
Abstract
Recombinant human erythropoietin (EPO), a glycohormone, is one of the leading biopharmaceutical products, while carbamylated erythropoietin (CEPO), an EPO derivative, is attracting widespread interest due to its neuroprotective effects without erythropoiesis in several cells and animal models. However, exogenous EPO promotes an angiogenic response from tumor cells and is associated with tumor growth, but knowledge of CEPO on tumor growth is lacking. Here we show that CEPO, but not EPO, inhibited Neuro-2a growth and viability. As expected, CEPO—unlike EPO—did not activate JAK-2 either in primary neurons or in Neuro-2a cells. Interestingly, CEPO did not induce GDNF expression and subsequent AKT activation in Neuro-2a cells. Before CEPO/EPO treatment, glial cell line-derived neurotrophic factor (GDNF) neutralization and GFR receptor blocking decreased the viability of EPO-treated Neuro-2a cells but did not influence CEPO-treated Neuro-2a cells. As compared to primary neurons, the expression of CD131, as a receptor complex binding to CEPO, is almost lacking in Neuro-2a cells. In BABL/C-nu mice, CEPO did not promote the growth of Neuro-2a cells nor extended the survival time compared to mice treated with EPO. The results indicate that CEPO did not promote tumor growth because of lower expression of CD131 and subsequent dysfunction of CD131/GDNF/AKT pathway in Neuro-2a cells, revealing its therapeutic potential in future clinical application.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although erythropoietin (EPO) treatment is widely accepted as safe for anaemic patients, and data accumulated during the last decade indicate its neuroprotective role, some concerns have been raised regarding the safety of EPO treatment in non-anaemic patients. EPO promotes the proliferation and survival of cancer cells expressing EPO receptor (EPOR) in various human tumors (Hedley et al. 2011). Thereby, it is postulated that EPO–EPOR plays a significant role in tumor growth, infiltration, and metastasis as well as treatment resistance (Wang et al. 2007). The down-regulation of EPOR expression on glioma cells reduces tumor growth and enhances animal survival (Kumar et al. 2012; Peres et al. 2011). On the other hand, constitutive activation of EPOR promotes cell proliferation in melanoma cells, increases cell proliferation and resistance to cisplatin treatment and results in tumor invasion of surrounding tissue in vivo (Peres et al. 2011). Meta-analysis of 53 trials of erythropoiesis-stimulating agents in 13,933 cancer patients revealed increased mortality during active therapy periods and worsened overall survival (Bohlius et al. 2009). However, it is also reported that EPO and EPOR were expressed in high-risk neuroblastomas (NB) but did not modify tumor cell proliferation, suggesting that EPO may be used safely in children with NB (Sartelet et al. 2007).
Carbamylation of lysine residues in EPO results in a derivative, carbamylated EPO (CEPO), which does not increase haematocrit and does not bind the haemopoietic EPO-R (Leist et al. 2004). Several studies reported that CEPO protects the brain from cerebral ischemia (Leist et al. 2004), hypoxia (Fantacci et al. 2006) and traumatic injury (Mahmood et al. 2007), spinal cord injury (King et al. 2007), diabetic peripheral nerve damage (Leist et al. 2004), and motor neuron degeneration (Mennini et al. 2006). CEPO promoted neurite outgrowth and neuronal spine formation in neurons (Choi et al. 2014). The beneficial effect of CEPO on spatial learning in rats subjected to traumatic brain injury is similar to that of EPO. Despite the 2009 clinical trial which showed that treatment with EPO was ineffective in stroke (Ehrenreich et al. 2009), CEPO is currently entering into a safety, tolerability, and pharmacokinetic clinical trial for the treatment of acute ischemic stroke and Friedreich’s ataxia (Leconte et al. 2011; Boesch et al. 2014).
Although the neuroprotective role of CEPO has received much attention, its role on tumor cells still lacks adequate understanding. In this study, we examined the possible risk of CEPO in tumor growth and explored its molecular pathway.
Materials and methods
Ethics statement
All experiments and animal care were approved by the Medical Ethics Committee of Huashan Hospital, Fudan University, Shanghai, China. Animals were housed under pathogen-free conditions, fed food and water ad libitum, and maintained on a reversed 12:12 (h) light/dark cycle for 1 week prior to experimental manipulation.
The preparation and identification of CEPO
CEPO was prepared from rhEPO (Chemo Wanbang Biopharma Co. Ltd., Shanghai) as described earlier (Leist et al. 2004). Briefly, one volume of EPO (1 mg/ml) was mixed with one volume of 1 M Na-borate (pH = 8.8), and recrystallized KOCN was added to a final concentration of 1 M. The mixture was incubated at 37 °C for 24 h. Samples were immediately dialysed against milli-Q water and subsequently against 20 mM sodium citrate in 0.1 M NaCl, pH = 6.0. After dialysis, the samples were concentrated by Centricon (Amicon). The content was determined by bicinchoninic acid Protein Assay Kit (Beyotime, Shanghai). The purity was verified, and the product of CEPO digested with endoproteinase Lys-C was identified by SDS-PAGE with low molecular weight staining.
CEPO and EPO (200 μg) were dried under vacuum and dissolved in 200 μl 6 M guanidinium-HCl, 250 mM Tris, pH = 9.5. Twenty-five microlitres of 0.1 M dithioerytritol was added, and incubation was continued in the dark at 37 °C. After 30 min, 25 μl of iodoacetamide (0.6 M) was added, and incubation was continued for 60 min at RT. Finally, the sample was desalted on a 5-ml HiTrap G25 column (Amersham-Biosciences, Little Chalfont, UK) into 50 mM NH4HCO3, 0.4 M urea, pH = 8.3. Ten micrograms of EPO or CEPO was mixed with 0.4 μg of Lys-C proteinase at 37 °C for 20 h. Because carbamylation targets lysine residues on rHuEPo, the product of CEPO digested with endoproteinase Lys-C was identified by SDS-PAGE and visualized by low molecular weight staining (Fig. 1).

The identification and erythropoiesis of CEPO. CEPO was prepared from rhEPO (Chemo Wanbang Biopharma Co. Ltd., Shanghai) as described earlier [12]. a After Lys-C digestion, EPO was degraded into about 16-kDa molecules. For CEPO, no degradation peptides were detectable (about 34 kDa). Lane 1 low molecular weight marker (LMW), lane 2 very low molecular weight marker (VLMW), lane 3 EPO without Lys-C digestion, lane 4 EPO with Lys-C digestion, lane 5 CEPO without Lys-C digestion and lane 6 CEPO with Lys-C digestion. b Hematopoietic bioactivity. EPO, CEPO, and saline were administered i.p. to C57/B6 female mice (Shanghai SLAC Laboratory Animal Co. Ltd) at dose of 50 μg/kg every other day until 30 days. The number of red blood cells was accounted at 30 days after injection. Quantitative value represents means ± SD from six to eight samples of three independent experiments. **P < 0.01
Primary neurons, Neuro-2a cells, and in vitro CEPO/EPO treatment
Embryonic day 18 mouse cortex (Shanghai SLAC Laboratory Animal Co. Ltd.) were dissected under a microscope and then dissociated in Neurobasal-A medium (Gibco) supplemented with 2 % B27 (Gibco), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco) by trituration. The cells were then plated in flanks coated with poly-d-lysine (0.1 mg/ml, Sigma) at a density of 1 × 106 cells/cm2. After 7 days, neurons were collected for experiments.
Neuro-2a cells (mouse neuroblastoma cells) were obtained from Shanghai Institute for Biological Sciences and grown as monolayers in Dulbecco’s modified Eagle’s medium (Gibco), supplemented with 10 % fetal bovine serum (Gibco), 100 U/ml penicillin and 100 μg/ml streptomycin.
Then, neurons were exposed to hypoxia for up to 3 h in a hypoxic incubator (Forma Anaerobic System, Marietta, OH, USA) containing 94 % N2, 1 % O2, and 5 % CO2 to establish neuronal oxygen-glucose deprivation (OGD). After OGD treatment, the cells were returned to a 37-°C normoxic incubator (95 % air and 5 % CO2) and incubated in Neurobasal-A medium for reoxygenation. CEPO/EPO (Chemo Wanbang Biopharma Co. Ltd., Shanghai, 5 ng/ml) was added to cultured cells.
Immunocytochemistry
Cells were incubated with primary anti-JAK-2 (1:200, Santa Cruz), anti-p-AKT (1:200, Chemicon), anti-CD131 (1:200; Santa Cruz), anti-MAP-2 (1:400, Chemicon), and anti-NeuN (1:1000, Chemicon) overnight at 4 °C. After washing, slides were incubated with a secondary antibody: Alexa Fluor 488 anti-rabbit (1:1000), Alexa Fluor 488 anti-rat (1:1000), Alexa Fluor 488 anti-mouse (1:1000), Alexa Fluor 555 anti-mouse (1:1000), Alexa Fluor 548 anti-rabbit (1:1000; all from Molecular Probes, Invitrogen). For double immunohistochemistry, appropriate primary and secondary antibodies were used. Hoechst 33342 (Sigma) was used to identify the nuclei. All brain sections were blindly examined under light microscopy (Olympus BX60) equipped for fluorescence.
MTT assay
Cell viability was detected by MTT assay. Briefly, 100 μl of MTT solution (0.5 mg/ml Duchefa) was added to cultured cells and incubated for an additional 4 h at 37 °C, until the media turned purple. Absorbance at 570 nm was measured by a microplate reader after addition of 100 μl DMSO. Each experiment was done in triplicate and repeated three times with separate cell preparations. Results were expressed as a percentage of PBS control.
Western blot analysis
Protein extracts (30 μg) were loaded on SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Immobilon-P; Millipore Corp.). The membranes were incubated at 4 °C overnight with antibodies against CD131 (1:200; Santa Cruz) followed by incubation with a secondary antibody. Bands of proteins were visualized with an enhanced chemiluminescence system (GE Healthcare Life Sciences). As loading control, antibody directed against β-actin (Sigma) was used.
GDNF ELISA
The supernatants from cultured cells were collected and centrifuged to remove cells. GDNF levels were quantified using commercial ELISA kits (Promega) following the manufacturer’s instructions. The concentrations of cytokines were quantified by reference to standard curves. Determinations were performed in triplicate, and results were expressed as pg/ml.
Antibody neutralization and receptor blocking
The interference of GDNF signaling was performed using well-characterized GDNF neutralization antibodies or GFR receptor antibodies. The neutralization of GDNF was performed using 0.5 μg/ml rabbit anti-mouse GDNF polyclonal antibodies (BioVision) for 24 h prior to EPO/CEPO treatment. Simultaneously, polyclonal rabbit anti-GFR receptor antibodies (Chemicon) were directly added to cultured cells for 24 h at a final concentration of 0.5 μg/ml before EPO/CEPO treatment, and cells were washed with PBS to remove blocking antibodies and then incubated with GDNF (0.5 μg/ml). As a control, affinity-purified rabbit IgG was used at the equivalent concentration.
CEPO/EPO treatment in Neuro-2a cell-injected mice
BALB/c-nu mice (Shanghai SLAC Laboratory Animal Co. Ltd.) were i.p. injected in the left posterior flank with 2 × 105 Neuro-2a cells and then treated with CEPO (50 μg/kg), EPO (50 μg/kg) or saline every other day for 14 consecutive days. Growth of tumor was followed every day by external caliper measurements. Surviving mice were recorded every day at 10:00 am.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 4 software. Comparisons were performed by Student t test, and multiple comparisons were by ANOVA followed by unpaired 2-tailed Student’s t test. P value less than 0.05 was considered significant.
Results
CEPO does not stimulate erythropoiesis
CEPO was obtained from rhEPO (Chemo Wanbang Biotech Pharma Co., Ltd., Shanghai) by chemical modification (Leist et al. 2004). CEPO and rhEPO were digested by Lys-C and analyzed by SDS-PAGE. After carbamylation of EPO, no degradation peptides were detectable for CEPO (Fig. 1a).
CEPO treatment, at a dose of 50 μg/kg every other day until 30 days, did not enhance the number of red blood cells compared to EPO-treated groups, suggesting that CEPO, unlike EPO, does not stimulate erythropoiesis (Fig. 1b). However, EPO stimulated erythropoiesis compared to saline (Fig. 1b; p < 0.01).
CEPO lacks proliferative activity and pAKT expression in Neuro-2a cells
Brdu (30 μg/ml) was added to cultured neurons immediately after OGD 3 h, and Brdu-positive cells were identified in cultured neurons treated with CEPO, EPO or saline. We noted that the number of Brdu-positive cells co-staining with MAP-2 was significantly higher in CEPO- or EPO-treated groups compared to normoxia and saline-treated groups (Fig. 2; p < 0.01 vs. saline). These results show that both CEPO and EPO increased neuron proliferation after OGD.

CEPO/EPO promotes proliferation in OGD model of primary neurons. Primary neurons were exposed to CEPO or EPO (5 ng/ml), immediately after starting OGD for 3 h. Brdu (30 μg/ml) was added to primary neurons for 48 h. Double staining of MAP-2 and Brdu was performed, showing that CEPO/EPO promotes neurogenesis. Microphotographs are representative of three independent experiments, and the quantitative value of Brdu+ neurons represents means ± SD from six to eight samples of two independent experiments. Scale bars 50 μm. **P < 0.01
To determine whether CEPO also has the capacity to promote tumor growth, we designed the following experiments: CEPO/EPO was added to Neuro-2a cells. The results revealed that the viability of Neuro-2a cells was increased in the EPO group than the CEPO group after 24 h of culture (Fig. 3a, b; p < 0.05), but there was no significant difference between CEPO and PBS treatment. Expression of phosphorylated AKT in CEPO-treated Neuro-2a cells was lower than in EPO-treated Neuro-2a cells (Fig. 3c; p < 0.01). As expected, the expression of phosphorylated AKT by Western blot was also lower in CEPO-treated cells than in EPO-treated Neuro-2a cells (Fig. 3d; p < 0.05).

CEPO lacks proliferative activity and pAKT expression in neuroblastoma cells. Neuro-2a cells were exposed to CEPO or EPO (5 ng/ml) for 24 h. a Cell viability was measured by MTT assay. b Representative micrographs of Neuro-2a cells exposed to EPO/CEPO. c Representative micrographs of pAKT expression and the number of pAKT-positive cells. d Representative bands of pAKT/AKT immunostaining and the ratio of pAKT/AKT expression. Quantitative values represent means ± SD from six to eight samples of two independent experiments. Microphotographs are representative of three independent experiments. *P < 0.05, ***P < 0.001. Scale bars 50 μm
CEPO did not activate JAK-2 and produce GDNF
Based on the evidence that CEPO does not promote the proliferation of Neuro-2a cells and that expression of phosphorylated AKT in CEPO-treated Neuro-2a cells is lower than in EPO-treated Neuro-2a cells, we asked whether the difference could be related to AKT activation. We measured the expression of JAK-2 in primary neurons and in Neuro-2a cells treated with CEPO or EPO. As expected, CEPO—unlike EPO—did not activate JAK-2 either in primary neurons or in Neuro-2a cells (Fig. 4a). We also noted that the GDNF level was reduced in Neuro-2a cells treated with CEPO compared to EPO (Fig. 4b). We also performed GDNF neutralization and GFR receptor blocking in Neuro-2a cells before CEPO/EPO treatment. Cell viability in the EPO-treated group was decreased, whereas no change was found in the CEPO-treated group (Fig. 4c). These results show that CEPO did not stimulate Neuro-2a cells to produce GDNF, which may partly explain the low expression of phosphorylated AKT and the inhibition of proliferation.

The production of GDNF in Neuro-2a cells exposed to EPO/CEPO. a Primary neurons and Neuro-2a cells were exposed to CEPO or EPO (5 ng/ml) for 24 h, and the expression of p-JAK-2 was detected by immunocytochemistry and Western blot. b Neuro-2a cells were exposed to CEPO or EPO (5 ng/ml) for 24 h, and the levels of GDNF were measured by ELISA. c GDNF neutralization and GFR receptor blocking did not affect cell viability in CEPO-exposed Neuro-2a cells. Interference with GDNF signaling in cultured cells via GDNF neutralization and GFR receptor blocking was performed as described in “Materials and methods”. Microphotographs are representative of three independent experiments. Quantitative values represent means ± SD from six to eight samples of two independent experiments. Scale bars 50 μm. *P < 0.05
The lack of CD131 expression in Neuro-2a cells
Then, we asked why CEPO can induce primary neurons to produce GDNF but does not stimulate Neuro-2a cells to generate GDNF. CEPO acts upon a heteroreceptor complex comprising of both EPO-R and CD131 which has been reported to be related to CEPO-enhanced neural progenitor cell proliferation and differentiation. We compared CD131 expression in primary neurons and Neuro-2a cells and found that CD131 was highly co-expressed with MAP-2 on primary neurons, but not in Neuro-2a cells (Fig. 5a, b). Western blot confirmed that CD131 expression is very low in Neuro-2a cells compared to primary neurons (Fig. 5c; p < 0.001).

Expression of CD131 in primary neurons and Neuro-2a cells. Primary neurons and Neuro-2a cells were cultured as described in “Materials and methods”. Cell number and viability were assessed, and cell cultures with a viability > 90 % were used. a Representative micrographs of CD131 (red) and MAP-2 (green) expression in primary neurons (up) and Neuro-2a (down) cells by immunocytochemistry. b Number of CD131+ cells in primary neurons and Neuro-2a cells. c Expression of CD131 in primary neurons and Neuro-2a cells by Western blot. Microphotographs are representative of three independent experiments. Quantitative values represent means ± SD from six to eight samples of two independent experiments. Scale bars 100 μm. ***P < 0.001
CEPO inhibits Neuro-2a cell growth in vivo
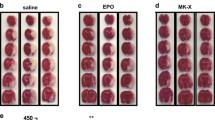
To further verify CEPO-mediated inhibition of Neuro-2a cell growth in vivo, BABL/C-nu mice were vaccinated by injection with 2 × 105 Neuro-2a cells in the left posterior flank. CEPO (50 μg/ml), EPO (50 μg/ml), and saline were given every other day at the same dose for 14 consecutive days. We observed that mice treated with EPO had prominent tumor growth compared to the saline- or CEPO-treated mice after 2 weeks (Fig. 6a). Survival curves showed that CEPO-treated mice had somewhat longer survival time compared to EPO-treated mice (44 vs. 38.5 days, p < 0.05; Fig. 6b). Taken together, our results demonstrate that CEPO did not promote the growth of Neuro-2a cells compared to EPO, revealing its therapeutic potential in future clinical application.

CEPO extends survival days in BALB/c-nu mice injected with CEPO. BALB/c-nu mice were injected with CEPO, EPO or saline (NS) every other day for 14 consecutive days as described in “Materials and methods”. a Representative photograph of tumor size at 2 weeks after injection of Neuro-2a cells. b Survival curves of mice from 12 mice in each group from three independent experiments. Scale bars 1 cm. p < 0.05, CEPO vs. NS
Discussion
The recent evidence showing that CEPO lacks erythropoietic effect while being neuroprotective suggests that the neuroprotective effect of CEPO is not linked to erythropoietic function. However, mechanisms underlying the neuroprotection of CEPO have not been fully characterized. It has been stated that EPO first activates JAK-2, which then engages secondary signaling pathways that involve MAPK and PI3K/AKT, while CEPO appears to require the common β chain of IL-3/IL-5/GM-CSF receptor (also known as CD131) (Leconte et al. 2011). Recent reports propose that CD131 is expressed at low levels in the brain (Wang et al. 2007). In studies of EPO, many possible anti-apoptotic signaling pathways have been reported, including JAK-2, STAT5, MAPK, and/or AKT signaling (Brines and Cerami 2005). Consistent with a previous finding (Leist et al. 2004), our results show that CEPO did not activate JAK-2, while AKT activation was observed in both CEPO-/EPO-treated cells.
The question is how CEPO results in AKT activation without activating JAK-2. In a mouse model of diabetic autonomic neuropathy, CEPO exhibited neurotrophic function via NGF (Schmidt et al. 2008). In traumatic brain injury, CEPO promoted neurogenesis and increased BDNF expression (Mahmood et al. 2007). Other investigations showed that GDNF can trigger the activation of the PI3K/AKT pathway (Anitha et al. 2006), resulting in cell survival and stimulation of neurogenesis. GDNF, as an important neurotrophic factor for the survival and differentiation of neurons (Airaksinen and Saarma 2002), prevents neuronal cell death in CHP134 neuroblastoma cell line (Takada et al. 2001) and enhances the migration of U87 and U251 glioma cells but not C6 cells through the increase of MMP-13 production (Lu et al. 2010). GDNF overexpressed in glioblastoma cell lines and human gliomas may contribute to the increased cellular survival via AKT activation and JNK inhibition (Ng et al. 2009). A recent study showed that, in the absence of conventional receptors, a novel receptor for GDNF (heparan sulfate proteoglycan syndecan-3) is provided for GDNF-induced cell growth (Bespalov et al. 2011).
Unlike EPO, CEPO does not bind the classical homodimeric EPOR, and its neuroprotective action appears to require the common β chain of IL-3/IL-5/GM-CSF receptor (also known as CD131). The blockage of CD131 resulted in reduced GDNF production, and that addition of GDNF to cultured neurons increased phosphorylated AKT, suggesting that CD131 participates in CEPO-induced GDNF production and that CEPO protects neurons via the CD131/GDNF/AKT pathway (unpublished data). However, knockout of the common β chain fully abolished the tissue-protective properties of EPO in the nervous system (Brines and Cerami 2006). EPO prevented staurosporine-induced apoptosis of differentiated human neuroblastoma SH-SY5Y cells and activated the STAT5, AKT, and MAPK signaling pathways, comprising the “classical” Epo receptor signaling complex (Um and Lodish 2006; Um et al. 2007). Thus, further studies on CEPO/EPO signaling on neuroprotection are still needed.
In addition, we noted that the level of GDNF was lower in CEPO-treated Neuro-2a cells than in EPO-treated cells, showing that CEPO did not stimulate Neuro-2a cells to produce GDNF, which in turn explains the low expression of phosphorylated AKT and the inhibition of cell proliferation. The AKT signaling network is considered a key determinant of the biological aggressiveness of these tumors. Specific inhibition of AKT has been studied, such as with LY249002, with significant effects on cell progression and apoptosis in tumoral cells (Sartelet et al. 2008).
CEPO acts upon a heteroreceptor complex comprising both EPO-R and CD131 which is reported to be related to CEPO-enhanced neural progenitor cell proliferation and differentiation. When comparing CD131 expression in primary neurons and Neuro-2a cells, we found that CD131 was highly co-expressed in MAP-2-positive primary neurons, but not in Neuro-2a cells. It has been stated that CEPO signals through EPO-R and CD131 heteroreceptor (Leist et al. 2004; Wang et al. 2007). The deficiency of CD131 expression in Neuro-2a cells should obstruct the CEPO-mediated signaling pathway, thereby explaining why CEPO did not promote the growth of Neuro-2a cells in vitro and in vivo.
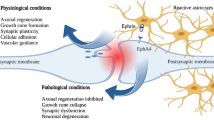
In conclusion, CEPO did not promote the growth of Neuro-2a cells because of low levels of CD131 and GDNF in Neuro-2a cells (Fig. 7). Overall, these findings highlight the potential usefulness of CEPO in the treatment of neurodegenerative and neuropsychiatric diseases without triggering adverse effects such as thrombosis or tumor growth.

Diagram of different biological roles for EPO/CEPO in primary neurons and Neuro-2a cells. In primary neurons and Neuro-2a cells, EPO can bind homodimeric EPO-R to first activate JAK-2, which then engages secondary signaling pathways that involve AKT. AKT pathway might participate in neuroprotection and/or tumor growth. EPO has also been implicated in other signaling pathways that are not shown in this diagram. CEPO does not bind classic homodimeric EPO-R to activate JAK-2 but appears to require a heteroreceptor complex comprising of both EPO-R and CD131, which can trigger the GDNF-AKT pathway, and mediate neuroprotection in primary neurons. However, CEPO does not promote growth of Neuro-2a cells because of low levels of CD131 expression that hinder CD131-GDNF-AKT pathway
References
Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–94.
Anitha M, Gondha C, Sutliff R, Parsadanian A, Mwangi S, Sitaraman SV, et al. GDNF rescues hyperglycemia-induced diabetic enteric neuropathy through activation of the PI3K/Akt pathway. J Clin Invest. 2006;116:344–56.
Bespalov MM, Sidorova YA, Tumova S, Ahonen-Bishopp A, Magalhaes AC, Kulesskiy E, et al. Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J Cell Biol. 2011;192:153–69.
Boesch S, Nachbauer W, Mariotti C, Sacca F, Filla A, Klockgether T, et al. Safety and tolerability of CEPO in Friedreich’s ataxia. Mov Disord. 2014;29:935–9.
Bohlius J, Schmidlin K, Brillant C, Schwarzer G, Trelle S, Seidenfeld J, et al. Recombinant human erythropoiesis-stimulating agents and mortality in patients with cancer: a meta-analysis of randomised trials. Lancet. 2009;373:1532–42.
Brines M, Cerami A. Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci. 2005;6:484–94.
Brines M, Cerami A. Discovering erythropoietin’s extra-hematopoietic functions: biology and clinical promise. Kidney Int. 2006;70:246–50.
Choi M, Ko SY, Lee IY, Wang SE, Lee SH, Oh DH, et al. Carbamylated erythropoietin promotes neurite outgrowth and neuronal spine formation in association with CBP/p300. Biochem Biophys Res Commun. 2014;446:79–84.
Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009;40:e647–56.
Fantacci M, Bianciardi P, Caretti A, Coleman TR, Cerami A, Brines M, et al. Carbamylated erythropoietin ameliorates the metabolic stress induced in vivo by severe chronic hypoxia. Proc Natl Acad Sci U S A. 2006;103:17531–6.
Hedley BD, Allan AL, Xenocostas A. The role of erythropoietin and erythropoiesis-stimulating agents in tumor progression. Clin Cancer Res. 2011;17:6373–80.
King VR, Averill SA, Hewazy D, Priestley JV, Torup L, Michael-Titus AT. Erythropoietin and carbamylated erythropoietin are neuroprotective following spinal cord hemisection in the rat. Eur J Neurosci. 2007;26:90–100.
Kumar SM, Zhang G, Bastian BC, Arcasoy MO, Karande P, Pushparajan A, et al. Erythropoietin receptor contributes to melanoma cell survival in vivo. Oncogene. 2012;31:1649–60.
Leconte C, Bihel E, Lepelletier FX, Bouet V, Saulnier R, Petit E, et al. Comparison of the effects of erythropoietin and its carbamylated derivative on behaviour and hippocampal neurogenesis in mice. Neuropharmacology. 2011;60:354–64.
Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M, et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004;30:239–42.
Lu DY, Leung YM, Cheung CW, Chen YR, Wong KL. Glial cell line-derived neurotrophic factor induces cell migration and matrix metalloproteinase-13 expression in glioma cells. Biochem Pharmacol. 2010;80:1201–9.
Mahmood A, Lu D, Qu C, Goussev A, Zhang ZG, Lu C, et al. Treatment of traumatic brain injury in rats with erythropoietin and carbamylated erythropoietin. J Neurosurg. 2007;107:392–7.
Mennini T, De Paola M, Bigini P, Mastrotto C, Fumagalli E, Barbera S, et al. Nonhematopoietic erythropoietin derivatives prevent motoneuron degeneration in vitro and in vivo. Mol Med. 2006;12:153–60.
Ng WH, Wan GQ, Peng ZN, Too HP. Glial cell-line derived neurotrophic factor (GDNF) family of ligands confer chemoresistance in a ligand-specific fashion in malignant gliomas. J Clin Neurosci. 2009;16:427–36.
Peres EA, Valable S, Guillamo JS, Marteau L, Bernaudin JF, Roussel S, et al. Targeting the erythropoietin receptor on glioma cells reduces tumour growth. Exp Cell Res. 2011;317:2321–32.
Sartelet H, Fabre M, Castaing M, Bosq J, Racu I, Lagonotte E, et al. Expression of erythropoietin and its receptor in neuroblastomas. Cancer. 2007;110:1096–106.
Sartelet H, Oligny LL, Vassal G. AKT pathway in neuroblastoma and its therapeutic implication. Expert Rev Anticancer Ther. 2008;8:757–69.
Schmidt RE, Green KG, Feng D, Dorsey DA, Parvin CA, Lee JM, et al. Erythropoietin and its carbamylated derivative prevent the development of experimental diabetic autonomic neuropathy in STZ-induced diabetic NOD-SCID mice. Exp Neurol. 2008;209:161–70.
Takada N, Isogai E, Kawamoto T, Nakanishi H, Todo S, Nakagawara A. Retinoic acid-induced apoptosis of the CHP134 neuroblastoma cell line is associated with nuclear accumulation of p53 and is rescued by the GDNF/Ret signal. Med Pediatr Oncol. 2001;36:122–6.
Um M, Lodish HF. Antiapoptotic effects of erythropoietin in differentiated neuroblastoma SH-SY5Y cells require activation of both the STAT5 and AKT signaling pathways. J Biol Chem. 2006;281:5648–56.
Um M, Gross AW, Lodish HF. A “classical” homodimeric erythropoietin receptor is essential for the antiapoptotic effects of erythropoietin on differentiated neuroblastoma SH-SY5Y and pheochromocytoma PC-12 cells. Cell Signal. 2007;19:634–45.
Wang L, Zhang ZG, Gregg SR, Zhang RL, Jiao Z, LeTourneau Y, et al. The Sonic hedgehog pathway mediates CEPO-enhanced proliferation and differentiation of adult neural progenitor cells. J Biol Chem. 2007;282:32462–70.
Acknowledgments
We would like to thank Chemo Wanbang Biopharma Co. Ltd., Shanghai, for providing the rhEPO. This work was supported by project grants from the National Natural Science Foundation of China (Code 30572164, Code 30972715, Code 81371414).
Disclosures
None of the authors has any conflict of interest related to this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ding, J., Li, QY., Yu, JZ. et al. The lack of CD131 and the inhibition of Neuro-2a growth by carbamylated erythropoietin. Cell Biol Toxicol 31, 29–38 (2015). https://doi.org/10.1007/s10565-015-9292-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-015-9292-y