
Abstract
Generating large amounts of recombinant protein in transgenic animals is often challenging and has a number of drawbacks compared to cell culture systems. The baculovirus expression vector system (BEVS) uses virus-infected insect cells to produce recombinant proteins to high levels, and these are usually processed in a similar way to the native protein. Interestingly, since the development of the BEVS, the virus most often used (Autographa californica multi-nucleopolyhedovirus; AcMNPV) has been little altered genetically from its wild-type parental virus. In this study, we modified the AcMNPV genome in an attempt to improve recombinant protein yield, by deleting genes that are non-essential in cell culture. We deleted the p26, p10 and p74 genes from the virus genome, replacing them with an antibiotic selection cassette, allowing us to isolate recombinants. We screened and identified recombinant viruses by restriction enzyme analysis, PCR and Western blot. Cell viability analysis showed that the deletions did not improve the viability of infected cells, compared to non-deletion viruses. However, expression studies showed that recombinant protein levels for the deletion viruses were significantly higher than the expression levels of non-deletion viruses. These results confirm that there is still great potential for improving the BEVS, further increasing recombinant protein expression yields and stability in insect cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transgenic animals (including sheep, goats, cows, pigs, rabbits, mice and insect larvae) are currently being developed and used for the production of recombinant proteins in milk, egg white, blood, urine, seminal plasma and silk worm cocoons (Houdebine 2009). However, the use of these animals has a number of drawbacks, including the length of time required to assess protein production (Chew 1993). Additionally, separation of recombinant proteins from the host may be difficult, and the product must be screened for the presence of potentially harmful pathogens. Indeed, the recombinant protein itself may be harmful to the animal expressing it (Houdebine 2009).
Cell culture-based expression systems offer a viable alternative to the use of transgenic animals. The baculovirus expression vector system (BEVS) is widely used to express recombinant proteins to high levels in insect cells (Kitts et al. 1990; Kitts and Possee 1993), producing authentic proteins which are normally functionally active (Luckow and Summers 1988; Miller 1988). The system is based on the deletion of the virus polyhedrin gene (polh), which is replaced by the gene to be expressed, under the control of the powerful polh promoter. In recent years, the incorporation of bacterial replicons into the baculovirus genome has greatly improved this system, making it much simpler and faster to generate recombinant viruses (Luckow et al. 1993; Possee et al. 2008). The baculovirus most often used as an expression vector is Autographa californica multi-nucleopolyhedrovirus (AcMNPV). AcMNPV has a genome size of 133,894 nucleotides (NC_001623), containing 156 predicted protein-coding genes (Ayres et al. 1994). The functions of many of these genes are still unknown, and in some instances, they may play no role in the ability of the virus to be propagated in cell culture (Wang et al. 2007; Carpentier et al. 2008; Goenka and Weaver 2008; Li et al. 2008). Commercially available BEVs usually only lack the polh gene and otherwise have a complete genome (i.e. Bac-to-Bac, Invitrogen; BacPAK6, BD-Biosciences). Consequently, there is significant potential to improve this system by removing genes that may be deleterious to recombinant protein production or are a burden to viral replication in cell culture. However, the deletion of baculovirus genes to improve recombinant protein expression is poorly studied. Deletion of chitinase (chiA) and a cysteine protease (cath) from the AcMNPV genome has been shown to enhance secreted recombinant protein stability (Kaba et al. 2004). These modifications were primarily designed to facilitate the movement of recombinant proteins through the cell's secretory pathway and to prevent their degradation once they are released into the culture media. Another gene deletion that has been described as potentially beneficial to recombinant protein production is removal of p10. P10 is a 10-kDa protein of unknown function, expressed concurrently with polh late in infection, and is non-essential in cell culture (Carpentier et al. 2008). However, to date, the effect of p10 deletion on recombinant protein expression has not been demonstrated conclusively. Two other auxiliary genes are non-essential to virus infectivity in cell culture, flank p10, namely p26 and p74. P74 is essential for oral infectivity of occlusion-derived virus (ODV) in the host, where it plays a role in midgut attachment and fusion (Faulkner et al. 1997), but it has been shown to have no effect on virus production in vitro (Kuzio et al. 1989). P26 is an early gene that codes for a 240-amino acid polypeptide of unknown function (Liu et al. 1986) and has the same 5′ terminus as p10 (Rankin et al. 1986). Deletion of the 3′-end of p26 and fusion to lacZ or p10 have previously been shown to have no effect on virus replication in vitro (Chaabihi et al. 1993; Rodems and Friesen 1993).
The aim of this study was to construct a bacmid-based BEV that had deletions of all five of these non-essential genes, on the basis that this combination of deletions should have an additive beneficial effect on recombinant protein expression. A BEV was constructed that was deficient in chiA, cath, p26, p10 and p74, and recombinant protein production was compared to vectors with no deletions, or single or multiple deletions of the above genes.
Materials and methods
Materials
Electrocompetent DH10B cells were obtained from Invitrogen. Antibiotics were obtained from Sigma. Primers were synthesised by MWG. Plasmid extraction reagents kits were purchased from Qiagen. Restriction enzymes, molecular-mass markers and PCR reagents were purchased from New England Biolabs. Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) reagents were obtained from Sigma, National Diagnostics and Generon. His-tag antibody was purchased from AbD Serotec. Secondary antibodies (goat anti-rabbit and anti-guinea pig IgG) conjugated with alkaline phosphatase (AP) were purchased from Sigma. Membranes were obtained from GE Healthcare. Strataclean resin for protein concentration was obtained from Stratagene and Bradford assays were obtained from BioRad. Protein purification kits were supplied by EMD Biosciences. Insect cell culture plasticware was supplied by IWAKA and EX-CELL 420 cell culture media was obtained from SAFC Biosciences. Lipofectin was purchased from Invitrogen and Trypan blue was obtained from Sigma. Baculovirus titration was carried out using baculoQUANT (Oxford Expression Technologies) and β-galactosidase assays were purchased from Promega.
Gene deletions
The baculovirus DNA AcRP30.AvrII2.bac (Possee et al. 2008) was mixed with a transfer plasmid based on pBS.SK containing the lacZ coding region under the control of the polyhedrin gene promoter and flanked by approximately 1,000 bp from the AcMNPV genome (Ayres et al. 1994) corresponding to nucleotides 104161–105207 and 107954–109080. These regions border the chiA and cath genes in AcMNPV. The virus and plasmid DNA mixture were used to transfect Sf21 cells and recombinants subsequently identified by plaque assay in the same cell type and with X-gal staining. Genetically homogeneous viruses were isolated by further rounds of plaque isolation and amplified to derive virus genomic DNA. This DNA was digested with Bsu36I, which cleaved once in the lacZ region and mixed with a pBS.SK variant containing only the two regions flanking the chiA and cath genes. These DNAs were used to transfect Sf21 cells and viruses without lacZ sequences were isolated and designated AcchiA−/cath−. This virus was then modified subsequently to remove p26, p10 and p74 genes. A slightly different approach was used where AcMNPV 1,000-bp regions flanking the genes were placed on either side of a neomycin cassette to direct insertion in lieu of the native genes. The transfer plasmid was mixed with AcchiA−/cath− and used to transfect Sf21 cells. One day post-transfection, total DNA was purified from the cells and used to electroporate DH10B cells to recover virus genomes containing the neomycin cassette. These were selected on nutrient agar plates containing 15 µg/ml chloramphenicol and 15 µg/ml kanamycin. Individual colonies were amplified in liquid medium containing 15 μg/ml chloramphenicol and 15 μg/ml kanomycin and bacmid DNA was isolated by alkaline lysis before being purified on caesium chloride gradients. The virus DNA was then further modified by digesting with AvrII and religating to remove part of ORF1629 (Possee et al. 2008).
Confirming deletion of p26, p10 and p74
Recombinant bacterial colonies were initially identified by restriction enzyme (REN) analysis using HindIII and EcoRI to digest 500 ng of virus DNA. Agarose gels (0.6%) were stained with SYBR Green I and scanned using a Typhoon Variable Mode Imager (GE Healthcare). PCR was carried out using the forward primer P10-neoF (CGATCGATTGATGAGTCATTTG) and a reverse primer P10-neoR (GCTTTGAAATGCACAACGAA). The PCR was performed in a volume of 50 μl containing 100 ng of template DNA, 0.2 mM of each of the primers, 5 μl of Taq polymerase 10× reaction buffer, 5 μg of bovine serum albumin (BSA), 1.5 mM MgCl2, 200 μM dNTPs and five units of Taq DNA polymerase. The PCR was performed at 55°C for annealing, 72°C for elongation, and 95°C for denaturation.
Virus propagation and insect cell culture
Genes were cloned into suitable baculovirus transfer plasmids using standard cloning techniques (Sambrook and Russell 2001) and co-transfected with virus DNA to generate recombinant viruses. The virus DNAs used were BacPAK6 (contains chiA, cath, p26, p10 and p74) or bacmid-based derivatives of this virus, which have different gene deletions including AcchiA− (deficient in chiA) and AcchiA−/cath− (deficient in chiA and cath). Co-transfections were carried out in 1 × 106 Spodoptera frugiperda 9 (Sf9) cells in 35-mm2 cell culture dishes as described by King and Possee (1992) or in 24-well plates (Possee et al. 2008). Five days post-transfection, 0.5 ml co-transfection medium was added to 50 ml of Sf9 cells (2 × 106 cells/ml) and incubated for a further 5 days to produce a high-titre virus stock. Recombinant viruses were routinely propagated in Sf9 cells maintained in EXCELL 420 serum-free medium in shaker flasks at 150 rpm. Cell viability assays were carried out in triplicate shake culture flasks, seeded with Sf9 insect cells at 2 × 106 cells/ml and infected at multiplicity of infection (moi) of five plaque-forming units (pfu) per cell. Samples were then harvested at 1, 6, 12, 24, 48, 72 and 96 h post infection (hpi) and clarified by low-speed centrifugation. Recombinant viruses were titrated using quantitative PCR (QPCR) as described by Hitchman et al. (2007), or by plaque assay (King and Possee 1992). Cell viability (ratio of viable cells to total cells) was determined using the Trypan blue exclusion method, by staining cells with 0.2% Trypan blue and measuring the percentage of viable cells in a Neubauer hemocytometer.
SDS-PAGE and Western blotting
Sf9 cells (2 × 106 cells/ml) were infected at a moi of 5 pfu per cell, or were mock-infected with culture medium in shaker flasks and harvested at different time-points. Supernatant fractions were harvested and clarified at 200×g and protein was concentrated using Strataclean resin as described by the manufacturer. Pellet fractions were separated by centrifugation and washed twice with phosphate-buffered saline (PBS) before being quantified by Bradford assay. Total protein (25 µg) from the supernatant or crude lysate was mixed with loading buffer and boiled for 3 min. SDS-PAGE and Western blot analysis were carried out as described previously (King and Possee 1992). Membranes were probed with primary antiserum (1:10,000) to the 6 × His-tag or the anti-P10 (1:750) (Patmanidi et al. 2003) followed by a secondary IgG antibody conjugated to alkaline phosphatase (1:10,000). Blots were then developed with 5-bromo-4-chloro-3-indoyl phosphatase (BCIP) and nitro blue tetrazolium (NBT).
βeta-galactosidase and enhanced green fluorescent protein assays
Sf9 cells were inoculated with virus at moi 5 pfu per cell. The virus was allowed to adsorb for 1 h at room temperature. The inoculum was then removed and replaced with EX-CELL 420 medium, and incubation was carried out at 27°C for the time indicated (see Fig. 3). The cells were then harvested, washed with PBS and pelleted at 200×g for 5 min. After the cellular debris had been pelleted, the extracts were assayed for β-galactosidase activity using the o-nitrophenyl-β-d-galactopyranoside with the β-galactosidase enzyme assay system (Promega), and an ELX 808 Ultra Microplate Reader (Bio-Tech Instruments). Briefly, reporter lysis buffer (100 µl) was added to a cell pellet and incubated at room temperature for 15 min, vortexed and centrifuged at 200×g for 2 min. The supernatant was then transferred to a fresh tube. Assays were carried out in 96-well plate format, where dilutions of cell lysate (10−1, 10−2 and 10−3) were prepared in reporter lysis buffer in a total volume of 50 µl. Fifty microlitres of 2× assay buffer was then added to the plate and mixed on a blood tube rotator, before incubating the plate at 37°C for 30 min. Each plate also contained a negative control (reporter lysis buffer only) and a standard dilution of β-galactosidase (1:10,000). The reaction was stopped with 100 µl of 2 M sodium carbonate and the absorbance was read immediately at 405 nm.
To analyse intracellular enhanced green fluorescent protein (EGFP), Sf9 cells in 24-well plates were infected using viruses generated in a 24-well plate format, as described previously (Possee et al. 2008). Briefly, 100 µl of each co-transfection from a 24-well plate was added to each well of a fresh 24-well plate and incubated at 27°C for 72 h. Cells were then harvested, washed with PBS and pelleted at 200×g for 5 min. Pelleted cells were lysed in 50 mM NaH2PO4, pH 8.0 containing 300 mM NaCl, 10 mM imidazole, 1% v/v Tween 20 and centrifuged at 10,000×g for 10 min. Supernatants were distributed into 96-well black/clear polystyrene assay plates (BD Biosciences, Oxford, UK) and EGFP fluorescence for each individual plate well measured at 535 nm following excitation at 485 nm in an InFinite F200 microplate reader (Tecan Group, Theale, UK).
Statistical analysis
Levels of protein expression were analysed in a semi-quantitative manner by Western blot visualisation and scanning. The density of each band was normalised by subtracting the background intensity, and the average intensities of three scans of each of the bands were then determined using GeneTools (Syngene, UK) and compared to neighbouring bands. Statistical analysis was carried out using analysis of variance calculator (www.danielsoper.com). The averages of all measurements taken were used for the construction of the graphs, and standard errors were calculated using Microsoft Excel 2003.
Results
Construction and testing AcchiA−/cath−/p10−
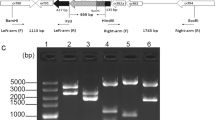
As described in the “Materials and methods” section, we constructed a BEV that was deficient in chiA, cath, p26, p10 and p74 (AcchiA−/cath−/p10−). We initially screened potential deletion clones by REN digestion (Fig. 1). Successful recombination replaces a 3,733-bp fragment containing p26, p10 and p74 with a ~1-kb neomycin cassette. Deletion of the 3,733-bp fragment also removes two HindIII restriction sites from the virus backbone, fusing the ~2.2-kb Q, 2.2-kb P and ~8.4-kb G fragments (Ayres et al. 1994). Therefore, after digestion with HindIII, the deletion clones should lack the Q, P and G fragments (Q−, P−, G−) but gain a ~10-kb fragment (QPG+). Figure 1a shows three of the candidate deletion clones (Fig. 1a, lanes 4, 5 and 6), which had a new ~10-kb fragment (indicated by an arrow, QPG+) after digestion with HindIII, compared to the parental virus (Fig. 1a, lane 3). The missing Q, P and G fragments are also indicated (Q−, P−, G−). The G fragment likely co-migrates with other fragments, and its absence is, therefore, less obvious. Digestion with EcoRI also removes two restriction sites, fusing the ~0.5-kb Z, ~2-kb P and ~13.4-kb fragments to produce a new ~13-kb fragment (Fig. 1b, ZPB+). The missing ~2-kb P fragment (P−) is indicated for three clones in Fig. 1b (lanes 4, 5 and 6), compared to the parental virus shown in lane 3. There is no actual increase in size for the EcoRI B fragment, and so it migrates to a similar position as the parental virus (indicated by an arrow, ZPB+). The SYBR stain at the base of the gel obscures the absence of the Z fragment. These deletions corresponded to the expected sizes after deletion of the fragment containing p26, p10 and p74 and insertion of neomycin. We then used PCR to further confirm that the fragment had been deleted from these clones. Figure 1c shows PCR amplification of the target region using primers that bind upstream of p26 and downstream of p74. Using these primers, no product was amplified for the negative control (Fig. 1c, lane 2), but a 3,733-bp fragment was amplified for the AcchiA−/cath− parental virus (Fig. 1c, lane 3), corresponding to the presence of p26, p10 and p74. A ~1.5-kb-sized band was amplified for a positive control virus containing a neomycin cassette (Fig. 1c, lane 4), and a similar-sized band was also amplified for the three candidate AcchiA−/cath−/p10− deletion clones (Fig. 1c, lanes 5, 6 and 7), suggesting that neomycin had replaced p26, p10 and p74.

a Agarose gel (0.7%) of HindIII and b EcoRI digested DNA stained with SYBR Green I. Lane 1 shows a 10-kb molecular weight marker. Lane 2 contains AcchiA− DNA. Lane 3 shows AcchiA−/cath− DNA. Lanes 4, 5 and 6 show DNA purified from three p26, p10 and p74 deletion clones (AcchiA−cath−p10−). The presence and absence of DNA bands are indicated by the arrows. c Agarose gel electrophoresis of PCR amplification products using primers flanking p26, p10 and p74. Lane 1 shows a 10-kb molecular weight ladder. Lane 2 shows no-template control. Lane 3 shows positive control for p26, p10 and p74 (AcchiA−/cath−). Lane 4 shows positive control for ~1-kb neomycin cassette. Lanes 5, 6 and 7 show three p26, p10 and p74 deletion clones (AcchiA−cath−p10−). d Coomassie-stained SDS-PAGE gels showing P10 and β-galactosidase expression. Lane 1 shows a kDa molecular weight marker. Lane 2 shows non-infected cells. Lane 3 shows expression for BacPAK6. Lane 4 shows expression for AcchiA−/cath− + lacZ. Lane 5 shows expression levels for AcchiA−cath−p10− + lacZ. P10 and β-galactosidase bands are indicated with arrows. e Corresponding Western blot for P10 expression. Lane 1 shows a molecular weight marker. Lane 2 shows non-infected cells. Lane 3 shows expression for BacPAK6. Lane 4 shows expression for AcchiA−/cath− + lacZ. Lane 5 shows expression levels for AcchiA−cath−p10− + lacZ
One of the deletion clones was then chosen and used to make a recombinant virus containing lacZ. We then tested this virus for the absence of the P10 protein by SDS-PAGE and Western blot. Protein was extracted from virus-infected cell pellets and separated by SDS-PAGE as shown in Fig. 1d. A band corresponding to P10 was visible in the cells infected with BacPAK6 (Fig. 1d, lane 3) and the parental AcchiA−/cath−-infected cells (Fig. 1d, lane 4), but not in the mock-infected cells (Fig. 1d, lane 2) or the AcchiA−/cath−/p10−-infected cells (Fig. 1d, lane 5). All recombinant viruses (Fig. 1d, lanes 3, 4 and 5) showed the presence of β-galactosidase (indicated by an arrow), which was absent in the mock-infected cells (Fig. 1d, lane 2). Figure 1e shows the corresponding Western blot, and, as expected, P10 was detected for BacPAK6 and the parental AcchiA−cath− using an anti-P10 antibody (Fig. 1e, lanes 3 and 4), but no P10 was detected in the mock-infected cells or the AcchiA−/cath−/p10−-infected cells, as shown in Fig. 1e, lanes 2 and 5.
Cell viability
Deletion of p10 has been suggested to improve cell viability (Thomas et al. 1998). To test this idea, Sf9 cells were infected with AcchiA-/cath−, AcchiA−/cath−/p10− or BacPAK6 at moi 5 pfu per cell, and samples were harvested over 6 days. As shown in Fig. 2a, the viability of BacPAK6, AcchiA−/cath− and AcchiA−/cath−/p10−-infected cells remained high until 24 hpi. After 24 hpi, the viability started to decrease, and after 48 hpi, it declined rapidly for AcchiA−/cath− and AcchiA−/cath−/p10−-infected cells and was approximately 38% and 28%, respectively, at 72 hpi. BacPAK6-infected cells showed a slower decrease in viability and were ~66% viable at 72 hpi

a Comparison of cell viability between BacPAK6 (triangles), Ac−chiA/−cath (diamonds) and Ac−chiA/−cath/−p10-infected Sf9 cells (squares). Triplicate shake cultures of Sf9 cells were infected with recombinant baculoviruses at moi 5. The percentage of surviving cells was determined from 0 to 120 hpi by Trypan blue exclusion. Results shown represent means and +/− standard errors of the mean for triplicate flasks. b Temporal AcchiA−/cath− and AcchiA−/cath−/p10− or BacPAK6 β-galactosidase expression. Sf9 cells were infected with AcchiA−/cath− (diamonds) and AcchiA−/cath−/p10− (squares) or BacPAK6 (triangles) at moi 5. The specific activity of β -galactosidase in the infected cells was determined at various times p.i. Results shown represent means and +/− standard errors of the mean for triplicate flasks
Recombinant protein expression
We compared AcchiA−/cath− and AcchiA−/cath−/p10− β-galactosidase expression with BacPAK6 over time. Cell pellets were lysed and β-galactosidase activity was determined using a plate reader. Figure 2b shows that β-galactosidase activity rose from 12 hpi and then rapidly increased for AcchiA−/cath− and AcchiA−/cath−/p10−, peaking at 48 hpi. It then declined rapidly from 48 to 72 hpi. This sudden decline in activity corresponds to the decline in cell viability observed in Fig. 2a. The highest AcchiA−/cath−/p10− β-galactosidase activity occurred at 48 hpi when the cells were ~82% viable. The increase in BacPAK6 activity was much more gradual and peaked at 72 hpi, when the cells were ~73% viable, before declining slowly. The observed specific activity at 48 hpi of AcchiA−/cath−/p10− was nearly 2× that of the highest BacPAK6 activity (72 hpi) and was slightly higher than AcchiA−/cath−.
We then compared expression levels of EGFP, a stable and easily quantifiable protein, in a high-throughput format using 24-well plates and a CAS-1200 liquid handler (Possee et al. 2008). At 72 hpi, EGFP expression levels across 24 wells of a plate infected with AcchiA−/cath−/p10− were observed to be significantly higher (F = 25.326, df = 70, p = <0.005) than 24 wells of a plate infected with either AcchiA−/cath− or AcchiA− (Fig. 3).

EGFP expression analysis. EGFP was expressed using either AcchiA−, AcchiA−/cath− or AcchiA−/cath−/p10− viruses and expression levels determined at 72 hpi in a plate reader. Results shown represent means and +/− standard errors of the mean for 24 replicate samples from a 24-well plate experiment
We then compared expression levels of five different proteins (β-galactosidase (β-gal), heat-shock 90-KD protein 1, alpha (HSPCA1), polo-like kinase 1 (Plk1), phosphatase and tensin homolog (PTEN) and KappaB-Ras2 using AcchiA−, AcchiA−/cath− and AcchiA−/cath−/p10−. Sf9 cells were infected at moi 1 and cells and supernatants were harvested at 72 hpi. Proteins were purified using Ni-NTA His-Bind® or Strep-Tactin® Resin following the manufacturer's protocol and equal quantities were separated by SDS-PAGE. Figure 4a shows individual gels for each of the genes expressed using AcchiA− (lanes 1, 4, 7, 10 and 13), AcchiA−/cath− (lanes 2, 5, 8, 11 and 14) and AcchiA−/cath−/p10− (lanes 3, 6, 9, 12 and 15), respectively. Lane M shows a 10–225-kDa molecular weight marker. For each gene, there was an increase in the amount of protein expressed using the deletion virus (AcchiA−/cath−/p10−), and densitometry analysis (Fig. 4b) confirmed that AcchiA−/cath−/p10− expression levels (lanes 3, 6, 9, 12 and 15) were higher than either AcchiA− (lanes 1, 4, 7, 10 and 13) or AcchiA−/cath− viruses (lanes 2, 5, 8, 11 and 14). The increase in expression was dependent on the gene being expressed and was greater for β-gal, HSPCA1 and Plk1 than PTEN and KappaB-ras2. However, generally, there was a clear trend of increasing protein expression with increasing number of gene deletions.

a Comparative Western analysis of five recombinant proteins. Sf9 cells were infected with AcchiA−, AcchiA−/cath− and AcchiA−/cath−/p10− virus for 72 hpi. After infection, the cells were harvested, recombinant protein purified and then subjected to SDS-PAGE and Western blot analysis. Lane M contains a 10–225-kDa molecular weight marker. Lanes 1, 4, 7, 10 and 13 contain fractions of cells infected with AcchiA−; lanes 2, 5, 8, 11 and 14 in all panels contain fractions of cells infected with AcchiA−/cath−; lanes 3, 6, 9, 12 and 15 contain fractions of cells infected with AcchiA−/cath−/p10−. The names of the recombinant proteins are indicated below each individual gel. b Band intensities from the protein gel shown in a were quantified by densitometry. Lanes 1, 4, 7, 10 and 13 contain fractions of cells infected with AcchiA−; lanes 2, 5, 8, 11 and 14 in all panels contain fractions of cells infected with AcchiA−/cath−; lanes 3, 6, 9, 12 and 15 contain fractions of cells infected with AcchiA−/cath−/p10−. The names of the recombinant proteins are indicated below each individual graph. Results shown represent means and +/− standard errors of the mean for three densitometry readings
To test the stability of proteins produced using AcchiA−/cath−/p10−, we used a protein that was more sensitive to proteolysis, p21-activated kinase (PAK5). We tested kinase expression levels and cell viability over a 5-day time course, comparing AcchiA−, AcchiA−/cath− and AcchiA−/cath−/p10−. Figure 5 shows SDS-PAGE and Western blot analysis. At 24 hpi, protein was detected for all viruses and levels were highest in the AcchiA−/cath−/p10− lanes (Fig. 5a, lanes 10, 11 and 12). The AcchiA− lanes showed signs of protein degradation (lanes 3, 4 and 5). AcchiA− continued to produce PAK5 at 48 and 72 hpi but with significant degradation (Fig. 5b, c, lanes 4, 5 and 6). At 96 hpi, the PAK5 appeared to be completely degraded (Fig. 5d, lanes 4, 5 and 6). Neither AcchiA−/cath− nor AcchiA−/cath−/p10− showed evidence of degradation, and protein levels continued to increase until 72 hpi (Fig. 5a–c, lanes 7, 8 and 9 and 10, 11 and 12, respectively). At 96 hpi, the protein appeared to decline but was still stable (Fig. 5d, lanes 7, 8 and 9 and 10, 11 and 12, respectively). Cell viability followed a similar trend as previously (Fig. 2a), where AcchiA−/cath−/p10− viability was lower than AcchiA− or AcchiA−/cath− (data not shown). However, despite displaying lower cell viability, PAK5 expression was higher for AcchiA−/cath−/p10− compared to AcchiA− and AcchiA−/cath−.

Time course of AcchiA−, AcchiA−/cath− and AcchiA−/cath−/p10− PAK5 expression. Sf9 cells were infected with AcchiA−, AcchiA−/cath− or AcchiA−/cath−/p10−. Cell lysates were prepared from equivalent numbers of cells at various times after infection, and each was assayed in triplicate. Total protein was determined and equal quantities of protein were loaded onto 12% SDS-PAGE. Western analysis was then carried out using a 6 × His-tagged antibody. a 24 hpi, b 48 hpi, c 72 hpi, d 96 hpi. Lanes 1 and 2: molecular weight marker. Lane 3: mock-infected cells. Lanes 4, 5 and 6: AcchiA−. Lanes 7, 8 and 9: AcchiA−/cath−. Lanes 10, 11 and 12: AcchiA−/cath−/p10−
Discussion
We aimed to construct a BEV that was deficient in five non-essential genes and test if this modified vector improved recombinant protein expression. To do this, we deleted a fragment containing p26, p10 and p74 from the genome of a virus already deficient in chiA and cath. The deletions were confirmed by REN analysis, PCR, SDS-PAGE and Western blot. The virus DNA was also passaged through multiple rounds of bacterial replication as described previously (Possee et al. 2008), to confirm the stability of the DNA within bacteria (data not shown). This was important for the future routine generation of virus.
Previous studies suggest that deletion of p10 may improve cellular and nuclear stability (Thomas et al. 1998) and, hence, the infected insect cells might be expected to remain more viable, providing a longer protein production window and improved post-translational modifications. In this study, AcchiA−/cath− and AcchiA−/cath−/p10−-infected cells showed reduced viability compared to BacPAK6-infected cells, and their decline in viability appeared to be reflected in a decrease in β-galactosidase activity, between 48 and 72 hpi. It has been reported that infection with baculoviruses containing different recombinant genes results in different cell death kinetics (Wu et al. 1994). Therefore, cell viability may be specific to the recombinant protein being expressed. However, we have also observed decreased cell viability for AcchiA−/cath−/p10−-infected insect cells producing a kinase and a secreted glycoprotein, compared to BacPAK6-infected cells (data not shown). This result was unexpected, but determining the role of P10 has proved to be difficult and its function(s) still remain(s) a mystery. Recent work has shown that P10 forms extensive cytoskeleton-associated or cytoskeletal-like structures in the nucleus and cytoplasm, potentially de-stabilising the cells' cytoskeleton (Carpentier et al. 2008), and possibly making it more fragile. It is unlikely though that the shear forces of the shake cultures were responsible for the decline in cell viability, as there was no difference in the quantity of cellular debris between cultures as observed by microscopy, and the cells appeared to be similarly intact. Additionally, cells infected in monolayer culture with AcchiA−/cath−/p10− or BacPAK6 also showed the same rate of decline in viability as observed for the shake cultures (data not shown). A similar reduction in cell viability was also observed for AcchiA−/cath−, suggesting that the p10 gene deletion alone was not responsible. Infected-cell viability was previously reported to be improved during expression of secreted urokinase using a p10 deletion virus compared to a wild-type virus (Thomas et al. 1998), although p26 and p74 were present in this virus. Therefore, deletion of p26 and p74 may also have an effect on cell viability. Indeed, a virus expressing the carboxy-terminal domain of P10 fused to the adjacent p26 orf (a delayed–early gene of unknown function) has been reported to induce premature cell lysis (Chaabihi et al. 1993; Goenka and Weaver 2008), although it is unknown whether this was due to the early expression of the P10 carboxy-terminal domain or interference with p26 function.
AcMNPV p10 is activated a few hours before polh (Roelvink et al. 1992) and has been demonstrated to compete with polh at a transcriptional level (Chaabihi et al. 1993). Inhibition and deletion of the p10 promoter has been shown to result in increased polh-controlled protein production (Vlak et al. 1988; Williams et al. 1989) and polh mRNA levels (Chaabihi et al. 1993). Therefore, removal of p10 should result in increased recombinant gene transcription from the polh promoter. To determine if the deletions improved recombinant protein production, we initially compared AcchiA−/cath− and AcchiA−/cath−/p10− β-galactosidase expression with BacPAK6 over time. β-galactosidase is a convenient reporter for stable intracellular expression, whose activity can be easily quantified. Despite having reduced cell viability, both AcchiA−/cath− and AcchiA−/cath−/p10− produced a much greater amount of β-galactosidase and at an earlier time point. Overall, β-galactosidase production was highest for AcchiA−/cath−/p10− and could be related to the viable cell density, where maximal protein production occurred when cell density was 80–90%. The sudden drop in specific activity at 48–72 hpi may have been due to a decrease in the rate of protein synthesis with the approach of cell death, or degradation of β-galactosidase.
We then compared expression levels of EGFP, and to generate a larger comparative data set, we constructed recombinant viruses in multi-well plates using a robotic platform. Again, the AcchiA−/cath−/p10− virus expressed EGFP to a significantly higher level than AcchiA− or AcchiA−/cath− recombinant viruses. EGFP is produced as an intracellular protein in the insect cell, but is later released into the medium as post-infection time increases, due to increasing lysis of the cells (Ho et al. 2004). For many proteins, it is critical to harvest the cells and extract the protein at the point of peak production before proteolysis starts having an effect on the protein yield. Both β-galactosidase and EGFP are very stable proteins, and so, they may not be good models for testing recombinant protein stability (Sander and Harrysson 2007). To assess if AcchiA−/cath−/p10− improved protein stability, we expressed a kinase, PAK5. AcchiA−-expressed PAK5 showed degradation from 24 hpi throughout the time course. However, no degradation was evident for the AcchiA−/cath− and AcchiA−/cath−/p10−-expressed PAK5. The degradation was likely due to the presence of cathepsin, a serine protease encoded by the virus (Gotoh et al. 2001). Later in the infection, the culture may also contain many lysed or leaking cells, liberating proteases and product into the medium, which may further degrade the recombinant proteins (Gotoh et al. 2001).
We then carried out comparative expression studies using a number of different genes. These all confirmed that the deletion virus constructed in this study gave higher expression yields than recombinant viruses that still contained p26, p10 and p74. The difference in expression levels was greater for some of the genes, compared to others, suggesting that the effects were protein-specific, although the results did not suggest which classes of proteins would be most affected.
In conclusion, deletion of p26, p10 and p74 removed a major competitor for limiting cellular resources and an unnecessary genetic burden from the recombinant virus genome. Deletion of chiA and cath has previously been shown to improve recombinant protein stability (Kaba et al. 2004), but, coupled with the removal of p26, p10 and p74, we have shown there is also an increase in cytosolic recombinant protein expression. Future studies will be directed towards testing this vector with secreted and membrane-targeted proteins.
Abbreviations
- AcMNPV:
-
Autographa californica multi-nucleopolyhedrovirus
- AP:
-
alkaline phosphatase
- BCIP:
-
5-bromo-4-chloro-3-indoyl phosphatase
- BEVS:
-
baculovirus expression vector system
- chiA:
-
chitinase A
- cath:
-
cathepsin
- dpi:
-
days post infection
- EGFP:
-
enhanced green fluorescent protein
- hpi:
-
hours post infection
- moi:
-
multiplicity of infection
- NBT:
-
nitro blue tetrazolium
- hr:
-
homologous region
- ODV:
-
occlusion derived virus
- PCR:
-
polymerase chain reaction
- pfu:
-
plaque-forming units
- PBS:
-
phosphate-buffered saline
- polh:
-
polyhedrin
- REN:
-
restriction enzyme
- RFU:
-
relative fluorescent units
- SDS-PAGE:
-
SDS-polyacrylamide gel electrophoresis
- Sf9:
-
Spodoptera frugiperda 9
References
Ayres MD, Howard SC, Kuzio J, Lopez-Ferber M, Possee RD. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology. 1994;202(2):586–605.
Carpentier DC, Griffiths CM, King LA. The baculovirus P10 protein of Autographa californica nucleopolyhedrovirus forms two distinct cytoskeletal-like structures and associates with polyhedral occlusion bodies during infection. Virology. 2008;371(2):278–91.
Chaabihi H, Ogliastro MH, Martin M, Giraud C, Devauchelle G, Cerutti M. Competition between baculovirus polyhedrin and p10 gene expression during infection of insect cells. J Virol. 1993;67(5):2664–71.
Chew NJ. Emerging technologies: transgenic therapeutics. Biopharm. 1993;6(3):24–6.
Faulkner P, Kuzio J, Williams GV, Wilson JA. Analysis of p74, a PDV envelope protein of Autographa californica nucleopolyhedrovirus required for occlusion body infectivity in vivo. J Gen Virol. 1997;78(Pt 12):3091–100.
Goenka S, Weaver RF. The p26 gene of the Autographa californica nucleopolyhedrovirus: timing of transcription, and cellular localization and dimerization of product. Virus Res. 2008;131(2):136–44.
Gotoh T, Miyazaki Y, Kikuchi K, Bentley WE. Investigation of sequential behavior of carboxyl protease and cysteine protease activities in virus-infected Sf-9 insect cell culture by inhibition assay. Appl Microbiol Biotechnol. 2001;56(5–6):742–9.
Hitchman RB, Siaterli EA, Nixon CP, King LA. Quantitative real-time PCR for rapid and accurate titration of recombinant baculovirus particles. Biotechnol Bioeng. 2007;96(4):810–4 (Mar 1).
Ho Y, Lo HR, Lee TC, Wu CP, Chao YC. Enhancement of correct protein folding in vivo by a non-lytic baculovirus. Biochem J. 2004;382(Pt 2):695–702.
Houdebine LM. Production of pharmaceutical proteins by transgenic animals. Comp Immunol Microbiol Infect Dis. 2009;32(2):107–21.
Kaba SA, Salcedo AM, Wafula PO, Vlak JM, van Oers MM. Development of a chitinase and v-cathepsin negative bacmid for improved integrity of secreted recombinant proteins. J Virol Methods. 2004;122(1):113–8.
King LA, Possee RD. The baculovirus expression system: a laboratory guide. London: Chapman and Hall; 1992.
Kitts PA, Ayres MD, Possee RD. Linearization of baculovirus DNA enhances the recovery of recombinant virus expression vectors. Nucleic Acids Res. 1990;18(19):5667–72.
Kitts PA, Possee RD. A method for producing recombinant baculovirus expression vectors at high frequency. Biotechniques. 1993;14(5):810–7.
Kuzio J, Jaques R, Faulkner P. Identification of p74, a gene essential for virulence of baculovirus occlusion bodies. Virology. 1989;173(2):759–63.
Li G, Wang J, Deng R, Wang X. Characterization of AcMNPV with a deletion of ac68 gene. Virus Genes. 2008;37(1):119–27.
Liu A, Qin JC, Rankin C, Hardin SE, Weaver RF. Nucleotide sequence of a portion of the Autographa californica nuclear polyhedrosis virus genome containing the EcoRI site-rich region (hr5) and an open reading frame just 5′ of the p10 gene. J Gen Virol. 1986;67(Pt 11):2565–70.
Luckow VA, Lee SC, Barry GF, Olins PO. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J Virol. 1993;67(8):4566–79.
Luckow VA, Summers MD. Signals important for high-level expression of foreign genes in Autographa californica nuclear polyhedrosis virus expression vectors. Virology. 1988;167(1):56–71.
Miller LK. Baculoviruses for foreign gene expression in insect cells. Biotechnology. 1988;10:457–65.
Patmanidi AL, Possee RD, King LA. Formation of P10 tubular structures during AcMNPV infection depends on the integrity of host-cell microtubules. Virology. 2003;317(2):308–20.
Possee RD, Hitchman RB, Richards KS, Mann SG, Siaterli E, Nixon CP, et al. Generation of baculovirus vectors for the high-throughput production of proteins in insect cells. Biotechnol Bioeng. 2008;101(6):1115–22.
Rankin C, Ladin BF, Weaver RF. Physical mapping of temporally regulated, overlapping transcripts in the region of the 10 K protein gene in Autographa californica nuclear polyhedrosis virus. J Virol. 1986;57(1):18–27.
Rodems SM, Friesen PD. The hr5 transcriptional enhancer stimulates early expression from the Autographa californica nuclear polyhedrosis virus genome but is not required for virus replication. J Virol. 1993;67(10):5776–85.
Roelvink PW, van Meer MM, de Kort CA, Possee RD, Hammock BD, Vlak JM. Dissimilar expression of Autographa californica multiple nucleocapsid nuclear polyhedrosis virus polyhedrin and p10 genes. J Gen Virol. 1992;73(6):1481–9.
Sambrook J, Russell DW. Molecular cloning : a laboratory manual New York. Cold Spring Harbour: Cold Spring Harbour Laboratory Press; 2001.
Sander L, Harrysson A. Using cell size kinetics to determine optimal harvest time for Spodoptera frugiperda and Trichoplusia ni BTI-TN-5B1–4 cells infected with a baculovirus expression vector system expressing enhanced green fluorescent protein. Cytotechnology. 2007;54(1):35–48.
Thomas CJ, Brown HL, Hawes CR, Lee BY, Min MK, King LA, et al. Localization of a baculovirus-induced chitinase in the insect cell endoplasmic reticulum. J Virol. 1998;72(12):10207–12.
Vlak JM, Klinkenberg FA, Zaal KJ, Usmany M, Klinge-Roode EC, Geervliet JB, et al. Functional studies on the p10 gene of Autographa californica nuclear polyhedrosis virus using a recombinant expressing a p10-beta-galactosidase fusion gene. J Gen Virol. 1988;69(Pt 4):765–76.
Wang Y, Wu W, Li Z, Yuan M, Feng G, Yu Q, et al. ac18 is not essential for the propagation of Autographa californica multiple nucleopolyhedrovirus. Virology. 2007;367(1):71–81.
Williams GV, Rohel DZ, Kuzio J, Faulkner P. A cytopathological investigation of Autographa californica nuclear polyhedrosis virus p10 gene function using insertion/deletion mutants. J Gen Virol. 1989;70(Pt 1):187–202.
Wu SC, Jarvis DL, Dale BE, Liao JC. Heterologous protein expression affects the death kinetics of baculovirus-infected insect cell cultures: a quantitative study by use of n-target theory. Biotechnol Prog. 1994;10(1):55–9.
Acknowledgments
The Oxford Protein Production Facility is supported by the MRC and BBSRC.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hitchman, R.B., Possee, R.D., Crombie, A.T. et al. Genetic modification of a baculovirus vector for increased expression in insect cells. Cell Biol Toxicol 26, 57–68 (2010). https://doi.org/10.1007/s10565-009-9133-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-009-9133-y