 cm−1 (bottom). The spectra of BC/CTS composite scaffolds in the middle showed combination of the spectral feature of BC and chitosan.

a ATR-FTIR spectra of BC/CTS porous scaffolds fabricated at freezing temperature of −30 °C. b Enlargement of 1,800–1,500 cm−1 region in (a)
In all spectra of BC/CTS scaffolds, the NH2 scissoring band of chitosan at 1,588 cm−1 diminished and a new sharp peak at 1,560 cm−1 (amide II) was established through EDC-mediated amide bond formation with surface carboxyl groups of BC nanofibrils (Fig. 3b). Moreover, lower frequency shift of amide I band initially present in chitosan spectrum and a sharp increase of amide III (~1,242 cm−1) band further confirmed the amide bond formation. Cross-linking using EDC involves the activation of carboxylic acid groups to give amine reactive intermediate O-acylisourea groups, which form cross-links after reaction with free amine groups. The crosslinking reaction results in formation of water-soluble urea as by-product. The extent of amide bond formation depends on the weight ratios of the BC and CTS. The reappearance of carboxylate peak at about 1,600 cm−1 in BC/CTS 50/50 (w/w%) indicated that the most available amide bond formation occurred below 50 wt% MFC.
Considering from moles value that shown in Table 1, all carboxyl groups have been involved in amide bond formation because the available surface carboxyl groups of BC are less than amino groups of chitosan in all compositions. A possible explanation of the reappearance of carboxylate peak in BC/CTS—50/50 and 60/40 is that the formation of additional cross-links is prevented owing to spatial limitations in this heterogeneous mixture with decreasing chitosan concentration.
Pore microstructure
Porous BC/CTS scaffolds can be successfully fabricated as variable sizes (Fig. 1b) by freezing and lyophilization method after chemical modification. The macromolecular morphology could be influenced by many parameters and the resulting scaffold pore microstructure is highly related with cell proliferation and ECM production in chondrocyte and osteoblast cell culture. Several studies have shown this point using various scaffold materials (Bhardwaj et al. 2001; Li et al. 2005; Malda et al. 2005; Yamane et al. 2007).
The effect of freezing temperature on mean pore size and the effect of fiber/matrix (BC/CTS) ratio on pore microstructure were investigated in the present study. FE-SEM images of BC/CTS scaffolds with various BC/CTS weight ratios prepared at two freezing temperatures are shown in Fig. 4. In general, the freezing and lyophilization process generated an open pore microstructure with a high degree of interconnecting pores in composite scaffolds. However, a few interconnecting pores were observed in pure BC scaffold. In particular, the composite scaffolds fabricated at −30 °C showed well-defined open pore with homogeneous size distribution, whereas presence of ill-defined pore structure and pore size distribution with increasing MFC content were observed in scaffolds prepared at −80 °C. They, however, can still keep their structural integrity for further analysis.

FE-SEM images of BC/CTS porous scaffolds with various weight ratios fabricated at −30 and −80 °C (scale bar = 100 μm)
FE-SEM images of pore wall surface at higher magnification are shown in Fig. 5. Figure 5a is a pore wall morphology of 20 wt% MFC content BC/CTS scaffold, where BC nanofibrils are well integrated within chitosan matrix. With increasing MFC content (60 wt% MFC), a surface-exposed network of BC fibrils oriented random-in-the-plane was observed as shown in Fig. 5b. The BC fibrils are exposed to the surface because of the concurrent decreasing chitosan concentration. No visible clumps of MFC aggregates further confirmed the uniform distribution. The fibrils are held two or three together rather than a single fibril and the mean width of these bundles are in the range of 40–100 nm with several micrometers in length. It is important that MFC used for the reinforcement should be well-distributed in the chitosan matrix to avoid the non-desirable stress transfer in the scaffolds.

FE-SEM images of the pore wall microstructure of BC/CTS scaffolds with a lower (20 wt%) and b higher (60 wt%) MFC content (scaffolds are prepared at −30 °C, scale bar = 100 nm)
The pore size distribution and porosity of the composite scaffolds are shown in Fig. 6 and Table 2. The mean pore diameter of pure chitosan (CTS/HCl) scaffolds prepared at −30 and −80 °C were 152 ± 34 and 101 ± 21 μm, respectively, whereas those of pure BC scaffolds were 296 ± 53 and 192 ± 28 μm, respectively. Pore size decreased with lowering the freezing temperature from −30 to −80 °C, normally observed in scaffolds prepared from freeze-drying and lyophilization process. This effect was more significant in pure BC scaffold than chitosan scaffold because of lower initial concentration of BC (6 mg/mL) than chitosan (10 mg/mL) as well as the viscosity difference between chitosan solution and water suspension of BC nanofibrils. During freezing process, the water will form ice crystals and separate from the solutes system, and the solutes will be confined to the interstitial regions between ice crystals (Jennings 1999). Larger ice crystal formation is expected in less viscous BC water suspension because the foam structure produced by freeze-drying and lyophilization method is directly related to the size and distribution of ice crystals in the frozen state.

Mean pore size of BC/CTS porous scaffolds at respective freezing temperature
However, decreased in pore size with lowering the freezing temperature is not straight forward to explain the mean pore size of the composite scaffolds because BC/CTS scaffolds were subjected to freeze-drying and lyophilization process after EDC-mediated cross-linking, which was different from most of the literatures reported. The covalent amide bond formation between BC nanofibrils and chitosan in BC/CTS mixture further confined the ice crystals formation during freezing process compared with non-cross-link condition in this heterogeneous system. As a result, the mean pore diameter of the BC/CTS scaffolds with MFC content between 30 and <50 wt% showed similar pore size at both freezing temperatures (Fig. 6) because the most available amide bond formation occurred at this range (Fig. 3) as described in previous section. The significant effect of freezing temperature on pore size was thus observed in lower and higher MFC content (BC/CTS-20/80 and BC/CTS-60/40) (Figs. 4 and 6), where the mean pore size decreased from 198 ± 26 to 121 ± 17 μm in BC/CTS-20/80 and from 284 ± 32 to 219 ± 29 μm in BC/CTS-60/40.
In terms of the effect of fiber content, the mean pores size increased with increasing MFC content (Svagan et al. 2008) in general. But a less effect was observed in the middle range of MFC content (between 30 and <50 wt%) in both freezing temperatures, the same reason as described above. The extent of chemical cross-linking before scaffold fabrication process has profound effect on the pore size rather than freezing temperature and MFC content in this range. The pore size, shape, and porosity of the scaffolds are important factors that affect cellular activity and tissue regeneration, that is, sufficient porosity facilitates diffusion of nutrients and clearance of wastes; adequate pore size influences growth behavior of particular tissue cells. Obviously, the composite scaffolds possess suitable pore size with interconnecting pores for tissue engineering application. In addition, the apparent porosity (Table 2) of the composite scaffolds was also high enough (>80%) for usage in tissue engineering.
In vitro degradability
The lysozyme, an enzyme with anti-bacterial action that is found in body fluids, saliva, sweat and tears as well as a widespread distribution in animals and plants (Klass et al. 1977; Kuettner et al. 1974; Mackie and Seal 1976; Powning and Davidson 1979). Naturally occurring biopolymer, chitin/chitosan could be degraded with lysozyme and the specificity of enzymatic hydrolysis towards the sequence of its N-acetylglucosamine unit has already been elucidated (Stokke et al. 1995).
As chitosan is one of the components of the BC/CTS scaffolds, the biodegradability of the BC/CTS −30 °C scaffolds were carried out in lysozyme containing PBS solution by incubating at 37 °C for 6 weeks. In order to know the homogeneity of both biopolymers through out the cylindrical scaffold, each scaffold was cut into two parts (upper 1/5 or air side and lower 4/5 or bottom side) and the percent weight loss of the scaffolds as a function of MFC content was determined. In general, the weight loss decreased with increasing MFC content (or decreasing CTS content) after 6 weeks as shown in Fig. 7. Noticeably, ca 1.7 times increased weight loss was observed in upper part of BC/CTS-20/80 and 30/70 scaffolds. This result indicated that the localized distribution of chitosan domain become increased in upper part than lower part of the scaffold during loading and freezing process particularly in lower MFC content. The reliable weight loss of pure BC scaffold could not be obtained at the end of incubation because the dissociation of BC nanofibrils occurred during incubation period of 4 weeks. Although porous CTS/HCl scaffold without crosslink can be prepared, it became soft and dissolved partially within 1 day once the scaffold was immersed into the enzyme containing PBS solution. As a consequence, it was difficult to make comparison among scaffolds made from BC only, CTS/HCl only, and BC/CTS composite.

Percent weight loss of BC/CTS scaffolds 6 weeks after incubation in lysozme solution as a function of MFC content (scaffolds are prepared at −30 °C)
The amount of reducing end groups formation by enzymatic degradation was also determined by Schales’ method using ferricyanide (Kurita et al. 1993). The method includes reading the absorbance value at 420 nm to detect reducing groups by enzymatic hydrolysis at predetermined time points. The differences in absorbance, ∆Abs, corresponding to the amounts of the reducing end groups were determined as a function of the enzyme contact time. The BC/CTS scaffolds of 20/80, 40/60, and 60/40 from the weight loss experiment were determined by this method. The scaffolds degraded much more quickly during the first 2 weeks. The degradation rate slowed down in later time period of until 6 weeks (data not shown).
Although there was a considerable amount of weight reduction with lysozyme treatment, the well-integrated BC nanofibrils stabilized the initial shape with a small size reduction after freeze-drying. The freeze-dried composite scaffolds were subjected to FE-SEM observation to determine the microstructure after lysozyme treatment (Fig. 8). There was a significant changes in the porous structure as well as the pore wall in lower MFC content (BC/CTS—20/80), where the smooth pore wall (Fig. 5a) became rough surface with exposed BC nanofibrils (Fig. 8c) after lysozyme treatment. However, scaffold of higher MFC content (BC/CTS—60/40) showed comparable microstructure with scaffold before lysozyme treatment (Figs. 5b and 8d).

FE-SEM images of BC/CTS porous scaffolds 6 weeks after incubation in lysozyme solution a pore and c pore wall microstructure of scaffold with lower MFC content (20 wt%) b pore and d pore wall microstructure of scaffold with higher MFC content (60 wt%) Scale bar = 100 μm (a and b), 100 nm (c and d)
Mechanical properties
Compressive mechanical properties are particularly important for scaffold used in tissue engineering because they are closely associated with the shape-persistence and durability in practical operations and applications, where the scaffolds are used to contact with culture medium in vitro or body fluid in vivo studies.
The compressive properties of cylindrical scaffolds with various MFC contents fabricated at two freezing temperatures were performed under dry and hydrated conditions. Stress-strain curves derived from the testing data of BC/CTS −30 °C group under dry condition are plotted in Fig. 9. The compressive stress-strain curves of the scaffolds were typical of cellular solids (Gibson and Ashby 1997), showing the three regimes of linear elasticity, collapse plateau and densification. The compressive modulus (E*) was measured from the slope of the linear elastic regime. The compressive strength (σ*) was determined from the intersection point of two regression lines drawn from linear elastic regime and collapse regime. The compressive modulus (E*) and compressive strength (σ*) calculated from stress-strain curves of all the composite scaffolds are listed in Table 3. In terms of testing condition, a dramatic decrease in the mechanical properties was observed in the hydrated state. The effect of water absorption, which was noticeable in the elastic region of the stress-strain curve (figure not shown) of the hydrated scaffolds, contributed to the lower elastic modulus value.

Compression stress-strain curves of BC/CTS scaffolds tested under the dry condition (scaffolds are prepared at −30 °C)
Effect of freezing temperature and mean pore size: As described earlier, the significant effect of freezing temperature on mean pore size was observed in scaffolds of lower MFC (20 wt%) content. A considerable decrease in pore diameter (121 ± 17 μm) was observed in BC/CTS- 20/80 scaffold freezing at −80 °C. As a result, increased value of E* and σ* were obtained in comparison with the same weight ratio of composite scaffold freezing at −30 °C (pore diameter-198 ± 26 μm) tested under the dry condition. However, an independent effect of the porosity and pore diameter on compressive modulus was observed in the hydrated condition because of the influence of hydrophilicity of higher chitosan (80 wt%) content. The similar mean pore size observed in scaffolds of MFC content between 30 and <50 wt% displayed similar E* and σ* value in the dry state.
Generally, E* and σ* value of scaffolds prepared at freezing temperature of −30 °C were higher than those of −80 °C in the hydrated state. The decrease E* and σ* values in scaffolds prepared at −80 °C was contributed from structural non-uniformities of ill-defined pore microstructure (Fig. 4) because the mechanical properties of porous scaffolds depend on the pore wall properties as well as the uniformity of pore microstructure (Harley et al. 2007; Svagan et al. 2008).
Effect of MFC content: Since the mechanical response of a porous scaffold is also determined by the properties of the material forming the walls between pores, the distribution and integration of BC nanofibrils play an important role in this respect. In general, the compressive modulus and strength increased with an increase in MFC content of composite scaffolds prepared at freezing temperature of −30 °C in both dry and hydrated states except similar E* value in two data points: (1) between 30 and 40 wt% in the dry state; (2) between 40 and 50 wt% in the hydrated state (Fig. 10, Table 3). The compressive modulus as well as compressive strength improved about twofold in scaffold integrated with 60 wt% MFC in comparison to those of scaffold integrated with 30 wt% MFC. The improved scaffold mechanical properties was resulting from closer packing of BC nanofibrils network with increased MFC content as well as an integration of network with chitosan matrix by EDC-mediated cross-linking. Because of the structural non-uniformities in scaffolds prepared at −80 °C, expected higher mechanical properties resulting from decreased mean pore size was unsuccessful. This may be due to either of following facts or combination: improper freezing temperature for present heterogeneous system; the influence of the cross-linking step before freezing process; the low volume fraction of the solid content; using preset freezing temperature rather than controlling the freezing rate.

Compressive modulus of BC/CTS scaffolds as a function of MFC content; a in the dry state, b in the hydrated state
It was interesting to note that no significant difference in mechanical properties (E*, σ*) was found between scaffold of pure MFC and composite scaffold of 40 wt% MFC in the dry state. In the hydrated state, similar compressive modulus was observed in pure MFC scaffold and composite scaffold of 20 wt% MFC. The result indicates the persistence of native mechanical property of BC nanofibrils although the BC pellicles are disintegrated and transformed into highly porous scaffolds. Since, the compressive modulus of never-dried native BC pellicle (Svensson et al. 2005) was ca.10 and ca.60 kPa for 0.3 and 3 mm thickness, respectively, the value of ca. 6 kPa of our highly porous pure BC scaffold in the hydrated condition was a reasonable result.
The compressive modulus of hydrated scaffolds BC/CTS −30 °C group (6.33 ± 1.5 to 33.22 ± 16 kPa) was higher than those of hydrated collagen-glycosaminoglycan scaffolds (208 ± 41 Pa, average mean pore size of 96–151 μm) (Harley et al. 2007) and hydrated cross-linked type II collagen scaffolds (0.88 ± 0.38 to 1.45 ± 0.84 kPa) (Pfeiffer et al. 2008). However, the value was lower than compressive modulus of human and animal cartilage specimens tested, which was varying between 200 and 1,200 kPa (Athanasiou et al. 1991; Eisenberg and Grodzinsky 1985; Jurvelin et al. 2003; Katakai et al. 2009; Mow et al. 1980). Since the compressive modulus is a material property that is strongly dependent on solid content, it is possible to improve solid content by optimizing the fabrication process and thus the compressive modulus applicable for tissue engineering of cartilage.
Conclusions
Bacterial cellulose/chitosan composite scaffolds of open pore microstructure with interconnecting pores were successfully fabricated through freezing and lyophilization method. The mean pore diameter of the composite scaffolds were in the range of 198 ± 26–284 ± 32 and 121 ± 17–219 ± 29 μm, respectively, for BC/CTS −30 °C group and BC/CTS −80 °C group. Because of the ill-defined and non-uniformities of the pore microstructure, BC/CTS −80 °C group showed lower mechanical properties than those of BC/CTS −30 °C group except 20 wt% MFC content. The compressive modulus of BC/CTS −30 °C group performed in the dry and the hydrated states displayed similar trend, that is, the mechanical properties was improved with increasing MFC content. The observed improvement in the scaffold rigidity was contributing from closer packing of BC nanofibrils as well as integration of BC nanofibrils web within chitosan matrix forming the pore walls, which was achieved by EDC-mediated covalent cross-linking. Because of using an excess amount of cross-linking agent in all compositions of the scaffolds as well as the variable weight ratios of BC and chitosan, the effect of cross-linking density on mechanical properties could not be evaluated. Further investigation is necessary to perform by selecting one or two weight ratios of BC/CTS with specific mole ratios of EDC/NHS in this regard. Moreover, the effect of cross-linking step, that is, at the slurry state before freeze-drying and after scaffold fabrication, on mechanical properties also needs to evaluate for this scaffold.
Alternatively, aqueous acetic acid (HAc) is used for chitosan dissolution in place of aqueous HCl and the dehydrothermal cross-linking method can be used in stead of EDC/NHS. Because ionic complex of chitosan with acetic acid undergoes a transformation to chitin, an amidized homologue of chitosan, at elevated temperature in solid state (Toffey et al. 1996), the combination of CTS/HAc and dehydrothermal cross-linking will be another way of processing method to modify the BC/CTS scaffold’s stiffness.
The polysaccharide based porous scaffold described in this study provides information for further development and optimization of a variety of nanofiber-polymer matrix porous scaffolds for potential applications as tissue scaffolds and regeneration aids.
References
Araki J, Kuga S, Magoshi J (2002) Influence of reagent addition on carbondiimide-mediated amidation for poly(ethylene glycol) grafting. J App Poly Sci 85:1349–1352
Athanasiou KA, Rosenwasser MP, Buckwalter JA, Malinin TI, Mow VC (1991) Interspecies comparisons of in situ intrinsic mechanical properties of distal femoral cartilage. J Orthop Res 9:330–340
Bhardwaj T, Pilliar RM, Grynpas MD, Kandel RA (2001) Effect of material geometry on cartilaginous tissue formation in vitro. J Biomed Mater Res 57:190–199
Czaja W, Krystynowicz A, Bielecki S, Brown RM Jr (2006) Microbial cellulose- the natural power to heal wounds. Biomaterials 27:145–151
Di Martino A, Sittinger M, Risbud MV (2005) Chitosan: a versatile polymer for orthopaedic tissue-engineering (review). Biomaterials 26:5983–5990
Eisenberg SR, Grodzinsky AJ (1985) Swelling of articular cartilage and other connective tissue: electromechanochemical forces. J Orthop Res 3:148–159
Freyman TM, Yannas IV, Yokoo R, Gibson LJ (2001) Fibroblast contraction of a collagen-gag matrix. Biomaterials 22:2889–2891
Gibson LJ, Ashby MF (1997) Cellular solids: structure and properties, 2nd edn. Cambridge University Press, Cambridge
Harley BA, Leung JH, Silva ECCM, Gibson LJ (2007) Mechanical characterization of collagen-glycosaminoglycan scaffolds. Acta Biomater 3:463–474
Helenius G, Bäckdahl H, Bodin A, Nannmark U, Gatenholm P, Risberg B (2006) In vivo biocompatibility of bacterial cellulose. J Biomed Mater Res 76A:431–438
Hestrin S, Schramm M (1954) Synthesis of cellulose by Acetobacter xylinum. 2. Preparation of freeze drized cells capable of polymerizing glucose to cellulose. Biochem J 58:345–352
Hsu YY, Gresser JD, Trantolo DJ, Lyons CM, Gangadharam PRJ, Wise DL (1997) Effect of polymer foam morphology and density on kinetics of in vitro controlled release of isoniazid from compressed foam matrices. J Biomed Mater Res 35:107–116
Ikada Y (2006) Tissue engineering: fundamental and applications, interface science and technology. In: Hubbard A (ed) vol 8 Academic Press, Elsevier Ltd, Oxford
Jennings TA (1999) Lyophilization: introduction and basic principle. CRC, Boca Raton
Jonas R, Farah LF (1998) Production and application of microbial cellulose. Polym Degrad Stab 59:101–106
Jurvelin JS, Buschmann MD, Hunziker EB (2003) Mechanical anisotropy of the human knee articular cartilage in compression. Proc Inst Mech Eng [H] 217:215–219
Katakai D, Imura M, Ando W, Tateishi K, Yoshikawa H, Nakamura N, Fujie H (2009) Compressive properties of cartilage-like tissues repaired in vivo with scaffold-free, tissue engineered constructs. Clinic Biomech 24:110–116
Klass HJ, Hopkins J, Neale G, Peter TJ (1977) The estimation of serum lysozyme: a comparison of four assay methods. Biochem Med 18:52–57
Klemm D, Schumann D, Udhardt U, Marsch S (2001) Bacterial synthesized cellulose-artificial blood vessels for microsurgery. Prog Polym Sci 26:1561–1603
Kosher RA, Lash JW, Minor RR (1973) Environmental enhancement of in vitro chondrogenesis: stimulation of in vitro somite chondrogenesis by exogenous chondromucoprotein. Dev Biol 35:210–220
Kuettner KE, Sorgente N, Croxen RL, Howell DS, Pita JC (1974) Lysozyme in preosseous cartilage. VII. Evidence for physiological role of lysozyme in normal endochondral calcification. Biochim Biophys Acta 372:335–344
Kurita K, Yoshino H, Nishimura SI, Ishii S (1993) Preparation and biodegradability of chitin derivatives having mercapto groups. Carbohydrate Poly 20:239–245
Lahiji A, Sohrabi A, Hungerford DS, Frondoza CG (2000) Chitosan supports the expression of extracellular matrix proteins in human osteoblasts and chondrocytes. J Biomed Mater Res 51:586–595
Li Z, Ramay HR, Hauch KD, Xiao D, Zhang M (2005) Chitosan-alginate hybrid scaffolds for bone tissue engineering. Biomaterials 26:3919–3928
Li J, Yun H, Gong Y, Zhao N, Zhang X (2006) Investigation of MC3T3–E1 cell behavior on the surface of GRGDS-coupled chitosan. Biomacromolecules 7:1112–1123
Mackie IA, Seal DV (1976) Quantitative tear lysozyme assay in units of activity per microlitre. Brit J Ophthal 60:70–74
Madhihally SV, Matthew HWT (1999) Porous chitosan scaffolds for tissue engineering. Biomaterials 20:1133–1142
Malda J, Woodfield TB, van der Vloodt F, Wilson C, Martens DE, Tramper J, van Blitterswijk CA, Riesle J (2005) The effect of PEGT/PBT scaffold architecture on the composition of tissue engineering cartilage. Biomaterials 26:63–72
Mao JS, Liu HF, Yin YJ, Yao KD (2003) The properties of chitosan-gelatin membranes and scaffolds modified with hyaluronic acid by different methods. Biomaterials 24:1621–1629
Mow VC, Kuei SC, Lai WM, Amstrong CG (1980) Biphasic creep and stress relation of articular cartilage in compression: theory and experiments. J Biomech Eng 102:73–84
Nehrer S, Breinan HA, Ramappa A, Young G, Shortkroff S, Louie LK, Sledge CB, Yannas IV, Spector M (1997) Matrix collagen type and pore size influence behavior of seeded canine chondrocytes. Biomaterials 18:769–776
Nge TT, Sugiyama J (2007) Surface functional group dependent apatite formation on bacterial cellulose microfibrils network in a simulated body fluid. J Biomed Mater Res 81A:124–134
O’Brien FJ, Harley BA, Yannas IV, Gibson LJ (2005) The effect of pore size on cell adhesion in collagen-gag scaffolds. Biomaterials 26:433–441
Olde Damink LHH, Dijkstra PJ, van Luyn MJA, van Wachem PB, Nieuwenhuis P, Feijen J (1996) Cross-linking of dermal sheep collagen using water-soluble carbodiimide. Biomaterials 17:765–773
Parikh SJ, Chorover J (2006) ATR-FTIR spectroscopy reveals bond formation during bacterial adhesion to iron oxide. Langmuir 22:8492–8500
Pfeiffer E, Vickers SM, Frank E, Grodzinsky AJ, Spector M (2008) The effects of glycosaminoglycan content on the compressive modulus of cartilage engineered in type II collagen scaffolds. Osteoarthritis Cartilage 16:1237–1244
Powning RF, Davidson WJ (1979) Studies on insect bacteriolytic enzymes III. Lytic activities in some plant materials of possible benefit to insects. Comp Biochem Physiol 63B:199–206
Roberts GAF (1992) Chitin chemistry. Macmillan, London
Saito T, Isogai A (2004) TEMPO-mediated oxidation of native cellulose. The effect of oxidation conditions on chemical and crystal structures of the water-insoluble fractions. Biomacromolecules 5:1983–1989
Schulz-Torres D, Freyman TM, Yannas IV, Spector M (2000) Tendon cell contraction of collagen-gag matrices in vitro: effect of cross-linking. Biomaterials 21:1607–1619
Stokke BT, Vårum KM, Holme HK, Hjerde RJN, Smidsrød O (1995) Sequence specificities for lysozyme depolymerization of partially N-acetylated chitosan. Cana J Chem 73:1972
Suh JKF, Matthew HWT (2000) Application of chitosan-based polysaccharide biomaterials in cartilage tissue engineering (review). Biomaterials 21:2589–2598
Svagan AJ, Samir MASA, Berglund LA (2008) Biomimetic foams of high mechanical performance based on nanostructured cell walls reinforced by native cellulose nanofibrils. Adv Mater 20:1263–1269
Svensson A, Nicklasson E, Harrah T, Panilaitis B, Kaplan DL, Brittberg M, Gatenholm P (2005) Bacterial cellulose as a potential scaffold for tissue engineering of cartilage. Biomaterials 26:419–431
Toffey A, Samaranayake G, Frazier CE, Glasser WG (1996) Chitin derivatives. I. Kinetics of the heat-induced conversion of chitosan to chitin. J Appl Polym Sci 60:75–85
Vigano C, Manciu L, Buyse F, Goormaghtigh E, Ruysschaert JM (2000) Attenuated total reflection IR spectroscopy as a tool to investigate the structure, orientation and tertiary structure changes in peptides and membrane proteins. Biopolymers (Pept Sci) 55:373–380
Yamanaka S, Watanabe K, Kitamura N, Iguchi M, Mitsuhashi S, Nishi Y, Uryu M (1989) The structure and mechanical properties of sheet prepared from bacterial cellulose. J Mater Sci 24:3141–3145
Yamane S, Iwasaki N, Kasahara Y, Harada K, Majima T, Monde K, Nishimura SI, Minami A (2007) Effect of pore size on in vitro cartilage formation using chitosan-based hyaluronic acid hybrid polymer fibers. J Biomed Mater Res 81A:586–593
Zeltinger J, Sherwood JK, Graham DA, Muller R, Griffith LG (2001) Effect of pore size and void fraction on cellular adhesion, proliferation, and matrix deposition. Tissue Eng 7:557–572
Acknowledgments
This research was supported by Japan Society for Promotion of Science (JSPS) under the Grant-in-Aid for Scientific Research (Grant number16004160) and RISH-Mission project (2007 fiscal year), Center for Exploratory Research on Humanosphere, Research Institute for Sustainable Humanosphere, Kyoto University.
The authors would like to acknowledge Prof. Yamanaka from Shinshu University for a gift of Acetobacter strain used in this study, and Prof. Isogai and Dr. Saito from The University of Tokyo for their guidance to conduct conductimetric titration.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nge, T.T., Nogi, M., Yano, H. et al. Microstructure and mechanical properties of bacterial cellulose/chitosan porous scaffold. Cellulose 17, 349–363 (2010). https://doi.org/10.1007/s10570-009-9394-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-009-9394-x