
Abstract
Hydrotalcite-like compounds (HTlcs) can be used as the catalysts as it is since they contain various transition metal cations as the catalytically active species well dispersed on the basic support materials. Moreover, increasing numbers of the applications of HTlcs after the heat treatment have been found since the oxides with very small crystal size, stable to thermal treatments, are obtained after the calcination. The oxides possess interesting properties such as high surface area, basic properties and further form small and thermally stable metal crystallites by reduction. Moreover, the calcined oxides show a unique property, i.e., “memory effect,” which allows the reconstitution of the original hydrotalcite structure. We have developed the catalytic applications of hydrotalcites as it is and moreover the mixed oxides derived from hydrotalcites for various catalytic reactions, i.e., oxidation, dehydrogenation and reforming of hydrocarbons, and even for the reforming of methanol and the CO shift reaction. Aerobic oxidation of alcohols, Baeyer−Villiger oxidation of ketones and O3 oxidation of oxalic acid have been successfully carried out with the Mg−Al hydrotalcites containing Ni, Fe and Cu, respectively, as the catalysts in liquid phase. In the O3 oxidation of oxalic acid, the catalytic activity was enhanced by the “memory effect,” i.e., Mg(Cu)–Al hydrotaclite was reconstituted on the surface of Mg(Cu,Al)O periclase particles and oxalic acid was incorporated as anions in the hydrotalcite layer, resulting in an enhanced oxidation of oxalic acid. As the catalysts in the vapor phase reactions, Mg/Fe/Al mixed oxides prepared from Mg–Al(Fe) hydrotalcites and effectively catalyzed the dehydrogenation of ethylbenzene. Supported Ni metal catalysts have been prepared from Mg(Ni)–Al hydrotalcites and successfully used in the steam reforming and the oxidative reforming of methane and propane. Moreover, the Ni catalysts have been improved by combining a trace amount of noble metals by adopting the “memory effect” and used in the production of hydrogen for the PEFC under the daily startup and shutdown operation. Also starting from aurichalcite or hydrotalcite precursor as the precursor, Cu/Zn/Al catalysts with high Cu metal surface area have been prepared and successfully applied in the steam reforming of methanol and dimethyl ether, and moreover in the CO shift reaction.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrotalcite-like compounds (HTlcs) shown by a general formula: [M(II) 1 − x M(III) x (OH)2]x+(An− x/n ) · mH2O, contain various cartions (M(II) and M(III)) and anion (An−) [1]. The most popular HTlcs is composed of Mg–Al combination. The structure of HTlcs is basically composed of brucite, Mg(OH)2, where octahedral of Mg2+ (6-fold coordinated to OH−) share edge to form infinite sheets. These sheets are stacked on top of each other and are held together by hydrogen bonding (figure 1) [2]. When Mg2+ ions are substituted by a trivalent ion having a radius not too different from that of Mg2+ (such as Fe3+ and Al3+ for pyroaurite and HT, respectively), a positive charge is generated in the hydroxyl sheets. This net positive charge is compensated for by CO 2−3 anions, which lie in the interlayer region between two brucite sheets. In the free space of this interlayer the water of crystallization also finds a place. The main feature of HTlcs structures therefore are determined by the nature of the brucite-like sheets, by the position of anions and water in the interlayer region and by the type of stacking of the brucite sheets. The sheets containing cations are built as in brucite, where the cations randomly occupy the octahedral holes in the close-packed configuration of the OH− ions. Generally M(II) and M(III) ions can be accommodated in the holes of the close packed configuration of OH group in the brucite-like layers as far as the ionic radii and the valence states are allowed. It is known that the insertion of copper ions into the brucite-like sheet is unfavored because of the Jahn–Teller effect, showed by these ions, which produces a distorted octahedral disposition of OH groups around the copper ion [3]. It has been however found that the presence of other divalent cations, such as Mg(II), Zn(II), and Co(II), favors the accommodation of Cu(II) into the sheet, giving rise to ternary HT phases [4].

Structure of HT.
The HTlcs as itself have been used as the catalyst since they contain various transition metal cations as the catalytically active species well dispersed on the basic support materials. Also they have been used as the catalysts mainly after the calcination since the oxides obtained by calcination possess interesting properties such as high surface area and basic properties. After the calcination, homogeneous mixtures of oxides are formed with very small crystal size, stable to thermal treatments, which further form small and thermally stable metal crystallites by reduction. Moreover, the calcined oxides show a unique property, i.e., “memory effect,” which allows the reconstitution of the original HT structure contacting the oxides after the calcination with aqueous solutions containing various anions.
The HTlcs derived oxides themselves are well known as a base catalyst and has been already reviewed [4,5]. We focused this report on our recent research works aiming the catalytic application of the HTlcs mainly obtained by introducing various metals as the active species. Also use of the aurichalcite or the HT as the precursor of the Cu/Zn/Al catalysts has been referred. The results have been discussed on in connection with the new catalyst preparation technologies [6].
Application of HTlcs as the catalysts
The HTlcs possess the structure basically composed of brucite-like layers and anions; various metal cations or metal-containing anions can be incorporated as far as ionic radii and valence state are permitted. These metal species are randomly distributed in the brucite phase and possibly show the catalytic activity for various types of reaction being assisted by the HT structure.
By adopting the “memory effect,” HT structure was reconstituted in the mixed oxide derived from the hydrotalcite. Such hydrotalcites reconstituted finely modify the surface of mixed oxides and also showed a unique catalytic activity in the oxidation reactions.
Oxidation by molecular oxygen
Catalytic oxidation of alcohols to carbonyl compounds has attracted much attention both in industrial processes and in organic synthesis. Alcohols have been traditionally oxidized by non-catalytic methods with stoichiometric oxidants such as dichromate and permanganate. These methods produce enormous amount of metal salts as wastes. Much effort has been made to develop homogeneous catalytic systems to solve these problems. However, most systems still required large quantities of additive such as NaOAc, NaOH, and K2CO3. There is little known about the oxidation of alcohols with molecular oxygen over heterogeneous catalysts. Catalytic oxidation with molecular oxygen is particularly attractive from an economical and environmental point of view. Moreover, heterogeneous catalysts in the liquid phase offer several advantages over homogeneous ones, such as an ease of recovery and recycling, atom utility, and enhanced stability in the oxidation reactions.
Activation of molecular oxygen on nickel in Ni–Al HTlcs anionic clay was reported to take place in the oxidation of alcohol [7]. The most common HT compounds consist of Mg–Al system, in which various metal cations can substitute both sites of Mg(II) and Al(III) depending on the valence state and ionic radii [3,4]. When Ni(II) substitutes the Mg(II) sites of Mg–Al HT, the Ni(II) cations are highly dispersed and octahedrally coordinated with oxygen. The Ni(II)/Mg–Al HT catalyzed the oxidation of alcohols by molecular oxygen [7,8]. Choudhary et al. [7] suggested that the Ni(II) species in the Ni–Al HT activates molecular oxygen in alcohol oxidation. If this hypothesis can work on the Mg(Ni)–Al HT, Mg as a base possibly assists the reductive activation of molecular oxygen resulting in the acceleration in the alcohol oxidation. Actually, we have observed that the Ni(II) substitution for the Mg(II) site in the Mg3–Al HT resulted in an appearance of the catalytic activity and the composition of Mg2.5(Ni0.5)–Al HT was the most effective [8]. The oxidation of primary and secondary alcohols afforded the corresponding carbonyl compounds mainly, and benzyl alcohol was most efficiently oxidized to benzaldehyde (1) (table 1). Turnover numbers were
calculated based on the number of the effective surface Ni sites which were evaluated by stoichimetric oxidation of benzyl alcohol to benzaldehyde using NaOCl as an oxidizing agent in N2 atmosphere; the amount of active Ni on the surface was calculated as 0.04 mmol of Ni for 0.5 g of Mg2.5(Ni0.5)–Al HT (total Ni was 0.647 mmol) from the amount of benzaldehyde formed [8]. The atomically isolated and octahedrally coordinated Ni(II) cations incorporated inside the framework of HT did not leach during the reaction and catalyzed efficiently the heterogeneous oxidation of alcohols with molecular oxygen (figure 2). It is considered that the Ni(II) sites work as the active sites by activating molecular oxygen assisted by the Mg(II) sites as a base and simultaneously alcohol was activated by the Al(III) sites as an acid, resulting in an enhancement of the activity of the Mg2.5(Ni0.5)–Al HT heterogeneous catalyst for alcohol oxidation.

Proposed mechanism for the aerobic oxidation of alcohol with Mg(Ni)–Al HT.
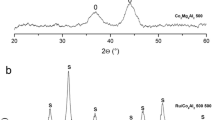
It has also been found that an Fe-containing Mg-A HT catalyst prepared by impregnation of Mg(Al)O periclase in an aqueous solution of Fe showed high activity for the Baeyer–Villiger oxidation of cyclic ketones to the corresponding lactones by using O2/benzaldehyde as an oxidizing agent (2) (table 2) [9]. Use of  Fe(NH4)2(SO4)2 · H2O was uniquely effective as the metal salt in the impregnation; Fe(III)–O–Fe(III) cluster-type compounds were produced as the active species on the surface of Mg(Al)O particles during the reconstitution of HTs by the “memory effect”, resulting in the highest activity around 6% of Fe loading. The reconstitution of HTs was clearly observed by the XRD (figure 3). During the reconstitution of HTs by the “memory effect,” Fe(II) cations from Fe(NH4)2(SO4)2 · 6H2O were likely incorporated into the HTs reconstituted on the surface of the particles of Mg(Al)O periclase. It is unlikely that either Fe(III) (0.645 nm) or Fe(II) (0.78 nm) directly replace the Mg(II) sites via the “memory effect” on Mg(Al)O periclase due to the difference in valence state or ionic radii; a combination of Mg(II)–Fe(III) alone was allowed to form HTlcs layered structure by co-precipitation starting from the nitrates of Mg(II) and Fe(III) [10]. Actually iron species observed for the Fe/Mg3(Al)O catalyst possessed the Fe(III) valence state although Mg3(Al)O was immersed in aqueous solution of Fe(NH4)2(SO4)2 · 6H2O containing Fe(II). Judging from Mössbauer (table 3), UV–vis and Fe K-edge XAFS spectra, the Fe species possess the Fe(III) valence state, are mainly octahedrally coordinated, and form Fe–O–Fe cluster-type structure on the Mg3(Al)O periclase. This clearly indicates that the Fe(III)–O–Fe(III) cluster-type compounds are more active for the Baeyer−Villiger oxidation than well dispersed Fe(III) species formed on the HTs prepared by co-precipitation [9].
Fe(NH4)2(SO4)2 · H2O was uniquely effective as the metal salt in the impregnation; Fe(III)–O–Fe(III) cluster-type compounds were produced as the active species on the surface of Mg(Al)O particles during the reconstitution of HTs by the “memory effect”, resulting in the highest activity around 6% of Fe loading. The reconstitution of HTs was clearly observed by the XRD (figure 3). During the reconstitution of HTs by the “memory effect,” Fe(II) cations from Fe(NH4)2(SO4)2 · 6H2O were likely incorporated into the HTs reconstituted on the surface of the particles of Mg(Al)O periclase. It is unlikely that either Fe(III) (0.645 nm) or Fe(II) (0.78 nm) directly replace the Mg(II) sites via the “memory effect” on Mg(Al)O periclase due to the difference in valence state or ionic radii; a combination of Mg(II)–Fe(III) alone was allowed to form HTlcs layered structure by co-precipitation starting from the nitrates of Mg(II) and Fe(III) [10]. Actually iron species observed for the Fe/Mg3(Al)O catalyst possessed the Fe(III) valence state although Mg3(Al)O was immersed in aqueous solution of Fe(NH4)2(SO4)2 · 6H2O containing Fe(II). Judging from Mössbauer (table 3), UV–vis and Fe K-edge XAFS spectra, the Fe species possess the Fe(III) valence state, are mainly octahedrally coordinated, and form Fe–O–Fe cluster-type structure on the Mg3(Al)O periclase. This clearly indicates that the Fe(III)–O–Fe(III) cluster-type compounds are more active for the Baeyer−Villiger oxidation than well dispersed Fe(III) species formed on the HTs prepared by co-precipitation [9].

Reconstitution of HT by the “memory effect” during the preparation Fe- supported on Mg(Al)O. (a) Mg3–Al HT after coprecipitation, (b) Mg3(Al)O after calcination at 850 °C for 5 h (0.83 °C min−1), (c) Fe(6.1 wt%)/Mg3(Al)O catalyst after dipping, followed by drying. •, HT; □, Mg(Al)O periclase.
As for the aerobic oxidation with the catalysts derived from HTlcs, the liquid-phase oxidation of a methylene group attached to an aromatic ring to carbonyl group has been studied; a MnO −4 exchanged Mg–Al HT was a highly active and selective, stable, and reusable catalyst for the oxidation of ethylbenzene to acetophenone and deiphenylmethane to benzophenone by molecular oxygen in the absence of solvent [11]. Various functionalized hydrotalcites have been prepared using the cation exchange ability and successfully used for the oxidation of alcohols in the presence of molecular oxygen, among which Co–Al–Ru–CO3 showed the highest activity for the oxidation of alcohols to ketones possessing conjugated double bonds [12]. A Ru-grafted Mg–Al HT was as excellent bi-functional catalyst for one-pot synthesis of quinolines from 2-aminobenzyl alcohol and various carbonyl compounds [13]; the reaction proceeds through aerobic oxidation of the alcohol by the Ru species, followed by aldol condensation on base sites of HT.
Oxidation by ozone
Ozone, due to its high oxidation and disinfection potential, has recently received much attention in water treatment technology. Ozone is known to be powerful oxidant, but it reacts slowly with some organic compounds such as inactivated aromatics. Moreover, in many cases, it does not cause the complete oxidation of organic compounds, which results in the formation of biodegradable organic matters (carboxylic acids and carbonyl compounds). Catalysis combined with the ozonation processes (catalytic ozonation) can improve the oxidation and degradation of organic contaminant compounds, especially small carboxylic acids that cannot be easily attacked by conventional oxidation processes (3). The catalytic ozonation provides a promising new  kind of water treatment method.
kind of water treatment method.
Mg–Al supported metal (Fe, Co, Ni and Cu) oxide catalysts were prepared by the co-precipitation of HTlcs clay minerals as the precursors, calcined, and used for the ozonation reaction of phenol and oxalic acid [14]. The reaction was carried out using the catalysts and an aqueous solution of phenol or oxalic acid in an O3/O2 mixed gas-flow at 20 °C. In the ozonation of phenol, the combination of ozone and the supported metal oxide catalysts was effective for the removal of total organic carbon (TOC). Also in the ozonation of oxalic acid as the main TOC component, Cu/Mg/Al catalysts showed the highest activity, followed by Ni/Mg/Al catalyst, while both Fe/Mg/Al and Co/Mg/Al catalysts were not active. Leaching of Cu and Ni, probably due to the chelation of metals by oxalic acid, was significantly observed at the beginning of the reaction. However the metal leaching disappeared at the end of the reaction possibly due to the entire consumption of oxalic acid during the reaction. The best result of oxalic acid mineralization was observed over the Cu/Mg/Al catalyst calclined at 600 °C, on which least leaching of the metal was detected. Moreover, the “memory effect” of HT accelerated the mineralization of oxalic acid over the Cu/Mg/Al catalyst; oxalate anions were captured and decomposed in the reconstituted HT interlayer space on the surface of the Cu/Mg/Al catalyst, resulting in a remarkable enhancement in the catalytic activity of the ozonation.
It was known that Mg2+(Cu2+)Al3+ system form ternary HT at the metal composition used in this work [15] and therefore the formation of periclase Mg(Al,Cu)O is considered at the calcination temperature above 400 °C. It was confirmed by TG-DTA that that the dehydration of Mg2.77(Cu0.23)–Al HT around 200 °C, followed by the decomposition to periclase around 400 °C, took place during the calcination. Moreover, by the calcination above 850 °C, spinel phase appeared whereas periclase Mg(Al, Cu)O was converted to pure MgO with sharpening reflection lines and to Mg(Cu)Al2O4 spinel. Effect of the calination temperature of the Mg2.77(Cu0.23)Al mixed oxide catalyst on the activity, the reconstitution of HT (A) and the Cu leaching (B) is shown in figure 4. In the ozonation of oxalic acid over the Mg2.77(Cu0.23)Al catalysts calcined at varying temperatures, the rate constant (k1/min−1) was calculated from the time course of ln(C/C 0), where C is the concentration of oxalic acid, and was plotted against the calcination temperature (figure 4a), assuming that the reaction proceeds by a quasi-first order with respect to oxalic acid. XRD patterns of the Mg2.77(Cu0.23)Al catalysts clearly showed the reconstitution of HT phase after the reaction, when the catalyst was calcined at 400, 600 and 850 °C. The intensity of the reflection by basal plane (003) of the HT formed on the catalysts by the reconstitution was plotted against the calcination temperature (figure 4a). Both peak intensity and first order rate constant showed the maximum value at 600 °C; a good correlation was observed in the dependences of both peak intensity and rate constant against the calcination temperature. This strongly suggests that the catalytic activity was enhanced by the reconstitution of HT phase on the Mg2.77(Cu0.23)Al catalysts. The HT reconstitution on the Mg2.77(Cu0.23)Al catalysts is probably due to the presence of oxalate anions compensating the positively charged layers, as is seen the infrared spectra of the material during the reaction (figure 5). The Mg2.77(Cu0.23)Al catalyst before calcination possessed Mg2.77(Cu0.23)–Al HT structure and showed the broad absorption bands around 3500 cm−1 ascribed to the stretching mode of hydroxyls attached to Al and Mg (figure 5a) [16]. A shoulder may be present around 3000 cm−1; this can be assigned to hydrogen bonding between interlayer water and interlayer carbonate [4]. An H2O bending vibration also occurred at 1650 cm−1, while the band around 1380 cm−1 can be assigned to vibration of interlayer carbonate anion [17]. After the calcination at 600 °C, all these bands were weakened (figure 5b). When the Mg2.77(Cu0.23)Al catalyst was used in the reaction for 15 min, the bands around 3500 and 1650 cm−1 were intensified, whereas the band at 1380 cm−1 was not, suggesting that interlayer space was filled with water molecules and, at the same time, brucite phase was regenerated (figure 5c). Moreover a new band at 1640 cm−1 and a shoulder at 1320 cm−1 appeared. In general, interlayer anion absorption bands are found around 11,200 and 1700 cm−1 [4]. None of information concerning these bands in HT with interlayer oxalate is available in the literature. It is likely that the band at 1640 cm−1 and the shoulder at 1320 cm−1 are ascribed to interlayer oxalate since these absorptions also appeared when the sample was dipped in aqueous solution of oxalic acid.

Effect of the calcination temperature of the Mg2.77(Cu0.23)Al catalyst on both activity and reconstitution of HT (a) and Cu leaching (b). Catalyst, 0.4 g; oxalic acid, 3.2 mmol; water, 400 mL; reaction temperature, 20 °C; (a) O3/O2 mixed gas, 1.4 vol% (200 mL h−1); (b) O2, 200 mL h−1. •, quasi-first order rate constant of oxalic acid consumption; ■, intensity of diffraction by basal planes (003) of HT reconstituted; ▲, quasi-first order rate constant of Cu leaching.

Infrared spectra of the Mg2.77(Cu0.23)Al catalyst during the reaction. (a) before calcination; (b) after the calcination at 600 °C; (c) after the reaction for 15 min; (d) after the reaction for 4 h.
The intercalation by oxalate anions can also be confirmed by the XRD observations (table 4). Interlayer distance was calculated assuming that the thickness of the brucite-like layer is 0.48 nm [18]. When the Cu0.23Mg2.77Al catalyst calcined at 600 °C was dipped in distilled water (table 4, Sample I), the basal interlayer spacing of HT reconstituted was calculated as 0.757 nm, which is close to the value reported for the Mg–Al carbonate HT [19]. After the reaction for 15 min (table 4, Sample II), the basal spacing slightly increased to 0.778 nm. The sample further prepared by being dipped in an aqueous solution of oxalic acid (table 4, Sample III) as a reference showed the value 0.782 nm. It was reported that the basal spacing was 0.960 nm for the Mg–Al oxalate HT [20]. It is therefore likely that the intercalation by oxalate anions was not completed in the present samples after the reaction or after the dipping in oxalic acid solution. This is partly due to the rapid decomposition of oxalate in the interlayer space of the catalysts. Actually both absorption bands around 1640 cm−1 and shoulder around 1320 cm−1 observed in the infrared spectra disappeared after the reaction for 240 min, suggesting that the oxalate anions were totally consumed by the mineralization reaction with ozone (figure 5d).
Palomares et al. [21] reported that the “memory effect” of HTs accelerated the catalytic reduction of nitrates in water. Pd was wet impregnated on the Mg(Cu)Al mixed oxide prepared from the Mg(Cu)–Al HT as the precursor and was used in the catalytic hydrogenation of nitrates in water. The “memory effect” took place on the calcined HT, and the nitrates are forced to be located between the positively charged layers of the HT and therefore close to the reductive active sites. As a result, the nitrates are reduced to nitrites that remain in the same position, and then are further reduced to nitrogen. These final products due to their inadequate charge are released to the solution, reducing the problems related with the diffusion limitations. Also in the present reaction, a similar effect is considered during the catalytic ozonation of citric acid; the citrate anions were forced to be located between the positively charged layers of the HT and were effectively oxidized to carbon dioxide, which is released to the solution due to the inadequate charge.
We have reported that, in the aerobic oxidation of alcohols with Mg(Ni)–Al HTs catalysts, ca. 10% of alcohol was absorbed and remained in the HT layer during the reaction [8]. However, in the aerobic Baeyer–Villiger oxidation of ketones with improved Fe/Mg–Al HT catalyst, such adsorption phenomena of ketones was not observed probably due to too thin layer of the HT reconstituted on the Mg(Al)O periclase [9]. In the ozonation of citric acid, the reconstitution of HT and the oxidation reaction concurrently occurred on the catalysts surface and moreover the latter reaction proceeded rather rapidly. As a result, the effect of adsorption of substrate in the layered structure on the total reaction could not be observed clearly.
Application of mixed oxides derived from HTlcs as the catalyst
Mixed oxides resulting from thermal treatment of the HTlcs present several interesting properties [4]. Thermal decomposition of these materials by calcination results in the formation of mixed oxides with a high thermal stability and a large surface area. In addition, well-dispersed metallic particles are obtained after the reduction treatment [4,5]. Moreover the structure can accommodate a wide variation of the different metals, leading to different catalytic properties.
Application as the dehydrogenation catalysts
Styrene, an important basic chemical as a raw material for polymers, is produced commercially by the dehydrogenation of ethylbenzene using a typical Fe–K–Cr oxide-based catalyst in the presence of a large quantity of steam at high temperatures of 600–700 °C, just below the temperature where the thermal cracking becomes significant. Although the commercial catalysts are very active and selective, they have some disadvantages; (i) The active oxidation state is unstable; hematite (α-Fe2O3) is preferred for styrene production, but tends to lower oxides and even to elemental iron, both of which catalyze coke formation and dealkylation. (ii) The catalysts have low surface area. (iii) They are deactivated with time, being susceptible to poisoning by halides and residual organic chloride impurities. The most serious deactivation is caused by the loss of potassium promoter, which migrates as the catalyst ages. Also, the toxicity of the chromium compounds causes damage to humans and to the environment. Therefore, the search for new catalyst systems which have high surface areas and can stabilize the active state of iron, in the absence of potassium, is much needed. Aluminum was proved to be an excellent promoter, preventing sintering in the iron-oxide catalysts [22]. Stobbe et al. [23] have developed methods to support promoted iron oxide onto an MgO support material; a supported system can prevent the physical degradation of the catalyst particles.
The starting materials can also play a significant role in the catalytic performance [24]. A new catalyst for the process of ethylbenzene dehydrogenation to styrene under a CO2 or O2 atmosphere was obtained using vanadium-substituted Mg–Al HTs as precursors [25]. As for the dehydrogenation of ethylbenzene under a CO2 atmosphere, Fe–Mg [26], Fe–Mg–Al [27] and Fe–Zn–Al [28] mixed oxides were tested. The Fe–Mg catalysts possessed high surface area and were active for the dehydrogenation reaction while the incorporation of Zn and Al resulted in the highest ethylbenzene conversion of 53.8% and a styrene selectivity of 96.7% at 500 °C [26]. The activity losses over the Fe–Mg–Al catalysts with time-on-stream were completely restored by oxygen pulses [27]. The Fe–Zn–Al catalysts were effective and gave an areal rate of 4.15 μmol min−1 m−2 although the surface area was as low as 22.1 m2 g −1cat [28].
We have prepared supported iron catalyst from Mg–Fe–Al HTlcs as precursors, which have been successfully applied in the ethylbenzene dehydrogenation to styrene [29]. After the calcination, the iron-substituted HTlcs were converted to mixed oxides with a high surface area as well as a mesoporous character; the XRD analysis indicates the formation of periclase Mg(Fe,Al)O as a main phase (figure 6). Catalytic tests of the Mg2Fe x Al1−x catalysts showed that the styrene conversion increased with increasing the iron content up to x = 0.75 and then decreased, while the selectivity was the highest at x = 0.25. The optimum temperature for the reaction was 550 °C, which was lower than that used in the commercial process. Although ethylbenzene dehydrogenation has been carried out over Fe–Mg–Al catalysts under a CO2 atmosphere [27], no favorable effect of the addition of CO2 or O2 in the reaction medium was observed in this reaction. Actually, the pre-treatment with H2 resulted in an increase in the activity at the beginning of the reaction as well as a stable activity during the reaction. The ethylbenzene conversion of 60% and the styrene selectivity of 95% were kept for 3 h over the Mg3Fe0.5Al0.5 catalysts at 550 °C (table 5). After the reaction for 3 h, the iron species on the catalyst was partially reduced to the valence state between Fe2+ and Fe3+. The good catalytic performance can probably be attributed to the formation of partially reduced iron oxides on the surface of catalyst and to the high surface area along with the porous structure, which originated from the Mg–Fe–Al HT structure as the precursors.

XRD patterns of supported Fe catalysts. Effects of pre-treatment and the reaction-: (a) Mg3Fe0.5Al0.5 (after N2 treatment at 550 °C); (b) Mg3Fe0.5Al0.5 (after O2 treatment at 550 °C); (c) Mg3Fe0.5Al0.5 (after H2 treatment at 550 °C); (d) Mg3Fe0.5Al0.5 (after H2 treatment at 1000 °C); (e) Mg3Fe0.5Al0.5 (a, after the reaction); (f) Mg3Fe0.5Al0.5 (d, after the reaction). (□) Periclase Mg(Fe,Al)O; (■) Mg(Fe,Al)2O4 spinel; (+) Fe metal; (*) MgAl2O4; (•) Fe3C; (×) graphite.
In this connection, 2-octanol was dehydrogenated to 2-octanone at 300 °C over the Cu/Mg(Al)O catalysts prepared by the calcination of the Mg(Cu)–Al HT at 450 °C for 9 h, followed by the reduction at 300 °C for 2 h [30]. The reduced catalyst still contained Cu2+ and Cu+ together with Cu0 and the copper was present in a cluster-like species.
Ni/Mg(Al)O catalysts for the reforming of hydrocarbons
Hydrogen production for polymer electrolyte fuel cell (PEFC) is an emergent research area for solving global warming in the world. Steam reforming of hydrocarbons, especially of CH4, is the most popular and generally the most economical way to make H2 (4). This process still requires further advancements in the preparation
of suitable reforming catalysts. The authors have reported the preparation of highly dispersed and stable metal-supported catalysts starting from perovskite materials and HTlcs as the precursors [31].
Ni/Mg(Al)O catalysts were prepared by thermal treatment followed by reduction of ternary Mg(Ni)–Al HT with varying Mg/Al ratios [32–36] and were successfully applied for the partial oxidation [34], the steam reforming [35] and the autothermal reforming of CH4 [36]. Ni/Ca(Al)O catalyst was also prepared and used for the partial oxidation of CH4 [37]. The high and stable activity of these catalysts can be attributed to the formation of stable and well-dispersed Ni metal particles produced from Mg(Ni,Al)O periclase as solid solutions obtained by the calcination of Mg(Ni)-Al HT. The Mg(Ni,Al)O periclase was in situ reduced to produce fine Ni metal particles on the catalysts and showed the activity also in the dry reforming of CH4 [32]. After the pre-reduction treatment, the Ni/Mg(Al)O catalysts showed activity in the partial oxidation of CH4, in which the reaction proceeds via combustion (5), followed by steam
reforming (4) and dry reforming (6). The Ni0.5/Mg2.5(Al)O catalyst showed the highest
activity; CH4 conversion of the thermodynamic equilibrium was obtained even at the enormously high space velocity of 9 × 105 mL h−1 g −1cat (figure 7) [36]. This excellent value of the space velocity exceeded the value 7 × 105 mL h−1 g −1cat obtained in the CH4 partial oxidation over the 0.1 wt%Rh/MgO catalyst which was reported as the most active catalyst by Ruckenstein and Wang [38]. In the partial oxidation, Ni catalysts were frequently deactivated due to the surface oxidation of Ni metal particles and such deactivation was usually accompanied by drastic decrease in the CH4 conversion as seen for the commercial Ni/α-Al2O3 catalyst (FCR from Süd Chemie Co.) (figure 7). The FCR catalyst has been used in the steam reforming of CH4 in the industrial scale. The Ni0.5/Mg2.5(Al)O catalyst showed higher sustainability than the FCR catalyst in the steam reforming of CH4 (figure 8) [35]; the reaction was carried out using each 20 cc of the Ni0.5/Mg2.5(Al)O and FCR catalysts at 800 °C, with the steam to carbon ratio (S/C) = 1.6 and at the space velocity (SV) = 2500 h−1 for 600 h, during which SV was increased to 10,000 h−1 for evaluating precisely the deactivation of the catalyst. Under the latter conditions, the reaction temperature inevitably decreased to 660 °C due to the endothermic reaction. The Ni0.5/Mg2.5(Al)O always kept the CH4 conversion reaching the thermodynamic equilibrium of steam reforming, whereas the FCR showed clear decrease in the CH4 conversion, during 600 h of the reaction.

Partial oxidation of CH4 over supported Ni catalysts. (a) Ni0.5/Mg2.5(Al)O; (b) imp-16.3 wt% Ni/Mg3(Al)O-aq; (c) imp-16.3 wt% Ni/Mg3(Al)O-acac; (d) imp-16.3 wt% Ni/Mg3(Al)O-ac; (e) imp-16.3 wt% Ni/MgO; (f) imp-16.3 wt% Ni/γ-Al2O3; (g) imp-16.3 wt% Ni/α-Al2O3; (h) Commercial Ni/α-Al2O3 (FCR). A mixed gas of CH4/O2/N2 = 2/1/1 was used at 800 °C for 50 and 5 mg of the catalyst diluted by quartz beads at the low (dotted line) and the high (full line) space velocities, respectively.

Catalyst life test in steam reforming of CH4 over Ni0.5/Mg2.5(Al)O and commercial Ni/α-Al2O3 (FCR). Catalyst, 20 cc (17.3 g for Ni0.5/Mg2.5(Al)O); S/C = 1.6 (•,○), Ni0.5/Mg2.5(Al)O; (■,□), FCR.
When the Ni0.5/Mg2.5(Al)O catalyst was prepared by the co-precipitation from the nitrates of Ni(II), Mg(II) and Al(III), all Ni species distribute homogenously in the catalyst particles and therefore those located inside the particles cannot work as the active species. Actually the reduction degree obtained from the TPR measurements of the Ni0.5/Mg2.5(Al)O catalyst was 88% [39], indicating clearly that a part of Ni remains as Ni2+ in Mg(Ni,Al)O periclase particles even after the reduction. Effective Ni loading in the outer layer of the catalyst particles is preferable; such eggshell-type Ni loading on the Mg(Al)O periclase particles was achieved by adopting the “memory effect” of Mg–Al HT. Active Ni metal particles were effectively enriched in the surface layer of the catalyst particles by controlling the preparation conditions. The mechanism of reconstitution of Mg–Al HT leading to eggshell-type Ni loading on Mg(Al)O periclase was clarified (figure 9) [40]. The reconstitution of HT by the “memory effect” proceeded on micro-porous phase and formed “worm” like structures on Mg(Al)O particles. When the Mg–Al HT was calcined under mild conditions, the “worm” like structure quickly formed on the surface of the Mg(Al)O particles which finally constituted a dense layer and perfectly covered the surface of the particles (figure 9a, c and e). The dense layer hindered a further penetration of Ni2+ ions into the cores of the particles, resulting in eggshell-type Ni loading. On the contrary, high temperature calcination caused a formation of MgAl2O4 spinel on the particles, where the reconstitution proceeded slowly throughout the particles and simultaneously Ni2+ penetrated into the cores of the particles (figure 9b, d and f). A balance between the rate of the reconstitution of Mg–Al HT and the rate of penetration of the aqueous solution of nickel nitrate determined the loading type of Ni. Moreover the enhanced activity per unit amount of Ni due to the surface enrichment of active Ni species was successfully explained by the measurements of the effectiveness factor of the Ni/Mg(Al)O catalyst (figure 10) [40]. The eggshell-type Ni loaded catalyst was successfully applied not only for the steam reforming [41] but also for the autothermal reforming [42] of CH4. Such eggshell-type metal loading technique will be effective for the preparation of noble metal loaded catalysts on Mg(Al)O (vide infra).

TEM images of outer edges of Mg3(Al)O particles. (a) after the heating of Mg–Al(3/1) HT at a rate of 1.67° C min−1, followed by the calcination at 650 °C for 5 h; (b) after heating Mg−Al(3/1) HT at a rate of 1.67 °C min−1, followed by the calcination at 950 °C for 5 h; (c) after the dipping of (a) in 0.5 M Ni(II) nitrate aqueous solution for 1 h; (d) after the dipping of (b) in 0.5 M Ni(II) nitrate aqueous solution for 1 h; (e) high magnification image of a part in the circle in (c; f) high magnification image of a part in the circle in (d).

Effectiveness factor of Ni0.5/Mg2.5(Al)O catalyst in steam reforming of CH4. Reaction temperature, 700 °C; particle size of the catalyst, 0.15–0.48 mm in diameter. GHSV: ■, 7.2 × 105 mL h−1 g −1cat ; •, 9.0 × 105 mL h−1 g −1cat .
The HT-derived Ni catalysts were also successfully applied for the steam reforming of naphtha [43]. Ni loading can be decreased to 1.9 wt% on Mg(Al)O without both activity and stability loss for the CO2 reforming of propane to synthesis gas at 600 °C throughout a 34-day test [44]. The Ni particles are close contact with, and partially decorated by, the basic Mg(Al)O support, on which CO2 is associatively adsorbed and acts as a permanent source of oxygen species for the Ni metal. Propane reacts rapidly with Ni–O species to form CO and H2O.
Improvement of Ni/Mg(Al)O catalysts by the addition of noble metals
In contrast to the large-scale use of reformers in the industry under typical operating conditions, the temperature varied frequently at startup and shutdown, i.e., daily startup and shutdown (DSS), for the operation in hydrogen production for PEFCs in domestic use. Between shutdown and startup in each DSS operation, the catalyst bed in the reformer must be purged by a sufficiently inert and an economically available gas for preserving the safety. Thus, the catalyst must be able to withstand multiple cycles under those conditions and must also tolerate operation under such unusual transient conditions without deterioration. When the Ni/Mg(Al)O catalysts were applied in the DSS operation of the steam reforming of CH4 (figure 11), the Ni0.5/Mg2.5(Al)O catalyst was severely deactivated [45] although this catalyst showed high and stable activity in the steady state CH4 reforming (figures 7 and 8) [35,36]. The results obtained with the Ni/Mg(Al)O catalysts with various (Mg + Ni)/Al ratios together with commercial catalysts and those prepared by impregnation for three purge gases, i.e., steam, air and spent gas, are shown in figure 12 [45]. RUA is a commercial Ru/α-Al2O3 supplied from Süd Chemie Co., like as the FCR as Ni/α-Al2O3 catalyst. It must be emphasized that O2 in air damaged most severely all supported Ni catalysts. MgO-supported Ni catalysts were completely deactivated, whereas Al2O3-supported Ni catalysts showed stable activity for both steam and spent gas. The stability of the Ni/Mg(Al)O catalysts varied depending on the (Mg + Ni)/Al ratio. It is well known that Ni catalysts were frequently deactivated by sintering or coking during the long term reaction. However such deactivation was not significant in the present DSS operation. It was convinced that the deactivation proceeded rapidly due to the oxidation of Ni metal to Ni2+, which returned back into Mg(Ni,Al)O periclase, judging from the XRD observations; the reflection lines of Ni metal disappeared and those of Mg(Ni,Al)O periclase were strengthened after the deactivation. It is likely that the Ni oxidation occurred directly by the oxidation with O2 or H2O or was possibly induced by hydration of MgO to Mg(OH)2 brucite, since Mg(OH)2 is thermodynamically more stable than MgO under the present conditions (vide infra) [46].

DSS-like reaction mode.

Stabilities of catalysts during DSS-like operations. Stability was calculated by the following equation: Stability = CH4 conversion at 4th cycle/CH4 conversion at 1st cycle.
Noble metals, such as Rh, Ru and Pt, are also used as the active species in the catalysts prepared from Mg–Al HT as the precursors. A Rh5/Mg71(Al24)O catalyst showed a high and stable activity in the partial oxidation of CH4 under severe conditions [47]. The formation of Rh dissolved in a Mg and Al matrix is responsible for the highly dispersed and active Rh catalyst obtained after the reduction. The catalytic activity of Rh and Ru supported on Mg(Al)O was studied by changing the Mg/Al ratio in the precursors [48]; high Mg/Al ratio depressed the formation of the MgAl2O4 spinel and oppositely enhanced the formation of Mg(Al)O periclase, which in turn led to a narrow distribution of the particle size of both Rh and Ru metals, resulting in the high activity. Usually, Mg–Al HT is obtained with the carbonate as interlayer anion starting from metal nitrates since a NaOH/Na2CO3 couple is used as precipitating agent [49]. Various anions can be put in place of carbonates in the interlayer of Mg–Al HT. Starting from ternary Mg–Al(Rh) HT containing silicates as interlayer anions, new active and stable nano-structured catalysts were prepared by the calcination followed by the reduction [50]. The activity of these catalysts for the partial oxidation of CH4 was higher than those prepared from the conventional carbonate methods. Basile et al. [51] reported that synergy was observed in the partial oxidation of CH4 over Rh–Ni bimetallic catalysts prepared from quaternary Mg(Ni)–Al(Rh) HT precursors obtained by co-precipitation. Nagaoka et al. [52] carried out the reactions at very short residence times over the Rh–Ni bimetallic catalyst in the conditions where O2 is not totally converted; the high activity of the Rh/Ni catalyst is due to the fact that the Ni is maintained in the reduced state by the Rh. Rh–Ni bimetallic catalysts derived from the HT precursors were successfully tested in the autothermal reforming of CH4 in the presence of small amount of ethane [52]. Rh–Ni/Mg(Al)O catalyst showed a stable activity whereas Ni/Mg(Al)O catalyst was deactivated at very high space velocities (1.2 × 107 mL h−1 g −1cat ) through oxidation of metallic Ni. The enhanced stability of 25 wt%Ni–1 wt%Rh/Mg(Al)O catalyst against oxidation is attributed to H2 spillover from Rh in the NiRh alloy as well as to its high activity.
Improvement of Ni/Mg(Al)O catalysts by adopting the “memory effect”
Ni can also be incorporated by using nickel chelate as anion into interlayer of the Mg–Al HT [53–57], since Ni(II) has the ability to react with the anionic chelating agent of EDTA4−, producing the highly stable species [Ni(EDTA)]2−. We have studied on the effect of the preparation method of the precursors: (a) co-precipitation of Ni, Mg and Al nitrates with carbonate; (b) co-precipitation of Mg and Al nitrates with pre-synthesized nickel chelate and (c) anion exchange of NO −3 of HT with nickel chelate [53]. Ni/Mg17(Al2)O catalyst prepared by the method b was the most active in converting CH4, with a high H2 yield, in the steam reforming of CH4 probably due to the fact that they provided the best distribution of the active phase. When Mg(Al)O periclase was immersed in aqueous solution of the metal chelates, Mg–Al HT was reconstituted by the “memory effect” and the metal chelates were incorporated as anions in the surface HT structure. Rh, Ru and Pt were supported on Mg(Al)O by using noble metals pre-chelated with EDTA4−, which were incorporated in the interlayer of Mg–Al HT reconstituted by the “memory effect” [53]. The amount of noble metals could be lowered from 2.0 to 0.1 wt% of support without any decrease in catalytic activity or in selectivity to synthesis gas product in the CO2 reforming of CH4 [54]. The synthetic strategy applied for preparing noble metal catalysts, supported on Mg(Al)O, is summarized in figure 13 [56]. By calcining synthetic HT in air at 500 °C, Mg(Al)O was produced (step 1). Due to the “memory effect”, layered structure was then reconstituted, and negatively charged chelates of noble metals (MY−) were thus incorporated into the HT matrix (step 2). Calcination in air at 1000 °C converted HT to mixed metal oxide, mainly composed of Mg(Al)O periclase and of MgAl2O4 spinel (step 3). The metal introduced was supposed to be present in the form of fine oxide clusters (MO x ) bound to the surface of the support. Such surface-bound metal oxide clusters turned into metallic species (M0) under the reaction conditions (step 4) thus producing highly active and selective catalysts having a low coking capacity. Ru(0.1 wt%)–Ni(5.0 wt%)/Mg(Al)O catalyst revealed the highest activity and selectivity to synthesis gas, a stable durability and a low coking capacity in the CO2 reforming of CH4 [54–56].

Synthetic strategy of preparation of noble metal catalysts supported on Mg(Al)O.
Noble metals could be loaded on Ni/Mg(Al)O catalysts more simply by dipping the Mg(Ni,Al)O periclase particles in aqueous solution of noble metal nitrates. After the calcination followed by the reduction of the particles obtained, noble metal–Ni/Mg(Al)O catalysts have been prepared and successfully applied in the DSS operation of steam reforming of CH4 [58–60]. Small amount of noble metals, Rh, Pt and Ru, effectively suppressed the deactivation of the Ni0.5/Mg2.5(Al)O catalyst during the DSS operation of CH4 steam reforming. During the preparation of the noble metals–Ni0.5/Mg2.5(Al)O catalysts, the reconstitution of HT was clearly observed; XRD patterns of the samples during the preparation of Ru(0.1 wt%)–Ni0.5/Mg2.5(Al)O catalysts are shown in figure 14 [59]. The reflection lines of Mg2.5(Ni0.5)–Al HT were observed for the sample as deposited by co-precipitation (figure 14a) and those of Mg2.5(Al,Ni0.5)O periclase appeared after the calcination at 900 °C (figure 14b). After dipping the powders of Mg2.5(Al,Ni0.5)O periclase in aqueous solution of Ru(III) nitrate, Mg(Ni)–Al HT was reconstituted together with Mg(OH)2 brucite (figure 14c). The dipping treatment was followed by the drying using water bath at 100 °C, during which the reconstitution of HT was completed by the “memory effect” and no line of the periclase was observed. The formation of Mg(OH)2 brucite suggests that the reconstitution of HT from the periclase proceeds via the hydration of MgO, since MgO is thermodynamically unstable compared with Mg(OH)2 [46]. MgO reacts very easily even with moisture in the air, especially at low coordination atomic site, to form Mg(OH)2 brucite. The formation of Mg(OH)2 implies segregation of MgO from periclase, i.e., destruction of original periclase. However, Mg(OH)2 disappeared and the periclase phase regenerated after the calcination at 900 °C for 5 h (figure 14d). Moreover, the formation of Mg(OH)2 was suppressed by shortening the dipping time or by decreasing the amount of aqueous solution of Ru(III) nitrate. It must be noticed that Ru(III) was captured in the HT phase after the reconstitution although the detailed structure was not precisely determined. The lines of Ni metal appeared after the reduction of the sample calcined at 900 °C for 1 h (figure 14e). Interestingly after the 4th step of the DSS-like operation, the reflections of Ni metal were still observed intensively like as those just after the reduction and after the 1st step DSS-like operation (figure 14f) [59].

XRD patterns during the preparation of Ru(0.1 wt%)–Ni0.5/Mg2.5(Al)O catalysts. (a) Mg2.5(Ni0.5)–Al HT; (b) after calcination of (a) at 900 °C for 5 h (0.83 °C min−1); (c) after dipping (b) in an aqueous solution of Ru(III) nitrate, followed by drying; (d) after calcination of (c) at 900 °C for 5 h (1.7 °C min−1); (e) after reduction of (d) at 900 °C for 1 h (H2/N2 = 1/5); (f) after using (e) in the four cycled DSS operation followed by decreasing temperature to 200 °C under N2 purge. •, Mg(Ni)–Al HT; □, Mg(Al,Ni)O periclase; ▲, g(OH)2 brucite; ○, (Mg,Ni)Al2O4 spinel; ■, Ni metal.
Noble metal–Ni/Mg(Al)O catalysts for the DSS operation
The results of DSS-like operation over the supported Ru catalysts are shown in figure 15 [59]. The Ni0.5/Mg2.5(Al)O catalyst showed a clear deactivation just after the 1st steam purging, indicating that Ni metal was immediately oxidized by steam. The Ni oxidation was confirmed by XRD analyses; reflection of neither Ni metal nor NiO was observed, whereas those of Mg(Ni,Al)O periclase was sharpened, suggesting that Ni metal was oxidized to Ni(II) and was incorporated into the periclase. The 13.5 wt%Ni/γ-Al2O3 catalyst showed also the deactivation during the DSS operation. By the addition of 0.5 wt%Ru on 13.5 wt%Ni/γ-Al2O3, the catalytic behavior was improved, but the catalyst was slowly deactivated in the 2nd and 3rd runs and the activity was lost totally after the 3rd steam purging in the DSS-like operation. Such drastic deactivation is certainly due to the oxidation of Ni metal. The addition of Ru on the Ni0.5/Mg2.5(Al)O catalyst was quite effective for keeping the high activity during the 4 cycled DSS-like operation; even when the Ru loading decreased from 0.5 wt% to a tracel amount of 0.05 wt% (figure 15). With the Ru loading of 0.01 wt%, however, the catalyst was totally deactivated after the 3rd cycle. Usually supported metal catalysts have been prepared by incipient wetness method using an aqueous solution, the volume of which corresponds to pore volume of the support materials. The 0.1 wt%Ru–Ni0.5/Mg2.5(Al)O catalyst prepared by the incipient wetness method showed a clear deactivation after the 2 cycled DSS operation. No distinct HT reflection was observed in the XRD patterns of this catalyst, indicating that the “memory effect” plays important role in the present catalyst preparation. In the absence of Ni, the activity of 0.1–0.5 wt%Ru/Mg3(Al)O catalysts gradually decreased during the reaction independently on the cycle number, suggesting that the deactivation was not due to the Ru oxidation but probably due to the sintering or the coking on the catalyst. It must be emphasized that Ru alone was not enough active with such small loadings and the activity of the Ru–Ni0.5/Mg2.5(Al)O catalysts depended mainly on the Ni species (figure 15).

The results of DSS operation under steam purging in the steam reforming of CH4 over the Ru–Ni0.5/Mg2.5(Al)O catalysts. +, Ni0.5/Mg2.5(Al)O; ◊, 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O; Δ, 0.3 wt%Ru–Ni0.5/Mg2.5(Al)O; □, 0.1 wt%Ru-Ni0.5/Mg2.5(Al)O; ○, 0.05 wt%Ru–Ni0.5/Mg2.5(Al)O; *, 0.5 wt%Ru–13.5 wt%Ni/γ-Al2O3; •, 0.5 wt%Ru/Mg3(Al)O; ▲, 0.3 wt%Ru/Mg3(Al)O; ■, 0.1 wt%Ru/Mg3(Al)O.
The effects of Ru, Rh, Pd and Pt loading on the sustainability of the Ni0.5/Mg2.5(Al)O catalyst in the CH4 steam reforming under the steam purged DSS-like operation conditions are shown in figure 16 [58,59]. As noted previously, the Ni0.5/Mg2.5(Al)O catalyst was severely deactivated by the oxidation of Ni metal just after the 1st steam purging in the DSS operation [45]. The loading of small amount (0.05 wt%) of Ru, Rh and Pt brought about noticeable stability on this catalyst probably by suppressing the Ni oxidation with steam, whereas simple loading of the noble metal on Mg3(Al)O periclase showed only low activity. XRD patterns of 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O catalysts during the DSS operation in the steam reforming of CH4 are shown in figure 17. Just after the dipping, followed by the calcination, Ni was included as Ni(II) ions in the Mg2.5(Al,Ni0.5)O periclase (figure 17a). Ru was first trapped in the layered structure of Mg(Ni)–Al HT reconstituted and then, by the calcination, converted to the fine particles of RuO2 which cannot be observed in the XRD pattern. After the reduction, the Ni(II) ions in the Mg2.5(Al,Ni0.5)O periclase were reduced to Ni metal which came out on the surface of the periclase particles (figure 17b). Interestingly, such Ni metal species was still observed in the XRD throughout the DSS operation although the line intensity varied depending on the atmosphere (figure 17c and d). It is likely that Ni metal was partly oxidized by steam and was partly kept in the reduced state during steam purging, and moreover the oxidized Ni(II) was re-reduced during the steam reforming reaction at 700 °C. In the absence of noble metal, Ni0.5/Mg2.5(Al)O was completely deactivated and showed no reflection line of Ni metal after the 1st DSS operation [45]. All these results indicate that the active species is Ni metal even on the noble metal loaded Ni0.5/Mg2.5(Al)O catalysts and the noble metal, such as Ru, Rh and Pt, suppress the oxidation of Ni metal probably due to hydrogen spillover from the noble metals (vide infra) [59,60].

Effects of Ru, Rh, Pd and Pt loading on the sustainability of supported Ni catalysts in the CH4 steam reforming under steam purged DSS-like operation conditions a: ○, Ru–Ni0.5/Mg2.5(Al)O; •, Ru/Mg3(Al)O; b: ○, Rh–Ni0.5/Mg2.5(Al)O; •, Rh/Mg3(Al)O; c: ○, Pd–Ni0.5/Mg2.5(Al)O; D: ○, Pt–Ni0.5/Mg2.5(Al)O; •, Pt/Mg3(Al)O; - - -, CH4 conversion at thermodynamic equilibrium.

XRD patterns of 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O catalysts during the DSS operation in steam reforming of CH4. (a) after dipping Mg2.5(Al,Ni0.5)O periclase in aqueous solution of Ru(III) nitrate, followed by calcination at 850 °C for 5 h; (b) after reducing (a) at 900 °C for 1 h; (c) after using (b) in the 1st DSS operation at 700 °C, followed by keeping at 200 °C; (d) after heating (c) up to 700 °C; (e) after 4th DSS operation, followed by cooling to room temperature under N2 atmosphere. □, Mg(Al)O periclase; ■, Ni metal.
Noble metal–Ni/Mg(Al)O catalysts for the partial oxidation of propane
In contrast to CH4, only introductory studies concerning the partial oxidation of higher hydrocarbons have been limited to noble metal catalysts [61–70]. The extensive work by Schmidt and co-workers on short contact-time reactors [61,62] showed that Rh has high activity and selectivity, superior to that of other noble metals. Rh-impregnated alumina foams and metallic microchannel reactors were studied for production of hydrogen-rich syngas through short contact-time catalytic partial oxidation of propane [64,65]. However, these noble metal loaded catalysts do not seem acceptable for this process due to their high cost.
Only a few papers have been reported on Ni loaded catalysts for the partial oxidation of higher hydrocarbons [66,67]; Ni supported catalysts were modified by alkali metal or rare-earth metal oxides [66] and Ni/Mg(Al)O catalysts were prepared from HT as the precursors [67]. As a bimetallic catalyst, Pt–Ni/δ-Al2O3 was successfully devoted to the oxidative reforming of propane and butane [68–70]. The bimetallic systems showed superior catalytic performances compared to the monometallic systems probably due to the actions as micro heat exchangers; the heat generated by Pt sites during the exothermic total oxidation being readily transferred through the catalyst particles acting as micro heat exchangers to the Ni sites, which promote endothermic steam reforming [68–70]. We have found an improved behavior of Ru, Rh, Pd, Ir and Pt–Ni0.5/Mg2.5(Al)O bimetallic catalysts in the partial oxidation of propane under accelerated deactivation conditions [71,72].
The reaction was carried out with a temperature-recycled mode (figure 18) for testing the sustainability of the catalysts during the oxidative reaction atmosphere; the results of the partial oxidation of propane over the supported Ru, Rh, Pd, Ir and Pt catalysts are shown in figure 19 together with those over the Ni0.5/Mg2.5(Al)O, imp-Ni/γ-Al2O3, and the commercial RUA and FCR catalysts as controls [71]. The conversion of propane decreased with decreasing reaction temperature from 700 °C to 400 °C and again increased with increasing temperature to 600 °C; both decrease and recovery of the activity clearly depended on the metal species and the preparation method of the catalysts (figure 19A). The activities of both Ni0.5/Mg2.5(Al)O and imp-Ni/γ-Al2O3 significantly decreased at 400 °C and their recoveries in the activity at 600 °C were small. Among the commercial catalysts, the FCR showed a low activity together with a severe deactivation at 400 °C, and moreover the worst activity recovery at 600 °C. On the other hand, the RUA showed a considerable deactivation with decreasing the temperature from 700 °C to 400 °C, while a good recovery in the activity at 600 °C was observed. Among the noble metals–Ni bimetallic catalysts, the highest sustainability and the highest recovery were obtained over the Ru- and Rh–Ni0.5/Mg2.5(Al)O catalysts, followed by the Pd-, Pt- and Ir–Ni0.5/Mg2.5(Al)O catalysts. The rate of H2 production was 4.3 mol h−1 g −1cat for the Ru–Ni0.5/Mg2.5(Al)O catalysts at 700 °C [71].

Temperature-cycled operation mode for the partial oxidation of propane.

Partial oxidation of propane by temperature-cycled operation over noble metal-supported catalysts. a: Propane conversion; b: Selectivity to H2O. ○, 0.1 wt%Ru–Ni0.5/Mg2.5(Al)O; ◊, 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O; □, 0.1 wt%Rh–Ni0.5/Mg2.5(Al)O; Δ, 0.1 wt%Pd–Ni0.5/Mg2.5(Al)O; ■, 0.1 wt%Ir–Ni0.5/Mg2.5(Al)O; •, 0.1 wt%Pt–Ni0.5/Mg2.5(Al)O; ▲, Ni0.5/Mg2.5(Al)O; ×, imp-Ni/γ-Al2O3; +, FCR; *, RUA.
Partial oxidation of propane over Ni catalysts proceeds via combustion (7), followed by steam and dry reforming reactions (8 and 9). Direct partial oxidation of
propane (11) can proceed on Rh-supported catalysts as reported with short contact-time
reactors [73,74], and an excess loading of Ru on the Ni0.5/Mg2.5(Al)O catalyst resulted in an appearance of such activity (vide infra). However the contribution of direct partial oxidation is exceptional on the present Ni metal catalysts. Water–gas shift reaction (12), methanation (13), dehydrogenation of propane (14) and coke formation from propane (15) will play a role, depending on reactant composition, temperature and heat transfer
rate, residence time and the catalytic system involved. Additional side reactions, including cracking of propane (16) and carbon monoxide to carbon deposition (17)
must be considered; the latter is particularly unwonted and generally occurs when the O2/C3H8 ratio in the reaction mixture becomes too low.
In the present work, the excess O2/C3H8 molar ratio of 1.88 was adopted for accelerating deactivation compared with the molar ratio of 1.5 required for completing the syngas production by summing the reactions (7), (8) and (9). The selectivity to H2O inversely well correlated to the decrease in propane conversion (figure 19), suggesting that the deactivation takes place by the oxidation of Ni metal, resulting in an occurring of the combustion reaction (7) [71]. Assuming that the combustion reaction alone proceeded under the present reaction conditions, the conversion of propane is calculated as 37.5 % taking account of the molar ratio 1.88 of O2/C3H8 in the reaction (7). The Ni0.5/Mg2.5(Al)O catalyst was severely deactivated at 400 °C not only by the oxidation of Ni metal but also by the coking, judging from the propane conversion of 33% below 37.5% (figure 19A). The FCR showed only 0.6% propane conversion and negligible small H2 production at 400 °C (figure 19a); this is certainly due to both severe Ni oxidation and heavy coking (vide infra), resulting in a damaging of the active sites not only for the reforming but also for the combustion.
The product distributions in the first step reaction at 700 °C and the coke deposition after the total temperature-cycled operation of the propane oxidation are shown in table 6 [71]. Over both Ni0.5/Mg2.5(Al)O and iw-13.5 wt%Ni/γ-Al2O3 catalysts, a small amount of C2–C3 compounds, i.e., C2H4, C2H6 and C3H6, were produced together with H2, CO and CO2 as the main products. It must be emphasized that significant coke formation occurred on these supported Ni catalysts. Over both 0.1 wt%Ru/Mg3(Al)O and RUA catalysts, increasing selectivity to C2–C3 compounds was observed together with no significant coking, suggesting that Ru catalyzed the cracking of propane (16). The 0.1 wt% loading of noble metals on the Ni0.5/Mg2.5(Al)O catalyst remarkably suppressed both coking and formation of C2–C3 compounds except Ir loading; the effect was the most evident for the Ru loading as observed on the 0.1 wt%Ru–Ni0.5/Mg2.5(Al)O catalyst. Such properties of the Ru-loaded catalysts suggest that a remarkable synergy due to a strong interaction or a formation of alloy took place between Ru and Ni (vide infra).
Physico-chemical property of noble metal–Ni/Mg(Al)O catalysts
The BET surface areas, H2 uptakes, metal dispersion and particle sizes of Ni metal of the Ru, Rh, Pd, Ir and Pt-Ni0.5/Mg2.5(Al)O catalysts are shown in table 7 [71]. The surface area of the Ni0.5/Mg2.5(Al)O significantly decreased by the loading of noble metals. Contrarily, H2 uptake was drastically enhanced by the loading of noble metals on the Ni0.5/Mg2.5(Al)O catalyst; H2 uptake increased even with 0.01 wt% of Ru loading. At 0.1 wt% of noble metal loading, the Ru-, Rh- and Ir–Ni0.5/Mg2.5(Al)O catalysts showed higher values of the H2 uptake than the Pd- and Pt–Ni0.5/Mg2.5(Al)O catalysts. The contribution of noble metal itself to the H2 uptake on the noble metal-supported Ni0.5/Mg2.5(Al)O catalysts must be small, since the 0.1 wt%Ru/Mg3(Al)O catalyst showed an extremely small value of the H2 uptake. A strong synergy between noble metal and Ni metal is likely to occur on the catalysts, leading to an enhanced H2 uptake due to the formation of bimetal system such as alloy, etc.
TPR profiles of the Ru–Ni0.5/Mg2.5(Al)O catalysts with varying Ru loadings are shown in figure 20 [72]. In the absence of Ru, i.e., for the Ni0.5/Mg2.5(Al)O catalyst, a single and intensive peak appeared at 895 °C (figure 20a). The Ni reduction peak shifted toward lower temperature by the Ru loading (figure 20b–e); the shift became more significant with increasing the Ru loading on the Ni0.5/Mg2.5(Al)O catalyst and, additionally, a weak and broad peak appeared around 400 °C for the 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O catalyst (figure 20e). The decrease in the reduction temperature of Ni suggests either a formation of RuNi alloy or at least an occurring of strong interaction between Ru and Ni. The weak and broad peak observed around 400 °C for 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O catalyst can be ascribed to the reduction of RuO2 to Ru metal, since no other stable ruthenium oxides are known to exist in the solid state [75,76]. This indicates that a part of Ru was separated as prepared. The TPR of Ru/Al2O3 and Ru/MgO catalysts showed the Ru reduction peak around 250 and 235 °C, respectively [77,78]. However, a strong metal-support interaction was frequently observed on the catalyst having well-dispersed Ru particles, causing an increase in the reduction temperature. It seems that, in contrast to TiO2, Al2O3 has a tendency to stabilize ruthenium in the ionic state and the reduction temperature sometimes exceeds 700 °C [78]. It is likely that the RuO2 separated from the Ru–Ni bimetallic system exist as fine particles on the catalyst surface and showed a reduction peak around 400 °C in the TPR.

TPR profiles of supported Ru catalysts. (a) 0.1 wt%Ru/Mg3(Al)O; (b) 0.3 wt%Ru/Mg3(Al)O; (c) 0.5 wt%Ru/Mg3(Al)O; (d) Ni0.5/Mg2.5(Al)O; (e) 0.01 wt%Ru–Ni0.5/Mg2.5(Al)O; (f) 0.05 wt%Ru–Ni0.5/Mg2.5(Al)O; (g) 0.1 wt%Ru–Ni0.5/Mg2.5(Al)O; (h) 0.3 wt%Ru–Ni0.5/Mg2.5(Al)O; (i) 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O.
The 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O catalyst showed a serious deactivation exceptionally at 400 °C (figure 19a), however the activity was quickly recovered when the temperature was given back to 600 °C. Balint et al. [76] reported that the equilibrium exists between RuO2 and Ru metal around 450 °C on 12 wt%Ru/Al2O3 catalyst in the partial oxidation of CH4. The formation of RuO2 phase in the reaction mixture (O2/CH4 = 1.87), which is favored in the temperature below 450 °C, was found to be responsible for the combustion of CH4 to CO2 and H2O. The RuO2 ↔ Ru equilibrium is shifted to the formation of Ru metal when the reaction temperature increased above 450 °C. At the moment, the reaction rate increased considerably, and CO and H2 simultaneously produced as the primary reaction products by the direct partial oxidation (11). Propane conversion decreased on the 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O catalyst exceptionally at 400 °C compared with those above 500 °C (figure 19a). This phenomenon can be explained by the presence of RuO2 at 400 °C as observed in the TPR (figure 20); Ru catalyzed direct partial oxidation of propane (11) above 500 °C, whereas RuO2 catalyzed the combustion of propane (7) at 400 °C resulting in a decrease in propane conversion compared with the syngas formation by direct partial oxidation under the present molar ratio (1.87) of O2/C3H8 [71].
Role of noble metals in the bimetallic active species
XRD patterns of both 0.1 and 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O catalysts after the reduction are shown in figure 21 together with that of the Ni0.5/Mg2.5(Al)O catalyst as a control. The Ni0.5/Mg2.5(Al)O catalyst showed rather sharp and intensive reflection lines of Ni metal (figure 21Aa), which were broadened and shifted toward the lower reflection angles by the addition of 0.1 wt% Ru (figure 21Ab) [71]. Both line broadening and lower angle-shift became significant with increasing Ru loading up to 0.5 wt% (figure 21Ac) and were more clearly observed for the reflection of Ni (200) at 2 theta = 52.5° with increasing Ru loading (figure 21Ba, b and c). It is suggested that RuNi alloy was formed and/or the size of Ni metal particles decreased still keeping the strong interaction with the support by the Ru loading on the Ru–Ni0.5/Mg2.5(Al)O catalysts. Basile et al. [48] reported that Rh was completely soluble in the Mg(Al)O periclase phase, whereas Ru was not soluble in the Mg(Al)O periclase and remained as separated phase, when both were incorporated into the HT precursors with a large Mg/Al ratio. Such Ru species separated from the periclase reacted with Ni species liberated from the Mg2.5(Al,Ni0.5)O periclase during the reduction, resulting in the formation of RuNi alloy and/or well dispersed Ni metal particles on the Ni0.5/Mg2.5(Al)O catalysts.

XRD patterns of the supported Ru–Ni bimetallic catalysts after the reduction. (a) Ni0.5/Mg2.5(Al)O; (b) 0.1 wt%Ru–Ni0.5/Mg2.5(Al)O; (c) 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O. □, Mg2.5(Al,Ni0.5)O periclase; ■, Ni metal; ○, Mg(Ni)2Al2O4 spinel.
As shown in table 7, H2 uptake was drastically enhanced by the Ru loading on the Ni0.5/Mg2.5(Al)O catalyst. Particle sizes of Ni metal on the catalysts were calculated from the data of both XRD and H2 uptake [79,80]. In both cases, the Ni particle size decreased and the H2 uptake increased with increasing Ru loading. These can be well explained by the dependency of the H2 uptake on the metal particle size. The highest activity as well as the highest sustainability in the partial oxidation of propane was obtained with the 0.1 wt%Ru–Ni0.5/Mg2.5(Al)O catalyst. The contribution of Ru itself in the H2 uptake on the Ru–Ni0.5/Mg2.5(Al)O catalysts must be small, since the H2 uptake was significantly small on the 0.1 wt%Ru/Mg3(Al)O catalyst. The reforming activity of the Ru–Ni bimetallic catalysts is mainly due to the presence of Ni metal on the catalysts surface, since the selectivity to H2 was remarkably low on the 0.1 wt%Ru/Mg3(Al)O catalyst at 700 °C (table 6). The results of XAFS analyses of both Ru and Ni in 0.5 wt%Ru–Ni0.5/Mg2.5(Al)O catalysts suggest that Ru metal species were finely dispersed on or in the surface layer of the fine Ni metal particles [80]. Similar phenomena have been reported by Tomishige and co-workers [81]; surface modification of Ni catalysts with trace Pt has been much more efficiently achieved by the sequential impregnation method (Pt/Ni) than the co-impregnation method (Pt + Ni) on γ-Al2O3 as the support in the oxidative reforming of CH4. It is concluded that the catalytic property of Ni metal was enhanced by the loading of noble metal, probably first due to the formation of finely dispersed Ni metal particles and further due to the synergy between Ni metal and noble metal on the surface of fine Ni metal particles.
Self-activation of the 0.1 wt%Ru, Rh or Pt–Ni0.5/Mg2.5(Al)O catalysts were tested in the steam reforming of CH4; the catalysts as calcined at 900 °C were used in the reaction, i.e., without pre-reduction treatment with H2. Pt was the most effective, followed by Rh, and Ru was not so effective. However, not only the Pt–Ni and Rh–Ni systems but also the Ru–Ni system as calcined at 900 °C catalyzed the partial oxidation of propane, suggesting that noble metals worked for the C–H bond fission and liberated hydrogen atoms on the catalyst surface. As shown in figure 22 [72], noble metals quickly dissociate C–H bond of methane or propane to produce H atoms, which in turn rapidly migrate to Ni surface by spillover, reduce the oxidized Ni. As a result, the Ni species on the catalysts was kept in the reduced and active state during the reaction.

Sustainability of noble metal–Ni/Mg(Al)O catalysts due to spillover of hydrogen.
Application of Cu/Zn/Al mixed oxides derived from aurichalcite or HTlcs
Cu/ZnO catalysts derived from aurichalcite
We have prepared Cu/Zn/Al catalysts starting from aurichalcite or HT precursors obtained by the homogenous precipitation (hp-) using urea and successfully applied them in the steam reforming of methanol [82,83], dimethyl ether (DME) [84] and the CO shift reaction [85,86]. Malachite, Cu2(OH)2CO3, aurichalcite, (Cu,Zn)5(CO3)2(OH)6, and HT, (Cu,Zn)3Al2CO3(OH)16, all belonging to hydroxy carbonates were formed in the catalysts. The structure and the catalytic activity of hp-Cu/Zn/Al have been compared with those prepared by the co-precipitation (cp-) method. The pure formation of aurichalcite with rather low crystallinity in the precursors by the hp-method afforded high surface area of Cu metal after the calcination followed by the reduction, resulting in the high and sustainable activity for the catalytic reactions.
Cu/ZnO catalysts for methanol steam reforming
Hydrogen can be obtained directly from methanol according to three different processes: steam reforming (18), partial oxidation (19) and thermal decomposition (20)
[86–90]. Most fuel-processing technologies reported in the literature so far have been based on either steam reforming (18) or partial oxidation (19); both produce almost quantitatively H2 and CO2. While partial oxidation is exothermic, steam reforming is endothermic and produces a more favorable H2/CO2 ratio. Another option is to combine reactions (18) and (19) by co-feeding simultaneously oxygen, steam and methanol through the oxidative methanol reforming. In this process, the heat necessary to maintain steam reforming is supplied by partial oxidation by tuning the ratio of the three reactants as such that the overall reaction heat is nearly neutral.
Typical XRD patterns of the precursors of the hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts are shown in figure 23A and B, respectively [83]. In the hp-Cu/ZnO series, malachite, Cu2(OH)2CO3, phase was formed in the Cu-rich samples and was replaced by aurichalcite, (Cu,Zn)5(OH)6(CO3)2, phase with increasing Zn content (figure 23A a–c). In the hp-Cu/ZnO/Al2O3 series, malachite was obtained as the major component together with HT, (Cu,Zn)6Al2(OH)16CO3 · 4H2O, as a concomitant in the Cu-rich samples. With increasing Zn content, aurichalcite phase replaced malachite phase and became a major component (figure 23B a–c). In both hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 series, further increase in the Zn content caused the phase shift from aurichalcite to hydrozincite, Zn5(OH)6(CO3)2 (figure 23A c–e and B c–e). The mineral aurichalcite (Cu/Zn ratio is 0.67) crystallizes in the monoclinic space group P21/m (C 2 2h ) with lattice parameters: a = 13.82, b = 6.419, c = 5.29 Å and β = 101.04° (Z = 2) [91]. On the other hand, the mineral hydrozincite crystallizes in the monoclinic space group C2/m (C 3 2h ) with lattice parameters: a = 13.62; b = 6.30; c = 5.42 Å and β = 95.83° (Z = 2) [92]. Natural and synthetic aurichalcites are known with the Cu/Zn ratios in the range of 0.2–1 [93,94], whereas synthetic hydrozincite forms solid solution (Cu x Zn1−x )5(OH)6(CO3)2 (x = 0–0.3) [94]; phase shift from aurichalcite to hydrozincite seems to take place continuously by replacing the Cu sites with Zn [91, 92, 95].

XRD patterns of the precursors of the hp-Cu/ZnO (A) and hp-Cu/ZnO/Al2O3 (B) catalysts after the precipitation. (A): (a) hp-Cu90/Zn10; (b) hp-Cu70/Zn30; (c) hp-Cu50/Zn50; (d) hp-Cu30/Zn70; (e) hp-Cu10/Zn90. (B): (a) hp-Cu80/Zn10/Al10; (b) hp-Cu60/Zn30/Al10; (c) hp-Cu45/Zn45/Al10; (d) hp-Cu30/Zn60/Al10; (e) hp-Cu10/Zn80/Al10. •, (Cu,Zn)5(CO3)2(OH)6: aurichalcite; □, Zn5(CO3)2(OH)6: hydrozincite; ○, Cu2CO3(OH)2: malachite; ▲, Zn4CO3(OH)6 · H2O; ■, (Cu,Zn)6Al2(OH)16CO3 · 4H2O: HT.
It was reported that the presence of Cu(II) ions makes the synthetic procedures of pure HT more complex by conventional urea hydrolysis procedure [95,96], at least for two reasons. First, Cu(II) ions show the Jahn–Teller effect that favors the formation of distorted octahedral structures. As a result, the insertion of Cu(II) ions into the brucite-like sheet is unfavored, and preferentially gives rise to the precipitation of malachite phases. Secondarily, Cu(II) ions can be depleted by ammonia originated from urea hydrolysis. The first issue was confirmed in the present work by the preferential formation of malachite phase. However the second point was not so detrimental in the present work; no significant dissolution of Cu was detected in the absence of Al and the metal compositions determined after the calcination were actually same as those in the raw materials used in the preparation [85,86]. Moreover, it has been found [4] that the presence of other divalent cations, such as Mg(II), Zn(II) and Co(II), favors the accommodation of Cu(II) into the sheet, giving rise to ternary HT phases.
The line intensity of aurichalcite (figure 23) well correlates to the catalytic activity shown as CH3OH conversion (vide infra in figure 25), judging from their dependences on the Cu/Zn composition. Aurichalcite possesses closely located Cu(II) and Zn(II) ions which are bridged by oxygen atoms each other; such crystal structure results in the formation of highly dispersed CuO and ZnO particles which are strongly interacted each other after the calcination and finally form highly dispersed Cu metal particles after the reduction. SEM observation of the precursors of the hp-Cu50/Zn50 catalyst clearly showed a formation of spherical fine particles (ca. 10 μm in diameter) in the magnifications of ×500 and ×8500 (figure 24a and b) [83]. Spherical fine particle seems to be assembly of thin plate-like structures and the surface is not smooth in the magnification of ×20,000 (figure 24c). Spherical fine particles were most evidently observed for the samples showing the strong reflection of aurichalcite in the XRD (figure 23). Such spherical fine particle structure with high surface area was apparently maintained even after the calcination at 300 °C for 3 h in air, and is supposed to possess an important role in the catalytic activity. When the Cu content was increased up to 80 mol % (hp-Cu80/Zn20), some aggregates of fine particles were observed with both symmetry and sphericalness lost as seen in the magnification of ×2500 (figure 24d). This phenomenon well correlates with the low activity of hp-Cu80/Zn20 compared with that of hp-Cu50/Zn50 shown in figure 25A [83].

SEM images of the hp-Cu30/Zn70 and hp-Cu80/Zn20 catalysts. Catalysts and magnifications: (a) hp-Cu50/Zn50 ×500; (b) hp-Cu50/Zn50 ×8500; (c) hp-Cu50/Zn50 ×20,000; (d) hp-Cu80/Zn20 ×2500.

CH3OH steam reforming over the hp-Cu/ZnO (A) and hp-Cu/ZnO/Al2O3 (B) catalysts. CH3OH/H2O/N2 = 10/12/30 mL min−1; catalysts, 200 mg; pre-reduced with H2/N2 (5/30 mL min−1) at 350 °C for 20 min. •, CH3OH conversion (—, 250 °C; - - - -, 200 °C); □, BET surface area; ○, Cu(0) surface area.
The results of steam reforming of methanol over each series of the hp-Cu/ZnO and the hp-Cu/ZnO/Al2O3 catalysts are shown in figure 25A and B, respectively. In both cases, the activity well correlated with the surface area of Cu metal after the reduction, but not with the BET surface area, suggesting the formation of Cu(0) as the active species. The activity of both hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts increased with increasing Cu content up to the Cu/(Cu + Zn) atomic ratio of 0.5 (figure 25A and b) and the latter showed higher activity than the former. Temperature programmed reduction of hp-Cu/ZnO showed a decrease in the reduction temperature of Cu around the atomic ratio of 0.5 [85,86], indicating an easy reduction of Cu(II) by the formation of Cu/Zn binary system. Further increase in the Cu content resulted in a decrease in the activity over the hp-Cu/ZnO catalysts (figure 25A), whereas the activity shown by methanol conversion on the hp-Cu/ZnO/Al2O3 catalysts kept a high value even when the atomic ratio increased above 0.5 (figure 25b).
In the XRD patterns of the precursors of both hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 samples after the precipitation, various phases were observed together with aurichalcite depending on the Cu/Zn compositions (figure 23A and B). After the calcination at 300 °C for 3 h in air, all these phases disappeared in the XRD patterns and the reflection lines of both CuO and ZnO appeared for all Cu/Zn-based catalysts with varying intensities depending on the metal compositions and the preparation methods. After the reduction treatment, the lines of CuO were replaced by those of Cu metal. Particle sizes of CuO and ZnO after the calcination, Cu metal and ZnO after the reduction and those after the reaction were calculated from the line width in the XRD using Scherrer equation and are shown in table 8. Before the reaction the particle size of CuO could not be precisely determined for both hp-Cu50/Zn50 and hp-Cu45/Zn45/Al10 samples, since the CuO lines were too much broadened and feeble. The sizes of ZnO particles were also small for both hp-Cu50/Zn50 and hp-Cu45/Zn45/Al10 samples. After the reduction at 350 °C, the particle sizes of both Cu metal and ZnO were the smallest for both hp-Cu50/Zn50 and hp-Cu45/Zn45/Al10 samples among the catalysts tested, although the increasing was observed (table 8). The addition of Al2O3 was obviously effective for suppressing the size growth of the particles of CuO, Cu metal and ZnO for the hp-Cu/Zn-based catalysts. After the reaction between 200 and 300 °C, each particle size of Cu metal and ZnO slightly increased. Increase in the Cu metal particle size was more significant for the cp-catalysts than the hp-catalysts.
When stoichiometric amount of oxygen, i.e., O2/CH3OH = 1/2, for completing the partial oxidation (19) was present in the feed gas, the conversion of methanol increased at low temperatures but the selectivity to CO was kept low. It was reported that CH2O, (CH3)2O and CH4 were produced as the by-products together with H2, CO2 and CO in the oxidative steam reforming of methanol (CH3OH/H2O/O2 = 10/11/1.2) over Cu/ZnO/Al2O3 (Cu/Zn/Al = 18/33/49) catalyst derived from the HT precursors [83]. However, none of CH2O, (CH3)2O and CH4 was produced in the present reaction over both hp-Cu/Zn and hp-Cu/Zn/Al catalysts. Time course of the oxidative steam reforming reaction for 25 h over the hp-Cu45/Zn45/Al10, the hp-Cu50/Zn50, and the cp-Cu50/Zn50 catalysts, together with a commercial Cu/ZnO/Al2O3 (MDC-7) catalyst as a control, are shown in figure 26. The hp-Cu45/Zn45/Al10 catalyst showed high and stable activity, followed by the MDC-7. The hp-Cu50/Zn50 catalyst showed almost similar activity and sustainability to the MDC-7 as well as higher activity than the cp-Cu50/Zn50 catalyst. The selectivity to CO was always less than 1.0% over all catalysts tested. The hp-Cu50/Zn50 catalyst showed almost similar CO selectivity to that of the cp-Cu50/Zn50 catalyst, although the activity was far higher on the former than on the latter. Moreover, the CO selectivity gradually decreased during the reaction over the hp-Cu45/Zn45/Al catalyst, although no decline in the CH3OH conversion was observed, suggesting that the catalyst surface was improved by the Al2O3 addition to suppress the CO formation.

Oxidative steam reforming of CH3OH over the Cu/Zn-based catalysts. Catalyst, 0.2 g; CH3OH/H2O/O2/N2 = 10/12/5/30 mL-NTP min−1; reaction temperature, 200 °C. Full line, CH3OH conversion; dotted line, CO selectivity. •, hp-Cu45/Zn45/Al10; ■, MDC-7; ▲, hp-Cu50/Zn50; ×, cp-Cu50/Zn50
Judging from the results of TPD of both CH3OH and HCHO on the Cu/ZnO/Al2O3 catalysts, it is considered that CH3OH is dehydrogenated to form HCHO and H2 on Cu metal as the first step of steam reforming of methanol [82]. HCHO is then converted to HCOOCH3 by a nucleophilic attack of CH3OH under the steam reforming reaction conditions [97]. As a consequence, the reaction scheme is considered as shown in scheme 1: CH3OH is dehydrogenated to HCHO, HCHO is followed by a nucleophilic attack of H2O to form HCOOH, which in turn decomposed to H2 and CO2 on the Scheme 1 hp-Cu/Zn-based catalysts [83].

Moreover, hp-Cu/ZnO/Al2O3 catalyst was combined with ZSM-5 and successfully applied in the steam reforming of DME [84]. The reaction proceeded via a successive two-steps mechanism: hydrolysis of DME to methanol over solid acids, followed by steam reforming of methanol to produce H2 and CO2 over the Cu-based catalysts. The highest activity was obtained, when powders of both hp-Cu/ZnO/Al2O3 and ZSM-5 were kneaded in an agate mortar, pressed and sieved to the particles of composite catalysts. Either physical mixing of the particles of both catalysts or separate filling of ZSM-5 and hp-Cu/ZnO/Al2O3 resulted in the low activity, indicating that the closest packing of both active sites for the DME hydrolysis and the methanol steam reforming is required for the DME steam reforming. As the Cu-catalyst in the composite catalysts, the hp-Cu/ZnO/Al2O3 catalyst showed higher activity than the hp-Cu/ZnO catalyst and moreover than that prepared by co-precipitation. It is likely that the hp-preparation affords more finely dispersed Cu metal particles and moreover Al component stabilizes the finely dispersed Cu metal particles on the catalysts against sintering.
Cu/ZnO catalysts for the water–gas shift reaction
In the polymer electrolyte fuel cells (PEFCs) system, hydrogen is used as a fuel; it is supplied from steam reforming of hydrocarbons such as methane, propane and kerosene. The problem is that the reformed gas contains CO at the level of 1–10% which adsorbs irreversibly on the Pt electrode of the PEFC at the operating temperature (ca. 80 °C) and hinders the electrochemical reaction. Therefore, CO must be removed from the reformed gases to less than 10–20 ppm before feeding the gas mixture to the Pt electrode. The shift reaction (12) is desirable for removal of a large amount of CO since it is moderately exothermic reaction and the reaction temperature is easy to control. Lower temperature is favored for higher CO removal due to the exothermic shift reaction, whereas the reactant gases are not active enough to reach the chemical equilibrium at low temperature judging from the kinetics of the reaction. Therefore, a further development of the catalyst is required for breaking this bottleneck.
Ternary Cu/ZnO/Al2O3 catalysts have been widely employed commercially since the early 1960s in the water–gas shift reaction; the catalyst was usually prepared by co-precipitation method to afford the higher Cu metal dispersion in the resulting catalyst and, as a consequence, the higher catalytic activity [98]. We have prepared the hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts using urea hydrolysis and were successfully applied for hydrogen production by steam reforming of methanol [82,83]. Their good catalytic performances were due to both highly dispersed Cu metal species and high accessibility of the Cu metal species to methanol and steam. It is frequently pointed out that reverse water–gas shift reaction significantly contributes to the product selectivity in the steam reforming of methanol, since the reforming reaction produces actually CO2 at relatively high pressure [98,99].
Activities of the hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts for the shift reaction are shown in figure 27A and B, respectively, together with the BET surface area and the Cu metal surface area [85]. The marks with asterisk (*) show the reaction results and the surface areas obtained over the cp-Cu/ZnO (figure 27A) and cp-Cu/ZnO/Al2O3 (figure 27B) catalysts. The activity of the hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts was higher than that of the cp-Cu/ZnO and cp-Cu/ZnO/Al2O3 catalysts, respectively. The catalytic activity well correlated with the surface area of metallic copper which increased by the Cu–Zn binary system formation for both Cu/ZnO and Cu/ZnO/Al2O3 catalysts.

Activity and physical properties of hp-Cu/ZnO (A) and hp-Cu/ZnO/Al2O3 (B) catalysts. Catalyst, 200 mg; flow rate: CO/H2O/N2 = 0.7/2.2/47.1 mL-NTP min−1; reaction temperature, 150 °C. -•-, CO conversion; ···▲···, Cu(0) surface area; ···■···, catalyst surface area. The marks with sterisk show the results obtained over cp-Cu/ZnO (A) and cp-Cu/ZnO/Al2O3 (B) catalysts.
Turn over frequencies (TOF) over the hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts were obtained for the shift reaction at the reaction temperature of 110 °C and at GHSV = 60,000 mL h−1 g −1cat (figure 28) [85]. When the TOF values were calculated based on the Cu metal surface area (dotted lines), the values decreased with increasing Cu content in both hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts. In Auger-electron spectra of the hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts after the reduction (figure 29A and B), Cu(0) and Cu(I) species were observed with the following values of the kinetic energy (Cu-LMM): 918.5 and 916.5 eV, respectively [100]. A peak assigned to Cu(I) was clearly observed together with that of Cu(0) for both hp-Cu/ZnO (figure 29A) and hp-Cu/ZnO/Al2O3 (figure 29B) catalysts varying their intensities depending on the Cu/Zn ratio. Moreover a satellite peak characteristic of Cu(II) was unobservable around 945 eV in the XPS of Cu 2p3/2 on both hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts (data not shown) [101]; thus, the oxidation state of Cu could be zero- or mono-valent. It is therefore concluded that Cu(I) species was formed on both hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts after the reduction. The values of TOF were re-calculated based on the surface amount of Cu(I) estimated from Auger peak ratio of Cu(I)/Cu(0) assuming that Cu(I) species was localized on the catalyst surface (figure 28). The TOFs converged to constant values independently on the Cu content on both hp-Cu/ZnO (0.67 s−1) and hp-Cu/ZnO/Al2O3 catalysts (0.58 s−1), strongly supporting the hypothesis of the Cu(I) species as the active site [85].

Turn over frequencies of hp-Cu/ZnO and hp-Cu/ZnO/Al2O3 catalysts. ○ and •, hp-Cu/ZnO; □ and ■, hp-Cu/ZnO/Al2O3; ···, calculated based on Cu(0) obtained by N2O pulse method; –, calculated based on Cu(I) obtained from Auger-electron spectra.

Auger electron spectra of hp-Cu/ZnO (A) and hp-Cu/ZnO/Al2O3 (B) after. reduction. A: a, hp-Cu/ZnO (90/10); b, hp-Cu/ZnO (80/20); c, hp-Cu/ZnO (70/30); d, hp-Cu/ZnO (60/40); e, hp-Cu/ZnO (50/50); f, hp-Cu/ZnO (40/60); g, hp-Cu/ZnO (30/70); h, hp-Cu/ZnO (20/80); i, hp-Cu/ZnO (10/90). B: a, hp-Cu/ZnO/Al2O3 (80/10/10); b, hp-Cu/ZnO/Al2O3 (70/20/10); c, hp-Cu/ZnO/Al2O3 (60/30/10); d, hp-Cu/ZnO/Al2O3 (50/40/10); e, hp-Cu/ZnO/Al2O3 (45/45/10); f, hp-Cu/ZnO/Al2O3 (40/50/10); g, hp-Cu/ZnO/Al2O3 (30/60/10); h, hp-Cu/ZnO/Al2O3 (20/70/10); i, hp-Cu/ZnO/Al2O3 (10/80/10). All samples were pre-reduced in a mixed gas flow of N2/H2 (30/5 mL min−1).
When MgO was added in the hp-Cu/ZnO catalyst, Cu metal surface area increased, resulting in an increase in the activity (figure 30) [85]. The Mg contents actually obtained by ICP measurements were far smaller than those used in the raw materials, indicating that Mg could not be efficiently incorporated in the catalyst. This is probably due to the pH value at 7.0 in the catalyst preparation, since Mg easily dissolved in acidic or neutral aqueous solution. It was reported that the isoelectric point (pHIEP) of brucite (Mg(OH)2) was close to 11 and that steady-state dissolution rates of brucite were high in neutral to acid solution (pH ≤ 8), whereas the rates decreased abruptly in alkaline solution (pH > 8) [102]. Addition of 0.1 atomic % of Mg caused a significant increase in the CO conversion as well as in the Cu metal surface area (figure 30) [86].

Activity and physical properties of hp-Cu/MgO/ZnO catalyst. Catalyst, 200 mg; flow rate: CO/H2O/N2 = 0.7/2.2/47.1 mL-NTP min−1; reaction temperature, 150 °C. -•-, CO conversion; ···▲···, Cu(0) surface area; ···■···, BET surface area.
Two plausible mechanisms have been proposed in the shift reaction over Cu catalysts: one is the formate intermediate mechanism [103] and another is the surface reduction-oxidation mechanism [104,105]. In the formate intermediate mechanism, H2O dissociates to OH(a) and then OH(a) reacts with CO to form a formate intermediate which finally decomposes to CO2. In the reduction-oxidation mechanism, the surface is oxidized by H2O and subsequently reduced by CO to form CO2. In both cases, H2O dissociates, whereas CO is oxidized, on the catalyst surface. Frequently the O–H bond cleavage [106,107] and scarcely the CO oxidation [105,108] work as the rate-determining step of the shift reaction on Cu metal surface. In situ observation of Cu LMM Auger electron spectra of the hp-Cu/ZnO (45/55) catalyst during the steam treatment is shown in figure 31c; the hp-Cu/ZnO (45/55) pre-reduced was treated in a mixed gas flow of N2/H2O (45/2 mL-NTP min−1) at 150 °C for 60 min [86]. A peak at 918.5 eV assigned to Cu(0) disappeared, whereas a peak at 915.5 eV assigned to Cu(I) was strengthened; the kinetic energy was slightly lower than the value 916.5 eV reported by Batista et al. [100]. This clearly indicates an occurrence of the oxidation of Cu(0) to Cu(I) during the steam treatment, suggesting that the shift reaction proceeds via the reduction-oxidation mechanism expressed by equations (21) and (22):

Auger electron spectra of hp-Cu/ZnO (45/55) (a), hp-Cu/MgO/ZnO (45/2/53) (b) and hp-Cu/ZnO (45/55) treated by steam (c). c: hp-Cu/ZnO (45/55) pre-reduced in a mixed gas flow of N2/H2 (30/5 mL min−1) at 350 °C for 20 min was treated in a mixed gas flow of N2/H2O (45/2 mL min−1) at 150 °C for 60 min.
It is most likely that Cu(I) sites observed on the hp-Cu/MgO/ZnO catalyst are the active sites and locate at the surface boundary between Cu metal particles and ZnO crystals, where MgO assists their formations by stabilizing the cationic species. The cationic Cu(I) sites chemisorb CO as the first step or the reaction, followed by the reduction to Cu(0), which in turn provoke H2O dissociation as well as O–H bond cleavage, resulting in an acceleration of the surface oxidation-reduction reactions (21) and (22).
According to the results of Auger-electron spectroscopy of the hp-Cu/ZnO and hp-Cu/MgO/ZnO catalysts after the reduction, Cu(I) species has been clearly observed together with Cu(0) species (figure 31a and b) and possibly worked as the active site for the shift reaction. It is likely that the shift reaction was catalyzed via a reduction-oxidation mechanism between Cu(0) and Cu(I), in which Cu(I) sites chemisorbed and oxidized CO to CO2 to form Cu(0), whereas the reduced Cu(0) sites were reoxidized by H2O to form Cu(I) and H2 (figure 31c) [86]. However, a close similarity was still observed in the activity pattern and the exposed Cu metal surface area determined by N2O adsorption/decomposition test. In spite of the hypothesis of Cu(I) active sites, an important role Cu(0) cannot be perfectly denied since the reaction proceeded by the reduction-oxidation mechanism between Cu(0) and Cu(I). Moreover, the amount of Cu(i) sites reasonably depends on the particle size of Cu metal if Cu(I) forms at the boundary between Cu metal particles and ZnO particles, suggesting an apparent dependency of the activity on Cu(0) species.
Cu/ZnO/Al2O3 catalysts derived from HTs
Usually Cu/Zn catalysts contains Al component as the stabilizer in the industrially applied catalysts; ternary Cu/ZnO/Al2O3 catalysts have been widely employed commercially not only in the water–gas shift reaction but also in steam reforming of methanol, etc. The catalyst was usually prepared by co-precipitation method to afford the higher Cu metal dispersion in the resulting catalyst and, as a consequence, the higher catalytic activity, although the precursors usually contained several phases, such as malachite, bayerite, aurichalsite and HT [82, 98]. The insertion of copper ions into the brucite-like sheet is unfavored due to the Jahn–Teller effect producing a distorted octahedral disposition of OH groups around the copper ion [3,96]. However, the presence of other divalent cations, such as Mg(II), Zn(II), and Co(II), favors the accommodation of Cu(II) into the sheet, giving rise to ternary HT phases [4]. Actually, several Cu catalysts derived from HT precursors have been tested for the steam reforming of alcohols [96, 107, 109–111]. The Cu/Zn/Al catalysts derived from HTlcs precursors prepared by the homogeneous precipitation from metal chlorides using urea hydrolysis were successfully applied for the oxidative steam reforming of methanol, where Cu2O species was detected on the active catalyst surface by the Auger spectroscopy [96]. Cu/ZnO/Al2O3 catalysts similarly prepared showed total CH3OH conversion at 300–400 °C with GHSV = 0.6–1.2 × 105 h−1 as well as high selectivity giving H2 yield of 2.5–2.7 mol per mol CH3OH with CO concentration below the detection limit (500 ppm) [107,109]. The catalytic activity was almost independent on the copper content in the range 5–12% (atomic metal content) and the direct oxidation of CO to CO2 by Cu(II) ions is likely a key step both for oxidative steam reforming of methanol and water gas shift over the catalysts [111]. Cu1−x Ni x ZnAl mixed oxides catalysts derived from a HT precursor were tested for the oxidative steam reforming of ethanol [112]. Dehydrogenation of C2H5OH to CH3CHO was favored by Cu-rich catalyst, whereas introduction of Ni leaded to C–C bond rupture and producing CO, CO2 and CH4. H2 yield (selectivity) varied between 2.6 and 3.0 mol/mol of ethanol converted (50–55%) for all catalysts at 300 °C.
When 1–2% of metal loaded catalysts was prepared impregnating spinel-oxide mixture (ZnAl2O4/ZnO) obtained by thermal decomposition of Zn–Al HT at 900 °C with palladium nitrate solution, the catalysts showed a quite good catalytic activity with high methanol conversion and good selectivity to CO2 [113]. The high activity and selectivity were attributed to the prevailing presence, on the catalyst surface, of a highly dispersed 1:1 PdZn alloy phase. Pd-containing Cu/ZnO/Al2O3 catalysts were prepared by impregnation of the base Cu/ZnO/Al2O3 catalyst with an aqueous solution of Pd(NO3)2 and used for the methanol synthesis from CO2 and H2 mixture [114]. A dramatic decrease in the methanol yield was observed by Pd impregnation; during the impregnation the Cu/ZnO/Al2O3 catalyst was allowed the partial reconstitution of original HT structure by the “memory effect,” which improved the crystallinity of the copper oxide phase leading to the catalyst deactivation. However, such reconstitution of HT structure can oppositely improve the catalytic behavior of Cu/ZnO/Al2O3 catalysts as observed in the preparation of noble metal–Ni/Mg(Al)O catalysts. We have started the preparation of the Cu/ZnO/Al2O3 catalysts loaded with noble metals by adopting the “memory effect” on Zn(Cu)–Al mixed oxides obtained from Zn(Cu)–Al HT [115], expecting that such trial will be opening a new area of the catalyst preparation.
Conclusion
HTlcs possess the structure basically composed of brucite-like layers and anions; various metal cations or metal-containing anions can be incorporated. These metal species are randomly distributed in the brucite phase and possibly show the catalytic activity for various types of reaction. HTlcs after the heat treatment produced the oxides with very small crystal size, stable to thermal treatments, which can be applied as the catalysts possessing high surface area, basic properties and thermally stable metal crystallites by reduction. Moreover, by adopting the “memory effect,” HT structure was reconstituted in the mixed oxide derived from HT. Such HTs finely modify the surface of mixed oxides and also showed a unique catalytic activity in the various reactions.
Ni(II)/Mg–Al HT as itself efficiently catalyzed the oxidation of alcohols by activating molecular oxygen. Fe-containing Mg-A HT catalyst prepared by adopting the “memory effect” on Mg(Al)O derived from the HT showed high activity for the Baeyer−Villiger oxidation of cyclic ketones by using O2/benzaldehyde as an oxidizing agent. Cu/Mg/Al catalyst derived from Mg(Cu)–Al HT showed high activity for the mineralization of oxalic acid by O3 and moreover the “memory effect” of HT accelerated the mineralization by capturing oxalic acid in the reconstituted HT interlayer space on the Cu/Mg/Al catalyst. The iron catalyst derived from Mg–Fe–Al HT was successfully applied in the ethylbenzene dehydrogenation to styrene.
Ni/Mg(Al)O catalysts derived from Mg(Ni)–Al HT were successfully applied for the reforming of CH4. The high and stable activity of these catalysts was attributed to the formation of stable and well-dispersed Ni metal particles produced from Mg(Ni,Al)O periclase as solid solutions obtained by calcination of the Mg(Ni)-Al HT. Noble metals were incorporated in the surface layer of the Mg(Ni,Al)O periclase by adopting the “memory effect,” resulting in the creation of an efficient bimetallic catalysts for the H2 production for PEFC. Among these catalysts, Ru-Ni/Mg(Al)O was the most effective not only for the DSS steam reforming of CH4, but also for the oxidative reforming of C3H8. Cu/Zn/Al system produced some catalyst precursors such as “aurichalcite” and HT; the former obtained by homogeneous precipitation afforded efficient Cu catalysts for steam reforming of methanol and then CO shift reaction. It is expected that Zn(Cu)-Al HT also effective as the precursors for creating novel Cu catalysts for the H2 production in future.
The research works dealing with HTlcs are not only interesting for their industrial applications, but are also beautiful examples of the scientific preparation of catalysts. All the stages of the catalyst preparation based on a HTlcs precursor need precise chemical foundations in order to avoid inhomogeneous and/or chemical segregations, which would be detrimental to the properties of the final catalysts; choice of the optical composition, nature and amount of promoters, precipitation conditions, type of reagents, aging, washing, hydrothermal treatments, drying, calcination and activation significantly affected the properties of the final compounds. We have shown a little example in this contribution. Although a large amount of work has been done elsewhere, still more remains to be done. To exploit completely this potential and hopeful field of catalytic applications, a multidisciplinary approach is required, in which mineralogical, physicochemical, organic and industrial researchers collaborate, each contributing their specific knowledge. Exciting future will be prospected, including not only the conventional particle catalyst but also the use of monolithic catalyst, in liquid phase synthesis of fine chemicals, in the synthesis of large amount products in the vapor phase, and moreover in the new energy production with continuous processes and economic advantages.
References
Manasse E. (1915) Atti. Soc. Toscana Sci. Nat., Proc. Verb. 24: 92
Allmann R., Jepsen H.P. (1969) N. Jhb. Miner. Mh. 12: 544
Reichle W.T. (1986) Solid State Ionics 22: 135
Cavani F., Trifiro F., Vaccari A. (1991) Catal. Today 11: 173
Vaccari A. (1999) Appl. Clay Sci. 14: 161
Schmidt F. (2001) Appl. Catal. A 221: 15
Choudhary B.M., Kantam M.L., Rahman A., Reddy C.V., Rao K.K. (2001) Angew. Chem. Int. Ed. Engl. 40: 763
Kawabata T., Shinozuka Y., Ohishi Y., Shishido T., Takaki K., Takehira K. (2005) J. Mol. Catal. A 236: 206
Kawabata T., Fujisaki N., Shishido T., Nomura K., Sano T., Takehira K. (2006) J. Mol. Catal. A 253: 279
J.M. Fernández, Ulibarry M.A., Labajos F.M., Rives V. (1998) J. Mater. Chem. 8: 2507
Choudahry V.R., Indurkar J.R., Narkhede V.S., Jha R. (2004) J. Catal. 227: 257
Kaneda K., Yamaguchi K., Mori K., Mizugaki T., Ebitani K. (2000) Catal. Surv. Japan 4: 31
Motokura K., Mizugaki T., Ebitani K., Kaneda K. (2004) Tetrahedron Lett. 45: 6029
Shiraga M., Kawabata T., Li D., Shishido T., Komaguchi K., Sano T., Takehira K. (2006) Appl. Clay. Sci. 33: 247
Kannan S., Dubey A., Knozinger H. (2005) J. Catal. 231: 381
Roelofs J.C.A.A., Van Bockhoven J.A., Van Dillen A.J., Geus J.W., De Jong K.P. (2002) Chem. Eur. J. 8/24: 5571
Bish D.L., Brindley G.W. (1977) Am. Miner. 62: 458
Miyata S. (1975) Clays Clay Miner. 31: 305
Kahn A.L., O’Hare D. (2002) J. Mater. Chem. 12: 3191
Traversa E., Nunziante P., Chiozzini G. (1992) Thermochim. Acta 199: 25
Palomares A.E., Prato J.G., Rey F., Corma A. (2004) J. Catal. 221: 62
G.C. Araújo, Rangel M.C. (2000) Catal. Today 62: 201
Stobbe D.E., van Buren F.R., Stobbe-Kreemers A.W., van Dillen A.J., Geus J.W. (1991) J. Chem. Soc., Faraday Trans. 87: 1631
Oliveira A.C., Valentini A., Nobre P.S.S., Rangel M.C. (2002) React. Kinet. Ctal. Lett. 75:135
Carja G., Nakamura R., Aida T., Niiyama H. (2003) J. Catal. 218: 104
Ye X., Ma N., Hua W., Yue Y., Miao C., Xie Z., Gao Z. (2004) J. Mol. Catal. A 217: 103
Kuśtrowski P., Rafalska-Łasocha A., Majda D., Tomaszewska D., Djiembaj R (2001) Solid State Ionics 141–142: 237
Mimura N., Takahara I., Saito M., Sasaki Y., Murata K. (2002) Catal. Lett. 78: 125
Ohishi Y., Kawabata T., Shishido T., Takaki K., Zhang Q., Wang Y., Nomura K., Takehira K. (2005) Appl. Catal. A 288: 220
Crivello M., Pérez C., Herrero E., Ghione G., Casuscelli S., Rodoríguez-Castellón E. (2005) Catal. Today 107–108: 215
Takehira K. (2002) Catal. Survey Jpn. 6: 19
Shishido T., Sukenobu M., Morioka H., Furukawa R., Shirahase H., Takehira K. (2001) Catal. Lett. 73: 21
Shishido T., Sukenobu M., Morioka H., Kondo M., Wang Y., Takaki K., Takehira K. (2002) Appl. Catal. A 223: 35
T. Shishido, P. Wang, T. Kosaka and K. Takehira, Chem. Lett. (2002) 752
Takehira K., Shishido T., Wang P., Kosaka T., Takaki K. (2003) Phys. Chem. Chem. Phys. 5: 3801
Takehira K., Shishido T., Wang P., Kosaka T., Takaki K. (2004) J. Catal. 221: 43
Morioka H., Shimidzu Y., Sukenobu M., Ito K., Tanabe E., Shishido T., Takehira K. (2001) Appl. Catal. A 215: 11
Ruckenstein E., Wang H.Y. (2000) Appl. Catal. A 198: 33
Miyata T., Shiraga M., Li D., Atake I., Shishido T., Oumi Y., Sano T., Takehira K. (2007) Catal. Commun. 8: 447
Takehira K., Kawabata T., Shishido T., Murakami K., Ohi T., Shoro D., Honda M., Takaki K. (2005) J. Catal. 231: 92
Takehira K., Shishido T., Shoro D., Murakami K., Honda M., Kawabata T., Takaki K. (2005) Appl. Catal. A 279: 41
Takehira K., Shishido T., Shoro D., Murakami K., Honda M., Kawabata T., Takaki K. (2004) Catal. Commun. 5: 209
Melo F., Morlanes N. (2005) Catal. Today 107–108: 458
A Olafsen, Slagtern Å., Dahl I.M., Olsbye U., Schuurman Y., Mirodatos C. (2005) J. Catal. 229: 163
Ohi T., Miyata T., Li D., Shishido T., Kawabata T., Sano T., Takehira K. (2006) Appl. Catal. A 308: 194
Eun J.H., Lee J.H., Kim S.G., Um M.Y., Park S.Y., Kim H.J. (2003) Thin Solid Films 435: 199
Basile F., Fornasari G., Gazzano M., Kiennemann A., Vaccari A. (2003) J. Catal. 217: 245
Basile F., Fornasari G., Rosetti V., F. Trifirò, Vaccari A. (2004) Catal. Today 91–92: 293
M. Miyata, A. Okada, Clays Clay Miner. 25 (1977) 14
Arpentinier P., Basile F., Del Gallo P., Fornasari G., Gary D., Rosetti V., Vaccari A. (2005) Catal. Today 99: 99
Basile F., Fornasari G., F. Trifirò, Vaccari A. (2002) Catal. Today 77: 215
Nagaoka K., Jentys A., Lercher J.A. (2005) J. Catal. 229: 185
Tsyganok A.I., Suzuki K., Hamakawa S., Takehira K., Hayakawa T. (2001) Catal. Lett. 77: 75
Tsyganok A.I., Tsunoda T., Hamakawa S., Suzuki K., Takehira K., Hayakawa T. (2003) J. Catal. 213: 191
Tsyganok A.I., Inaba M., Tsunoda T., Suzuki K., Takehira K., Hayakawa T. (2004) Appl. Catal. A 275: 149
Tsyganok A.I., Inaba M., Tsunoda T., Uchida K., Suzuki K., Takehira K., Hayakawa T. (2005) Appl. Catal. A 292: 328
Fonseca A., Assaf E.M. (2005) J. Power Sources 142: 154
Miyata T., Li D., Shiraga M., Shishido T., Oumi Y., Sano T., Takehira K. (2006) Appl. Catal. A 310: 97
Miyata T., Shiraga M., Li D., Atake I., Shishido T., Oumi Y., Sano T., Takehira K. (2007) Catal. Commun. 8: 447
K. Takehira, T. Ohi, T. Miyata, M. Shiraga and T. Sano, Top. Catal. in press
Goetsch D.A., Schmidt L.D. (1996) Science 271: 1560
Bodke A.S., Bharadwaj S.S., Schmidt L.D. (1998) J. Catal. 179: 138
Ayabe S., Omoto H., Utaka T., Kikuchi R., Sasaki K., Teraoka Y., Eguchi K. (2003) Appl. Catal. A 241: 261
Aartun I., Gjervan T., Venvik H., O. Görke, Pfeifer P., Fathi M., Holmen A., Schubert K. (2004) Chem. Eng. J. 101: 93
Silberova B., Venvik H.J., Holmen A. (2005) Catal. Today 99: 69
Liu S., Xu L., Xie S., Wang Q., Xiong G. (2001) Appl. Catal. A 211: 145
Schulze K., Makowski W., R. Chyży, Dziembaj R., Geismar G. (2001) Appl. Clay Sci. 18: 59
Avci A.K., Trimm D.L., Aksoylu A.E., Önsan Z.İ. (2003) Catal. Lett. 88: 17
Avci A.K., Trimm D.L., Aksoylu A.E., Önsan Z.İ. (2004) Appl. Catal. A 258: 235
B.S. Çağlayan, Avci A.K., Z.İ. Önsan, Aksoylu A.E. (2005) Appl. Catal. A 280: 181
M. Shiraga, D. Li, I. Atake, T. Shishido, Y. Oumi, T. Sano and K. Takehira, Appl. Catal. A 318 (2007) 143
D. Li, M. Shiraga, I. Atake, T. Shishido, Y. Oumi, T. Sano and K. Takehira, Appl. Catal. A in press
Dias J.A.C., Assaf J.M. (2004) J. Power Sources 130: 106
Dias J.A.C., Assaf J.M. (2005) J. Power Sources 139: 176
Madhavaram H., Idriss H., Wendt S., Kim Y.D., Knapp M., Over H., Aβmann J., E. Löffler, Muhler M. (2001) J. Catal. 202: 296
Balint I., Miyazaki A., Aika K. (2003) J. Catal. 220: 74
Yin S.-F., Zhang Q.-H., Xu B.-Q., Zhu W.-X., Ng C.-F., Au C.-T. (2004) J. Catal. 224: 384
Elmasides C., Kondarides D.I., W. Grünert, Verykios X.E. (1999) J. Phys. Chem. B 103: 99
Bartholomew C.H., Pannell R.B., Butler J.L. (1980) J. Catal. 65: 335
D. Li, I. Atake, T. Shishido, Y. Oumi, T. Sano and K. Takehira, to be published
Li B., Kado S., Mukainakano Y., Miyazawa T., Miyao T., Naito S., Okumura K., Kunimori K., Tomishige K. (2007) J. Catal. 245: 144
Shishido T., Yamamoto Y., Morioka H., Takaki K., Takehira K. (2004) Appl. Catal. A 263: 249
T. Shishido, Y. Yamamoto, H. Morioka and K. Takehira, J. Mol. Catal. A in press
Kawabata T., Matsuoka H., Shishido T., Li D., Tian Y., Sano T., Takehira K. (2006) Appl. Catal. A 308: 82
Shishido T., Yamamoto M., Li D., Tian Y., Morioka H., Honda M., Sano T., Takehira K. (2006) Appl. Catal. A 303: 62
Shishido T., Yamamoto M., Atake I., Li D., Tian Y., Morioka H., Honda M., Sano T., Takehira K. (2006) J. Mol. Catal. A 253: 270
Agrell J., Boutonnet M., Fierro J.L.G. (2003) Appl. Catal. A 253: 213
Peters R., H.G. Düsterwald, Höhlein B. (2000) J. Power Sources 86: 507
Shiozaki R., Hayakawa T., Ishii T., Kumagai M., Hamakawa S., Suzuki K., Ito T., Shishido T., Takehira K. (1999) Catal. Lett. 58: 131
Shishido T., Sameshima H., Takehira K. (2003) Top. Catal. 22: 261
Marjorie M.H., Kariuki B.M. (1994) Acta Crystallogr. B 50: 673
Ghose S. (1964) Acta Crystallogr. 17: 1051
Jambor J.L. (1964) Acta. Crystallogr. 17: 1051
Couves J.W., Thomas J.M., Waller D., Jones R.H., Dent A.J., Derbyshire G.E., Greaves G.N. (1991) Nature 354: 465
Stoilova D., Koleva V., Vassileva V. (2002) Spectrochim. Acta A 58: 2051
Murcia-Masacrós S., Navarro R.M., L. Gómez-Sainero, Constatino U., Nocchetti M., Fierro J.L.G. (2001) J. Catal. 198: 338
Takezawa N., Iwasa N. (1997) Catal. Today 36: 45
M.J.L. Ginés, Amadeo N., Laborde M., Apesteguía C.R. (1995) Appl. Catal. A 131: 283
Mastalir A., Frank B., Szizybalski A., Soerijanto H., Deshpande A., Niederberger M., R. Schomäcker, R. Schlögl, Ressler T. (2005) J. Catal. 230: 464
Batista J., Pintar A., Mandrino D., Jenko M., Martin V. (2001) Appl. Catal. A 206: 113
Tanaka Y., Kikuchi R., Takeguchi T., Eguchi K. (2005) Appl. Catal. B 57: 211
Pokrovsky O.S., Schott J. (2004) Geochim. Cosmochim. Acta 68: 31
Ovesen C.V., Clausen B.S., B.S. Hammershøi, Steffensen G., Askgaard T., Chorkendorff I., J.K. Nørskov, Rasmussen P.B., Stolze P., Taylor P. (1996) J. Catal. 158: 170
Fujita S.-I., Usui M., Takezawa N. (1992) J. Catal. 134: 220
Koryabkina N.A., Phatak A.A., Ruettinger W.F., Farrauto R.J., Ribeiro F.H. (2003) J. Catal. 217: 233
Jakdetchai O., Nakajima T. (2002) J. Mol. Struct. 619: 51
Turco M., Bagnasco G., Constantino U., Marmottini F., Montanari T., Ramis G., Busca G. (2004) J. Catal. 228: 43
Ovesen C.V., Stolze P., J.K. Nørskov, Campbell C.T. (1992) J. Catal. 134: 445
Turco M., Bagnasco G., Costantino U., Marmottini F., Montanari T., Ramis G., Busca G. (2004) J. Catal. 228: 56
Constantino U., Marmottini F., Sisani M., Montanari T., Raims G., Busca G., Turco M., Bagnasco G. (2005) Solid State Ionics 176: 2917
Busaca G., Constatino U., Marmottini F., Montanari T., Patrono P., Pinzari F., Ramis G. (2006) Appl. Catal. A 310: 70
Velu S., Suzuki K., Vijayaraj M., Barman S., Gopinath C.S. (2005) Appl. Catal. B 55: 287
Lenarda M., Storaro L., Frattini R., Casagrande M., Marchiori M., Capannelli G., Uliana C., Ferrari F., Ganzerla R. (2007) Catal. Commun. 8: 467
Melián-Cabrera I., López Granadoz M., Fierro J.L.G. (2002) J. Catal. 210: 273
I. Atake, K. Nishida, D. Li, Y. Oumi, T. Shishido, T. Sano and K. Takehira, to be published
Acknowledgments
The authors are very grateful to Dr. T. Kawabata of Sumitomo Chem. Co., Japan, and Dr. A. I. Tsyganok of the University of Alberta, Canada, for their helpful collaborations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takehira, K., Shishido, T. Preparation of supported metal catalysts starting from hydrotalcites as the precursors and their improvements by adopting “memory effect”. Catal Surv Asia 11, 1–30 (2007). https://doi.org/10.1007/s10563-007-9016-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10563-007-9016-2