
Abstract
Catalytic wet air oxidation (CWAO) is a promising process for removing organic compounds from industrial aqueous streams. Over the last three decades a significant amount of research has been conducted on various aspects of the process including catalyst development, catalytic reaction mechanisms and oxidation reaction mechanisms, and the effects of important parameters such as temperature, pressure and pH on total organic carbon (TOC) removal. The Industrial Chemistry Group at RMIT University has conducted a significant amount of research on this process over the last several years focussing on organics removal from three very different industrial aqueous streams: Bayer liquor used in alumina refining; stripped sour water used in oil shale refining; and pulp and paper effluent used in paper manufacturing. Important results obtained on each of the aforementioned industrial aqueous streams are discussed in detail in this survey article.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The Industrial Chemistry Group at RMIT University has conducted a considerable amount of research on CWAO over the past several years, including three completed PhD theses [1–3]. The work has focussed predominantly on industrial waste stream treatment in close collaboration with the industrial partners Alcoa World Alumina for Bayer liquors, Southern Pacific Petroleum for stripped sour water (SSW) and CSIRO for paper and pulp effluent (PPE). The aim of the research was to obtain a thorough understanding of the processes and in particular critical process operating parameters such as pH, temperature and residence time for maximal organics removal. This understanding is necessary for developing industrially applicable waste stream treatment processes.
Organic pollutants must be removed from industrial aqueous streams, because of:
-
A lack of potable water and hence the need to recycle water
-
Discharging a polluted stream has significant detrimental effects on the environment
-
Organic or inorganic pollutants in an industrial aqueous stream may also negatively affect process productivity
Numerous approaches have been used to remove organic pollutants from industrial aqueous streams. The suitability of a process or method for treating a particular waste stream is highly dependent on factors such as type(s) and concentration of pollutants, pH, temperature, level of toxicity and volume of waste stream requiring treatment. Table 1 gives a summary of selected organics removal processes and their applicability.
Catalytic wet air oxidation (CWAO)—removal of organic pollutants in the liquid phase via complete oxidation using an oxidising gas such as oxygen or air in the presence of a catalyst—has received considerable interest over the last three decades [4]. CWAO has significant advantages over the more conventional processes incineration and biological treatment:
-
Reduced energy consumption compared to incineration due to the use of lower operating temperatures
-
Reduced gaseous emissions
-
Treatment of toxic streams that cannot be treated by biological processes
-
Low footprint requirements for CWAO compared to biological processes
Compared to other organics removal processes such as filtration and adsorption, CWAO produces no liquid or solid waste provided 100% organics oxidation is achieved and the catalyst is stable and/or can be easily removed.
The strong commercial potential of CWAO is reflected in a large volume of R&D studies on both fundamental and applied aspects of the process [5–14]. Catalyst development and catalytic mechanisms have received a considerable amount of attention but the discovery/development of suitable catalysts for use in industrial aqueous streams has proven difficult to date, and this is reflected by the low number of commercial CWAO units in operation as compared to non-catalysed WAO units [15].
Elucidation of catalytic reaction mechanisms and reaction sequences that take place during CWAO of industrial aqueous streams has proved an extremely difficult task as the streams can contain thousands or in some cases millions of different organic compounds. Similar to any other catalytic system, mechanistic studies are critical to the development of improved WAO catalysts. Thus numerous CWAO studies have been conducted using “synthetic” solutions containing only one or only a few representative organic compounds [10, 13, 16–22]. The model compounds are selected because:
-
They enable more detailed study of the role of the catalyst
-
They allow for studies on the effects of specific reaction conditions or species (in complex solutions it is usually very difficult to investigate the effects due solely to certain conditions or species)
Catalyst development for CWAO including catalyst preparation, characterisation, stability studies and catalyst re-usability has been studied extensively. Catalysts for operation under harsh conditions in industrial waste streams need to be robust but also cheap. A number of studies have confirmed the strong influence of catalyst preparation methods on catalytic activity and stability. Widely used methods for preparation of WAO catalysts include co-precipitation, sol–gel and incipient wetness impregnation, however, the field has recently been reviewed in detail [4] and hence will not be discussed in length here. In this survey we primarily present and discuss results from the research conducted by the RMIT Group on three different industrial aqueous streams; Bayer liquor, stripped sour water, and pulp and paper effluent.
2 Experimental
The materials and methods used for the majority of the experimental results discussed in this paper have been described in detail elsewhere (see references for different industrial streams below). Materials and methods that have not been given elsewhere are described in detail below.
2.1 Bayer Liquor and Synthetic Bayer Liquor Studies
2.1.1 Materials
The Bayer liquor used was provided by an Alcoa alumina refinery in Western Australia. The following chemicals were used to prepare synthetic Bayer liquors: NaOH, Al(OH)3, NaCl, Na2SO4, Na3PO4 and Si(OH)4 using a method given elsewhere [10].
2.1.2 Catalyst Preparation
The single metal heterogeneous catalysts were prepared by precipitation of appropriate metal salts by sodium hydroxide or other basic solutions while the bi-metal catalysts were prepared by co-precipitation method (co-precipitation of two metal salt mixtures). After precipitation, the catalysts were washed thoroughly, dried at room temperature and heat treated at 723 K for 12 h. The homogeneous catalysts were prepared by dissolving appropriate metal salt in water (dilute solution, pH>7.5) and were utilized as homogeneous catalysts.
2.1.3 CWAO Test Procedure
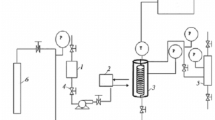
The procedure used for Bayer liquor CWAO experiments was as follows: 600 mL of Bayer liquor and solid catalyst were added to a 1.2 L inconel autoclave vessel (Parr Instruments IL.) which contained fittings for gas addition to the liquid phase and on-line liquid sampling. The vessel and contents were then pre-heated to the desired reaction temperature while stirred at 1,000 rpm. Upon reaching the desired reaction temperature oxygen was added to the system and maintained manually at a constant pressure throughout the test.
Methods used for total organic carbon (TOC) analysis and GC analysis of reaction products are given elsewhere [20]. TOC analysis was performed using an OI Analytical 1010 total carbon analyser which involves oxidation of samples using persulphate combined with UV light and detection of CO2 using an IR detector.
Details on the methods that were used for synthetic Bayer Liquor studies are given elsewhere [10, 21, 22]. A similar procedure to the Bayer liquor autoclave tests was used for synthetic Bayer liquor tests.
2.2 Stripped Sour Water Studies
Details on the materials and methods that were used for SSW studies are given elsewhere [23, 24]. The SSW used was obtained from an oil shale refinery in Gladstone, Queensland. SSW autoclave tests were conducted using a similar procedure to the Bayer liquor tests. TOC analysis was performed using an OI Analytical 1010 total carbon analyser.
2.3 Pulp and Paper (Ferulic Acid) Studies
2.3.1 Materials
The following chemicals were used as purchased: ferulic acid, (Sigma Aldrich) (>98% fulka), Orthophosphoric acid 85% Ajax Finechem, sodium peroxodisulfate purchased from Merck. MilliQ water was used for all prepared solutions. Catalysts used in these experiments were copper(II) chloride 2-hydrate, Iron(II) chloride 4-hydrate, Mangenese(II) chloride 2-hydrate, Cerium(III) chloride 7-hydrate, Cobalt(II) choride 6-hydrate, Magnesium(II) 6-hydrate, and Bismuth(III) chloride (all from BDH Chemicals Ltd).
2.3.2 Catalyst Preparation
The catalysts used in ferulic acid tests were prepared using the same procedures as outlined in the section on catalyst preparation for Bayer liquor studies.
2.3.3 Methods
The WAO/CWAO test procedure that was used is described in detail elsewhere [25]. A similar method as that outlined for the Bayer liquor studies was used. TOC analysis was performed using an OI Analytical 1010 total carbon analyser. FTIR spectra were obtained using a Perkin Elmer 1725X instrument.
3 Bayer Liquor
Bayer liquor, a highly alkaline aqueous stream which is used in the refining of alumina, contains a number of organic contaminants. The concentration of organic contaminants in Bayer liquor, which enter the process via the bauxite ore and additives used in the Bayer process, increases over time due to the cyclic nature of the Bayer process until a steady state level is reached. Organic contaminants hinder the productivity of the Bayer process in a number of ways [26, 27]. The two main detrimental effects of organic contaminants are a reduction in aluminium hydroxide yield due to poisoning of crystal growth sites, and a reduction in available sodium hydroxide which is required to digest the aluminium bearing compounds in the bauxite ore.
There are a few processes that are currently used for removing organic compounds from Bayer liquor. These processes include liquor burning, sodium oxalate removal via seeding/precipitation and sodium oxalate removal via co-precipitation with aluminium hydroxide. Although the aforementioned processes have been successfully implemented they do have some significant drawbacks, hence there is considerable interest in the development of alternative processes for removing organic compounds from Bayer liquor. CWAO has been considered by a number of alumina/aluminium companies [28] as a potential organics removal process. Alcoa World Alumina recently applied for a patent for a CWAO process for treating Bayer liquor [29].
3.1 CWAO Studies on Bayer Liquor
We have investigated low temperature CWAO of Bayer liquor, a complex aqueous solution that contains numerous organic and inorganic compounds (Table 2), using a variety of catalysts (Table 3). The Bayer liquor that was studied contained more than 30 g per litre of small and high molecular weight organic compounds.
Table 3 shows that the un-catalysed wet oxidation of Bayer liquor reduces the TOC content by 12%, while using catalysts the TOC removal is enhanced. The effective TOC removal just by using copper catalyst is enhanced by 110% while with the manganese catalyst, the TOC removal was enhanced by 25%. Interestingly, as compared with the TOC removal by the wet oxidation process, using bimetallic catalyst (the combination of copper and manganese) enhances the TOC removal up to 174%. The changes in concentrations of three Bayer organics from un-catalysed WO and CuO catalysed WO is given in Table 4. As can be seen in Table 4 CWAO of Bayer liquor leads to a significant increase in the sodium oxalate concentration. This is due to sodium oxalate being highly stable towards CWAO because of its high resonance stability.
CWO trials have shown that the Cu:Mn system is the most effective catalyst for TOC reduction (up to 33%) at lower temperature and the most stable to Bayer process conditions. The use of CWO process in Bayer liquor easily reduces high molecular weight organic compounds but generates low molecular weight organics (which are quite refractory in nature) proportionally.
Copper based catalysts have shown considerable promise in the low temperature oxidative reduction of Bayer organic species. Investigations into copper based catalytic wet oxidation system suggest that copper complexation plays a key role in the catalysis mechanism.
The influence of homogeneous catalysts of the removal of organics from Bayer liquor was investigated and the results of homogenous catalytic system are presented in the Table 5.
From the CWO results obtained by using bimetallic homogenous catalyst system it is observed that the removal of organics from Bayer liquor is higher than the bimetallic heterogeneous catalyst system. The removal of organics is approximately 8% higher in the bimetallic homogeneous catalytic system with major advantage of having smaller amount of catalyst in the reaction system.
The changes in the TOC with the reaction period were investigated during the CWO using heterogeneous and homogenous catalysts. The results of TOC changes with the reaction period are presented in Figs. 1 and 2.

TOC removal for various transition metal based catalysts. Conditions: T = 165 °C, oxygen pressure 500 kPa, t = 210 min, [catalyst] = 10 g/L (added as solid)

TOC removal for homogenous catalytic systems. Conditions: T = 165 °C, oxygen pressure 500 kPa, t = 210 min, [catalyst] = 800 mg/L
The results of the CWO experiments using heterogenous catalysts show that in case of the wet oxidation process (without catalyst) the reduction of TOC is 50% at the reaction period of 60 min with respect to the reaction period of 210 min, while using the single metal (Cu) catalyst results into the removal of 75% organics at the reaction time interval of 60 min and the bimetallic (Cu/Mn) reaction system results into the removal of more than 93% at the reaction period of 60 min.
The results of the CWO experiments for homogenous catalytic system show that the reduction of TOC at the reaction period of 60 min is higher than the wet oxidation process and the removal of TOC by the bimetallic homogenous catalyst is higher that the single metal homogenous catalyst at the same reaction time. The CWO (both heterogeneous and homogenous) results indicate that at the reaction period of 60 min majority of organics are destroyed during the CWO process.
3.2 CWAO Studies on Synthetic Bayer Liquor
CuO catalysed WO of a number of Bayer organics was studied in synthetic Bayer liquor (Table 6) to determine the reaction mechanism(s) occurring under Bayer like (highly alkaline) conditions.
One aspect of the catalytic mechanism investigated was the predominant phase of CuO responsible for the activity observed in Bayer liquor, as CuO is slightly soluble under Bayer like conditions and hence could act as both a homogenous and heterogenous catalyst. This was investigated by studying the effect of catalyst loading (above and below the solubility limit) on CuO catalysed WO of three different classes of organic compounds which are found in Bayer liquor.
The effect of CuO loading on CWO was studied for mixtures of monocarboxylates (sodium formate, acetate, priopionate and butyrate), dicarboxylates (sodium oxalate, malonate, succinate and glutarate) and hydroxycarboxylates (sodium lactate, malate, tartrate and citrate). The results for the monocarboxylates and dicarboxylates tests are shown in Figs. 3 and 4 respectively. The results for the monocarboxylates are not shown as the overall oxidation observed was too low (<2%) over the concentration range. From the results presented in Figs. 3 and 4 it can be seen that the extent of oxidation for the dicarboxylates and hydroxycarboxylates mixtures increases with increasing catalyst loading up until the solubility limit of CuO for each of the systems (The solubility limit of CuO for each system is different due to the formation of different copper–organic complexes which influence copper solubility). At CuO concentrations beyond the solubility limit no noticeable increase in oxidation is observed in both systems, indicating that soluble copper is predominantly responsible for the observed activity. In highly alkaline solution soluble copper can be represented by the following general molecular formula, [Cux(OH)y]2x−y.

Effect of CuO loading on WO of dicarboxylic acids. Conditions: T = 165 °C, oxygen partial pressure = 500 kPa, t = 2 h, [TOC] = 7.5 g/L (equal concentrations of each organic compound). *(% oxidation malonic + % oxidation succinic + % oxidation glutaric)/3. Sodium oxalate concentration increased in all tests, hence if any “original” sodium oxalate was oxidised it was not possible to determine the amount. Copper solubility calculated using average of two highest readings obtained in loading tests

Effect of CuO loading on extent of oxidation for mixture of hydroxy carboxylic acids. Conditions; T = 165 °C, oxygen partial pressure = 500 kPa, t = 2 h, [TOC] = 7.5 g/L (equal concentrations of each organic compound) [22]. * (% oxidation citrate + % oxidation malate + % oxidation lactate + % oxidation tartrate)/4 Copper solubility calculated using average of two highest readings obtained during loading tests
The types of chemical reactions taking place were identified. In order to do this it was necessary to investigate Cu catalysed WO (i.e. soluble Cu catalysed WO) of the compounds used in the previous tests individually. The results of these tests are given in Table 7. Cu catalysed WO of the monocarboxylates was not studied as no appreciable oxidation was observed in the loading tests for the mixture of this class of organic compounds.
The results of the individual tests for the dicarboxylates revealed that only sodium malonate underwent Cu catalysed WAO. This was in contrast to results obtained in the mixture tests where sodium succinate and sodium glutarate underwent appreciable oxidation (22.7% and 34.8% removal respectively) using a CuO concentration of 1.0 g/L). The negligible oxidation result for sodium succinate when tested individually was in contrast to results obtained for sodium succinate in Bayer liquor, where appreciable oxidation was observed to occur (Table 4).
Based on the contrasting results obtained for the oxidation of sodium succinate and sodium glutarate these compounds must have been co-oxidised by sodium malonate in the mixture test, indicating that free radical intermediates are produced during Cu catalysed WO of sodium malonate. To confirm the aforementioned interaction the effect of sodium malonate on sodium succinate oxidation was investigated using different sodium malonate to succinate ratios (Fig. 5). From the results presented in Fig. 5 it can be seen that co-oxidation of sodium succinate by sodium malonate increased as the malonate to succinate ratio was increased from 1.3 to 5, while a decrease in co-oxidation of succinate occurred when the ratio was increased further to 11.9. This decrease was most likely due to a combination of:
-
(1)
increasing the initial malonate concentration does not increase proportionally the amount of malonate free radical intermediates produced over the malonate concentration range studied and
-
(2)
malonate free radical intermediates are able to react with unreacted malonate (i.e. an auto-oxidation mechanism occurs) and hence compete with succinate.

Effect of initial molar ratio of sodium malonate: sodium succinate on succinate removal. Conditions: T = 165 °C, oxygen partial pressure = 500 kPa, t = 2 h, [CuO] = 5 g/L, [sodium succinate] = 1.875 g OC/L in all tests [20]
Based on our earlier studies which identified that uncatalysed WO of sodium malonate increased significantly with increasing NaOH concentration, and the proposed mechanism for this reaction which suggested that at high alkalinity more free radical intermediates are formed from malonate via reaction with hydroxide instead of via propagation reactions [2], it was decided to investigate the effect of NaOH concentration on malonate induced co-oxidation of succinate. The results are presented in Fig. 6.

Effect of NaOH concentration on Cu catalysed malonate co-oxidation of sodium succinate. Conditions: T = 165 °C, oxygen partial pressure = 500 kPa, t = 2 h, [CuO] = 5 g/L, [sodium succinate] = 1.875 g OC/L in all tests. Initial malonate to succinate ratio = 5:1 in all tests
Figure 6 shows that increasing co-oxidation of sodium succinate occurs with increasing NaOH concentration. This supports the base catalysed mechanism proposed, which involves formation of more malonate free radical intermediates at high alkalinity as the reason for increased oxidation and not an increase in propagating reactions. The increased co-oxidation of succinate that was observed with increasing NaOH concentration could also have been due to increased soluble Cu.
The ability of malonate to co-oxidise some of the other organic compounds studied that did not undergo significant oxidation alone (acetate, propionate and butyrate) was investigated and the tests summarised in Table 8. Significant co-oxidation of all three compounds occurred in the presence of the Cu catalyst.
Detailed reaction/catalytic mechanisms for uncatalysed and Cu catalysed WO of malonate and malonate induced co-oxidation are presented in Tardio et al. [10] and [21].
The reaction/catalytic mechanisms occurring during Cu catalysed WO of the hydrocarboxylates listed in Table 7 were also investigated. Analysis of the soluble copper data for the individual tests showed a reasonable correlation between the initial concentration of soluble copper and the total oxidation hence tests were conducted using a higher initial organic concentration to investigate more closely the relationship between soluble copper (extent of copper–organic complexation) and the extent of oxidation. The results are presented in Figs. 7 and 8.

Soluble copper concentration during CWAO of various hydroxy carboxylates. Conditions: T = 165 °C, oxygen partial pressure = 500 kPa, t = 2 h, [CuO] = 5 g/L, [organic] = 1.875 g OC/L [22]

Relationship between soluble copper and catalytic activity for various hydroxy carboxylates. Conditions: T = 165 °C, oxygen partial pressure = 500 kPa, t = 2 h, [CuO] = 5 g/L, [organic] = 1.875 g OC/L in all tests [22]
Tests were conducted to determine if free radical reactions were occurring during Cu catalysed WO of sodium citrate, lactate, malate and tartrate. The co-oxidation method used to identify the free radical mechanism for Cu catalysed WAO of malonate was used to study Cu catalysed WAO of citrate, lactate, malate and tartrate. The results are presented in Table 9 which show that all of the hydroxycarboxylates were capable of co-oxidising succinate. However the extent of co-oxidation was quite low compared to that achieved by sodium malonate using very similar conditions. Based on the low extent of co-oxidation achieved using each of the hydroxycarboxylates it is unlikely that the predominant reaction mechanism for these compounds is free radical based. A complexation type mechanism that does not involve a high quantity of free radical reactions is most likely the predominant mechanism that occurs during Cu catalysed WAO of sodium citrate, lactate, malate and tartrate in highly alkaline solution. Further details of this mechanism are given in Tardio et al. [22].
4 CWAO Studies on Stripped Sour Water
Recent rising oil prices have led to increased interest in alternative sources of crude oil/petroleum products. One potential source of crude oil/petroleum products is oil shale. Oil shale is a sedimentary rock which contains a mixture of silica, clay minerals and organic matter (kerogen) which can be converted into crude oil. The sustainability of the oil shale refining industry depends greatly on the environmental impacts of the refining process. The oil shale refining process that was used in Gladstone, Australia (the Alberta Taciuk Process) produced an aqueous wastewater at a rate of approximately 20 m3/kTonne of product/hour, which had a high concentration of phenols, pyrroles, organic acids and a host of other inorganic and organic compounds that are ill-defined. This wastewater is steam stripped to remove ammonia and hydrogen sulphide after which the water is known as Stripped Sour Water (SSW). SSW has a total organic carbon (TOC) loading of ∼9 g/L and a chemical oxygen demand (COD) of 18 g/L and requires treatment prior to discharge. A selection of compounds present in SSW are listed in Table 10.
The removal of organics from SSW by CWAO was investigated using a number of mono-metallic and bi-metallic homogenous catalysts (Table 11) [23]. For the mono-metallic catalysts optimum TOC removal was achieved with Cu salts using an initial pH of 3.5, while Fe(NO3)2 was the most active mono-metallic catalyst when higher initial solution pH (6 and 13.5) was used. The high activity of the Cu based catalysts is consistent with other studies that have also shown homogenous Cu based catalysts are more active in CWAO than other homogenous transition metal salt catalysts [16, 17]. The higher activity of the Cu salts in acidic compared to neutral—basic conditions is most likely due to the low solubility of the Cu salts investigated at higher pH, due to precipitation of Cu(OH)2 which is readily converted to CuO under the reaction conditions (temperature) used. The higher activity of Fe(NO3)2 at pHi 6 compared to pHi 3.5 is most likely due to FeII being the preferred oxidation state at pH 3.5 compared to FeIII at pH > 4, while the lower activity of the Fe catalyst at pH 13 compared to pH 6 is most likely due to the reduced solubility of Fe at higher pH.
Of the bi-metallic catalysts studied, Cu/Pd (1:1) was the most active with ∼73% TOC removal being achieved with this catalyst under the reaction conditions used. The slightly improved TOC removal achieved with this catalyst would however not justify its use on a commercial scale due to the high price of Pd.
Based on the high activity of the Cu based salts, further studies were conducted on Cu(NO3)2 catalysed WO of SSW to gain some insight into the mechanism(s) of this catalyst.
Various tests were conducted to investigate the mechanism of the Cu(NO3)2 catalyst. The effect of a promoter (hydroquinone), inhibitor (t-butanol) and the reaction vessel surface material on Cu(NO3)2 catalysed WO of SSW was investigated. The results of these tests are presented in Fig. 9. TOC removal was slightly higher when using a glass vessel (compared to stainless steel), while a slight decrease in TOC removal occurred when an inhibitor was added to the system. These results indicate that free radical reactions are involved in Cu(NO3)2 catalysed WO of SSW. The effect of the inhibitor and promoter was however quite low, indicating that oxidation via free radicals is a minor mechanism for this system or the chosen promoter and inhibitor were not highly suitable compounds. Based on the improved TOC removal obtained using the glass vessel it was decided to use the glass vessel for future tests.

Effect of hydroquinone, t-butanol and reaction vessel surface on CWO of SSW. Conditions: T = 200 °C, t = 3 h, pO2 = 500 kPa, pHi = 3, [Cu(NO3)2] = 33.33 mmol/L. *Glass insert was placed inside SS autoclave reaction vessel
The effect of Cu(NO3)2 loading on CWO of SSW is presented in Fig. 10. From the data presented in Fig. 10 it can be seen that TOC removal increased with increasing Cu(NO3)2 concentration from 0 mmol/L to 67 mmol/L before plateauing at concentrations ≥67 mmol/L. The plateau in TOC removal at ∼75% is most likely due to either SSW containing organic compounds that are not catalytically wet oxidised under the conditions used and/or organic compounds formed from partial oxidation of the compounds present in SSW are not catalytically wet oxidised under the conditions used. Due to the high number of different organic compounds present in SSW, and the most likely high number of organics which would be present in CWO treated SSW, no attempt was made to determine the nature of the organics remaining in SSW. Based on the results of numerous CWO studies these compounds are most likely predominantly low molecular weight acids such as acetic and propionic acids.

Effect of Cu(NO3)2 loading on CWO of SSW. Conditions: T = 200 °C, t = 3 h, pO2 = 0.5 MPa, pHi = 3
A close examination of the data from the Cu(NO3)2 loading tests showed that significant TOC removal occurred during the heat-up period (Fig. 11), despite the lack of oxygen during this period. A further test was therefore conducted in the absence of oxygen to determine the maximum amount of TOC that could be removed from SSW using only Cu(NO3)2 and heat. The maximum amount of TOC that could be removed was ∼31% in ∼1 h in the presence of Cu(NO3)2 as opposed to ∼10% without added Cu(NO3)2 (Fig. 12). Hence ∼21% of the TOC in SSW was removed due to the presence of Cu(NO3)2, indicating that copper and/or nitrate are capable of direct oxidation of a significant amount of the TOC in SSW. Imamura et al. [30] have reported that Cu(NO3)2 can oxidise acetic acid in the absence of oxygen, and that the extent of oxidation increases with increasing Cu(NO3)2 concentration. Imamura et al. [30] attributed this to oxidation by nitrate ion, and reported no change in copper oxidation state during the reaction. The mechanism suggested by Imamura et al. [30] for Cu(NO3)2 induced oxidation however did not occur or was only a minor mechanism in Cu(NO3)2 induced oxidation of SSW based on visual observations of the colour of SSW before treatment (brown), immediately after treatment (yellow) and ∼1 h after treatment when left in air (green). The aforementioned colour changes strongly support a change in copper oxidation state from II to I during the reaction, and back to II when the solution was exposed to air. A test was also conducted using a CuO catalyst under the same reaction conditions which gave almost identical TOC removal as the Cu(NO3)2 system indicating that nitrate did not have a significant impact on TOC removal or that CuO and Cu(NO3)2 exhibit very different reaction mechanisms and the activity due to Cu in Cu(NO3)2 is significantly less than that for CuO. A possible mechanism that would account for the significant TOC losses that occurred due to direct oxidation of SSW organics by Cu(NO3)2 in the absence of oxygen is copper catalysed decarboxylation. Copper is a well known catalyst for decarboxylation reactions of compounds such as keto acids and aliphatic carboxylic acids [31, 32].

Effect of increasing Cu(NO3)2 loading on TOC removal in the absence of oxygen. Conditions: Final T = 200 °C, t = ∼30 min, pO2 = 0.5 MPa, pHi = 3

TOC removal with and without Cu(NO3)2 with no added oxygen. Conditions: T = 200 °C, pHi = 3, [Cu(NO3)2] = 67 mmol/L
The effect of reaction temperature on Cu(NO3)2 catalysed WO of SSW (Fig. 13) was investigated to determine if TOC removal could be modelled using empirical rate laws. Attempts to model TOC removal versus time for the temperatures studied using 1st, 2nd and 3rd order kinetics however revealed that these kinetic models could not be used to accurately represent the data obtained. Data in two distinct time regions was however able to be accurately represented by 1st order kinetics as shown in Fig. 14. The two distinct regions observed can be explained by the different types of organic compounds present in SSW and the types of organic compounds that form due to partial oxidation. During the initial fast region, a number of easily oxidised compounds and/or intermediates formed from partial oxidation are rapidly oxidised directly to carbon dioxide and water. In the second slower region the more recalcitrant compounds (those present initially in the SSW and those formed during the fast rate region) undergo oxidation.

Effect of temperature on CWAO of SSW. Conditions: PO2 = 500 kPa, [Cu(NO3)2] = 33.33 mmol/L, pHi = 3.5

Fast and slow rates of TOC removal during CWO of SSW at different temperatures. Conditions: PO2 = 500 kPa, [Cu(NO3)2] = 33.33 mmol/L
Based on the results obtained from the studies on Cu(NO3)2 catalysed WO of SSW, the two reaction mechanisms that are responsible for mineralisation of organics in SSW in the absence of oxygen are: (1) thermal oxidative degradation, (2) direct oxidation by copper (copper-induced oxidative degradation via for example Cu(II) catalysed decarboxylation).
Under CWAO conditions (oxidising atmosphere), a number of different reactions are possible during Cu(II) nitrate catalysed WO of SSW (Reaction Scheme 1) including: (1) oxidation of organics by a copper–oxygen species; (2) formation of copper–organic complexes and subsequent oxidation of these complexes by an active oxygen species; (3) copper-catalysed decomposition of hydroperoxides (which may form from uncatalysed or catalysed WO) and subsequent reaction of the hydroperoxyl radical formed with unreacted organic compounds (i.e. a free radical chain reaction mechanism) and (4) uncatalysed WO.

Proposed un-catalysed and catalysed reaction pathways during CWAO of SSW in the presence of Cu(II)
As SSW contains numerous different types of organic compounds it is not possible to study in detail the reaction mechanisms occurring during copper-catalysed WO of this solution, but the results of this study indicate that at least two or more of the mechanisms proposed occur during copper-catalysed WO of SSW. A summary of the aforementioned proposed pathways/mechanisms is given in Reaction Scheme 1.
The removal of copper from CWAO treated SSW was investigated as this would be required before the treated solution could be discharged. Removal was investigated using activated carbon. The treated SSW was treated for 2 h between pH 3 and pH 5 with the results for copper adsorption shown in Fig. 15. Cu adsorption increased with increasing pH over the pH range studied. The maximum copper removal achievable was approximately 64% at pH 4.8. As this quantity was below that required to reduce the soluble copper in treated SSW to an acceptable level, precipitation (and subsequent filtration) of copper (as Cu(OH)2) prior to treatment with activated carbon was attempted. Precipitation/filtration removed 94% of the copper and further treatment of the filtrate with activated carbon improved the removal to 99.8%.

Effect of pH on removal of Cu2+ from CWO treated SSW
A secondary significant benefit that was observed after treatment with activated carbon was the removal of odour from all samples. The TOC results after treatment with activated carbon are presented in Fig. 16. It was found that significant organic adsorption had occurred, corresponding with the loss of odour, with the carbon improving overall %TOC reductions by up to 21%.

Contribution of treatment with activated carbon to total TOC removal. Conditions (CWAO): T = 200 °C, oxygen partial pressure = 500 kPa, t = 3 h, [Cu(NO3)2] = 33.33 mmol/L. Adsorption conditions: pH 4.7, t = 2 h, [C] = 25 g/L, T = 25 °C
5 CWAO of Pulp and Paper Effluent
Pulp and paper mills produce wastewater from various steps, such as wood preparation, pulping, pulp washing, screening, bleaching, paper making and coating operations. All of these steps generate different grades of wastewater. Details about the type and strengths of pollutants produced from each of these steps are available in the literature [33]. The effluents from pulp and paper mills are highly toxic to marine life [34] and have an objectionable brown colour due to phenolic compounds formed from lignin degradation [35]. In general, pulp and paper mill effluents contain toxic organics which are too dilute to recover for commercial purposes or to incinerate, and too concentrated to treat with biological treatments [36, 37].
High temperature wet oxidation is a proven technology to treat pulp and paper mill effluents. At higher temperature (250–380 °C) and higher pressure (2–13.6 MPa), more than 90% reduction in Chemical Oxygen Demand (COD) of pulp and paper mills effluents has been reported [38, 39]. These extreme conditions are often complicated by severe technical difficulties as well as increased capital costs. The extreme conditions can however be reduced through the use of an appropriate catalyst. This has led to a significant amount of research on the development of WO catalysts.
Lignin, a complex polymer made up of coniferyl, sinapyl and p-coumaryl alcohol, is the main source of organics in pulp and paper mill effluents. In this section, the CWO of ferulic acid (a lignin model compound) is discussed. Ferulic acid has previously been studied as a model compound for lignin [40], while bio-synthesis of lignin from ferulic acid has been investigated elsewhere [41].
Catalytic wet oxidation of ferulic acid was investigated using several homogenous catalysts (chloride salts of Cu+2, Fe+2, Ce+3, Co+2, Mg+2, Bi+2, Mn+2, Ni+2 and Zn+2) at various concentrations (Fig. 17). Cu+2, Fe+2 and Mn+2 were found to be the most effective catalysts, removing more than 86%, 73% and 69% TOC respectively, at 100 °C after 120 min reaction for 0.1 mole/L catalyst concentration. The order of catalytic activity observed was Cu+2 > Fe+2 > Mn+2 > Ce+2 > Bi+2 > Co+2 > Zn+2 > Mg+2 > Ni+2. Ni+2 ions displayed the lowest catalytic activity, removing 26% of TOC at 100 °C after 120 min of reaction using 0.1 mole/L catalyst concentration, which was still considerably higher than the 3% TOC removal that occurred for the blank test.

Effect of catalyst loading on ferulic acid TOC removal using transition metal catalysts. Conditions: T = 100 °C, t = 2 h, 180 kPa oxygen partial pressure, [ferulic acid] = 1,000 ppm
Based on the high TOC removal achieved using Cu, further studies were conducted on:
-
(1)
The development of stable copper based heterogeneous catalysts and
-
(2)
Understanding catalytic mechanisms in copper catalysed WO of ferulic acid.
Several different copper based heterogenous catalysts were prepared using the methods described in Sect. 2 and their efficacy at catalysing WO of ferulic acid was investigated (Table 12). Cu–Ni–Ce/Al2O3 was the most effective catalyst tested with this catalyst being capable of removing 81% TOC using the following conditions: 100 °C, 300 kPa total pressure, 2 h, 800 rpm stirring speed, 200 mg/L catalyst loading. Based on the high activity of the Cu–Ni–Ce/Al2O3 catalyst it was decided to conduct further studies on this catalyst to determine its optimum loading and to gain an understanding of the catalytic mechanism.
A series of experiments were carried out to study the effect of catalyst loading on Cu–Ni–Ce/Al2O3 catalysed WO of ferulic acid at 100 °C (Fig. 18). It was observed that the use of even 100 mg/L could improve TOC removal significantly (by about 68%) as compared to the non-catalysed run (∼2% TOC removal). Catalyst performance steadily increased up to 600 mg/L loading, which gave 92% of TOC removal. A catalyst loading of 200 mg/L was chosen for further studies of the catalytic mechanism occurring in this system.

Effect of catalyst loading on TOC removal. Conditions: T = 100 °C, t = 2 h, 180 kPa oxygen partial pressure, [ferulic acid] = 1,000 ppm
The first aspect of the mechanism investigated was the proportion of activity due to the leached active components of the catalyst (Table 13). From the results presented in Fig. 19 it can be seen that the majority of catalytic activity of Cu–Ni–Ce–Al2O3 is due to the undissolved (heterogenous) portion of the catalyst. The leached component may have contributed up to ∼20% TOC removal based on the test conducted using the same amount of catalyst active components in homogenous form, and hence contributed up to ∼25% of the total TOC removal obtained using this catalyst (∼80%).

Effectiveness of homogeneous catalyst, heterogeneous catalyst and leached component for Cu–Ni–Ce/Al2O3 catalyst. Conditions: T = 100 °C, t = 2 h, 180 kPa oxygen partial pressure, [ferulic acid] = 1,000 ppm, [Cu–Ni–Ce/Al2O3] = 2,000 ppm
The next aspect of the reaction mechanism investigated was the possibility of TOC removal, via adsorption and/or catalytic oxidation via the catalyst’s lattice oxygen. This was thought to be a possibility based partly on the high TOC removals observed at time zero (prior to addition of oxygen). WO/CWO of ferulic acid at 100 °C and 150 °C is shown in Figs. 20 and 21 respectively. The TOC reduction after 2 h of CWO at 100 and 150 °C was 81% and 95% respectively. The time zero TOC removal at 100 and 150 °C was 42% and 60% respectively. The TOC reduction at time zero (in the absence of oxygen) at 150 °C is higher than 100 °C. If the TOC reduction in the absence of oxygen is assumed to be just due to adsorption, then it would be expected that this would decrease with increasing temperature. However, the opposite trend was observed. The increase in TOC reduction at time zero with increasing temperature may have been due to the following:
-
(1)
TOC removal occurred due to direct oxidation of organics using the catalyst’s lattice oxygen and/or catalytic decarboxylation of ferulic acid or intermediate products formed
-
(2)
Ferulic acid was converted into a product(s) that readily adsorbed on the catalyst and a higher amount of this product(s) was produced at higher temperature

TOC removal as a function of time at 100 °C. Conditions: T = 100 °C, t = 2 h, 180 kPa oxygen partial pressure, [ferulic acid] = 1,000 ppm, [Cu–Ni–Ce/Al2O3] = 2,000 ppm

TOC removal as a function of time at 150 °C. Conditions: T = 150 °C, t = 2 h, 180 kPa oxygen partial pressure, [ferulic acid] = 1,000 ppm, [Cu–Ni–Ce/Al2O3] = 2,000 ppm
To try and determine if the TOC removal occurring up to time zero was due to adsorption, the catalysts (at time zero) from the 100 and 150 °C tests were filtered and analysed for adsorbed carbon. The amount of TOC adsorbed on these catalysts was determined using the method described in Sect. 2. The amount of TOC adsorbed on the catalyst at time zero was 39% and 35% of the initial TOC for the 100 and 150 °C tests respectively. Thus all of the TOC reduction at time zero for the 100 °C test was due to adsorption. 60% of the observed TOC reduction at time zero at 150 °C is due to adsorption and the rest (∼40% of the observed TOC reduction) is due to either direct oxidation by the catalyst’s lattice oxygen or due to catalytic decarboxylation of ferulic acid and/or intermediate products formed.
Product analysis of time zero samples at 100 and 150 °C was performed using GC-MS and Electrospray MS. It can be seen that for the 100 °C test, at time zero, very little ferulic acid has been converted to low molecular organic compounds. This confirms that at 100 °C, ferulic acid did not oxidise using the oxygen during the heat up period. Combining these results with adsorption results (discussed in earlier paragraph) we can conclude that for 100 °C test, at time zero, all the observed TOC removal is due to adsorption of ferulic acid on the catalyst surface and there is no thermal degradation, decarboxylation or oxidation (using catalysts lattice oxygen).
For the 150 °C test, at time zero, most of the ferulic acid has been converted to vanillin (molecular weight 152 a.m.u) and 2-methoxy-4-vinayl phenol (molecular weight 150 a.m.u) (Fig. 23). Hence, we can conclude that ferulic acid was oxidised using the catalyst’s lattice oxygen during the heat up period up to 150 °C. The blank test at 150 °C did not show any thermal degradation during the heat up period. Combining these results with the adsorption results (discussed earlier), we can conclude that for the 150 °C test, at time zero, ∼60% of the TOC removal is due to adsorption of ferulic acid or products formed from ferulic acid on the catalyst’s surface, and the rest (∼40%) of the TOC removal is due to oxidation of ferulic acid using the catalyst’s surface oxygen and/or due to catalytic decarboxylation of ferulic acid.
To try and determine the fate of TOC adsorbed at time zero in CWO tests the following test was performed. The filtered time zero catalyst from a 100 °C test was added to milli-Q water without adding any additional catalyst or ferulic acid and a catalytic wet oxidation test was performed. Samples were taken at 15, 30 and 60 min reaction time. Nearly 50 ppm of constant TOC concentration was observed in the solution for the duration of the experiment. Figure 22 shows an FTIR spectra of filtered catalysts collected at different time periods. We can observe that the organic contents on the catalyst are decreasing over time by the decrease in intensity of various peaks in the FTIR spectra. This shows that catalyst releases organic over time, which is a good characteristic of the catalyst. In addition to this, the total organic carbon adsorbed on the catalysts’ surface was measured. The total organic carbon adsorbed on the catalyst was 39%, 29%, 14.9% and 12.2% of initial ferulic acid loading for time zero, 15, 30 and 60 min samples. This confirms that the catalyst releases TOC over time, while the lack of increase in TOC concentration in solution over time confirms that the TOC removed from the catalyst was mineralised.

FT-IR spectra of filtered catalysts
To understand the catalyst life-cycle, we re-used the same catalyst four times at 100 and 150 °C. The results are shown in Fig. 23. The catalyst was filtered from each test and used directly for the next test. So the organic content adsorbed on the catalyst surface is carried forward to the next test. The TOC reduction in the second test onwards is the oxidation due to oxidation of organics on the surface plus the TOC reduction in the solution. Here, the TOC reduction for 2nd, 3rd and 4th run are nearly 40% constant for 100 °C and 68% for 150 °C. This shows that the catalyst is quite active even after the 4th use.

%TOC reduction for re-used catalysts. Conditions: t = 2 h, 180 kPa oxygen partial pressure, [Ferulic acid] = 1,000 ppm, [Cu–Ni–Ce/Al2O3] = 2,000 ppm
6 Summary/Conclusions
Results on the CWAO of three different systems have been discussed. The main findings for each of these systems are summarised below.
6.1 Bayer Liquor/Synthetic Bayer Liquor
Copper is the most effective monometallic transition metal catalyst for the removal of organics from Bayer liquor, while Cu/Mn is the most effective bimetallic catalyst. Synthetic Bayer liquor studies showed that copper catalysed removal of a number of Bayer organics was predominantly due to dissolved copper. Synthetic Bayer liquor studies also showed that sodium malonate was capable of co-oxidising a number of stable organics and that this was also co-catalysed by copper. NaOH concentration was shown to have a significant effect on the co-catalysis mechanism.
6.2 Stripped Sour Water
Copper was the most effective monometallic transition metal catalyst for the removal of organics from stripped sour water, while Cu/Pd was the most effective bimetallic catalyst. Copper catalysed WO of stripped sour water was shown to involve free radical reactions. A significant amount of the TOC in SSW could be removed by copper in the absence of oxygen. This was proposed to be due to copper catalysed decarboxylation reactions. Kinetic studies showed that two distinct rate regions occurred during copper catalysed TOC removal from stripped sour water. These rate regions could be described using 1st order kinetics.
6.3 Ferulic Acid
Homogeneous copper was found to be among the most effective monometallic transition metal catalyst for oxidation of ferulic acid. Out of the nine tested nine multi-metallic copper catalysts, Cu/Ni/Ce supported on aluminium oxide was found to be the most active heterogeneous catalysts, followed by Cu/Mn supported on the same substrate. The activity of these catalysts can be attributed predominantly to the heterogeneous oxidation mechanism. The minimum copper leaching (% of actual loading) was observed for aluminium oxide supported Cu/Mn catalyst. Moreover, in order to understand the catalytic wet oxidation mechanism of ferulic acid degradation onto the surface of supported Cu/Ni/Ce catalyst, thermal degradation studies of ferulic acid were performed up to 150 °C, which show no thermal degradation of ferulic acid. However, heating of ferulic acid in the presence of a catalyst results in degradation of ferulic acid, which indicates an oxidative mechanism of degradation of ferulic acid onto the catalyst surface utilizing the catalyst’s lattice oxygen.
References
Eyer SL (2001) Investigation of catalytic wet oxidation of Bayer liquor. Ph.D. Dissertation, Department of Applied Chemistry, RMIT University, Melbourne, Australia
Tardio J (2002) Low temperature wet oxidation and catalytic wet oxidation of specific organic compounds in highly alkaline solution (synthetic Bayer liquor). Ph.D. Dissertation, Department of Applied Chemistry, RMIT University, Melbourne, Australia
Prasad J (2004) Wet oxidation based treatments for stripped sour water from an oil shale refining process. Department of Applied Chemistry, RMIT University, Australia
Bhargava SK, Tardio J, Prasad J, Föger K, Akolekar DB, Grocott SC (2006) Ind Eng Chem Res 45(4):1221
DeBellefontaine H, Foussard JN (2000) Waste Manage 20:15
Day DC, Hudgins RR, Silveston PL (1973) Can J Chem Eng 51(6):733
Robert R, Barbati S, Ricq N, Ambrosio M (2002) Water Res 36:4821
Rivas FJ, Kolaczkowski ST, Beltran FJ, McLurgh DB (1998) Chem Eng Sci 53(14):2575
Patterson DA, Metcalfe IS, Xiong F, Livingston GA (2001) Ind Eng Chem Res 40:5517
Tardio J, Bhargava S, Eyer S, Sumich M, Akolekar D (2004) Ind Eng Chem Res 43(4):847
Mantzavinos D, Lauer E, Hellenbrand R, Livingston AG, Metcalfe IS (1997) Wat Sci Tech 36:109
Stoffler B, Luft G (1999) Chem Eng Technol 22(5):409
Imamura S, Hirano A, Kawabata N (1982) Bull Chem Soc Jpn 55(11):3679
Santos A, Yustos P, Qunitanilla A, Ruiz G, Garcia-Ochoa F (2005) App Cat B: Environ 61:323
Larachi F (2005) Top Catal 33:109
Imamura S (1999) Ind Eng Chem Res 38:1743
Imamura S, Hirano A, Kawabata N (1982) Ind Eng Chem Prod Res Dev 21:570
Neri G, Pistone A, Milone C, Galvagno S (2002) App Cat B: Environ 38:321
Wu Q, Hu X, Yue PL, Zhao AS, Lu GQ (2001) App Cat B: Environ 32:151
Eyer S, Bhargava S, Tardio J, Akolekar D (2002) Ind Eng Chem Res 41:1166
Tardio J, Bhargava S, Eyer S, Akolekar D (2004) Ind Eng Chem Res 43(4):669
Tardio J, Bhargava S, Prasad J, Akolekar D (2005) Top Catal 32:193
Prasad J, Tardio J, Akolekar DB, Bhargava SK, Grocott SC (2004) Ind Eng Chem Res 43:6363
Prasad J, Tardio J, Jani H, Akolekar DB, Bhargava SK, Grocott SC J Hazard Mater (in press)
Jani H, Bhargava S, Tardio J, Akolekar DB, Hoang M Int J Environ Tech Manag (in press)
Grocott S, Rosenberg SP Soda in alumina: possible mechanisms for soda incorporation. Proceedings Alumina Quality Workshop, 1988, Darwin, NT
Coyne JF, Wainwright MS, Cant NW, Grocott SC (1994) Light Metals 39
Clegg RL, Armstrong LG (2005) Development of liquor purification at Alcan Gove. Proceedings of the 7th International Alumina Quality Workshop, Perth, WA
Baker C, Burnet S, Houghton P, Lewi C, Roach G WO 2006 /010218
Imamura S, Sakai T, Ikuyama T (1982) J Jpn Petrol Inst 25:74
Ragahavan NV, Leussing DL (1974) J Am Chem Soc 98:723
Agterberg FPW, Driessen WL, Reedijk J, Oevering H, Bujis W (1994) Stud Surf Sci Catal 82:639
Pokhrel D, Viraraghavan T (2004) Sci Total Environ 333(1–3):37
Jeney Z, Valtonen ET, Jeney G, Jokinen EI (1996) Arch Environ Contam Toxicol 30(4):523
An W, Zhang Q, Ma Y, Chang KT (2001) Catal Today 64(3–4):289
Babuna FG, Ince O, Orhon D, Simsek A (1998) Water Res 32(11):3490
Goring DAI (1971) Lignins. Interscience, New York
Prasad CVS, Joshi JB (1987) Indian Chem Eng 29(11):46
Teletzke GH, Pradt LA (1969) Proc Ind Waste Conf, Purdue Univ 139:1195
Conard DJ, Huck PM (1996) Water Res 30(11):2776
Pearl IA (1967) The chemistry of lignin. Marcel Dekker, New York
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bhargava, S.K., Tardio, J., Jani, H. et al. Catalytic Wet Air Oxidation of Industrial Aqueous Streams. Catal Surv Asia 11, 70–86 (2007). https://doi.org/10.1007/s10563-007-9020-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10563-007-9020-6