
Abstract
The development of sustainable chemical processes for dehydrogenative transformations yields promising results for the selective synthesis of nitrogen-containing compounds. We report the synthesis of Nb2O5/Co3O4 catalyst using co-precipitation, followed by the hydrothermal method. FESEM, NH3-TPD, PXRD, FT-IR pyridine adsorption studies, and XPS were used to characterize the chemical and structural properties of the synthesized material. The FESEM analysis revealed that Nb was distributed uniformly on the surface of the Co3O4. The 3.3 wt% Nb showed (2 5 0) and (0 0 1) facet on Co3O4, and these results are confirmed further by PXRD d-spacing. Whereas cobalt oxide exists in (2 2 2), (3 1 1), and (1 1 1) facet, which confirms the crystal cubic spinel structure of Co3O4. The TPD ammonia desorption Nb2O5/Co3O4 result confirms the presence of 0.441 mmol/g acidic sites. The synthesized Nb2O5/Co3O4 catalyst effectively transformed aromatic amines and benzyl alcohols to imines through a dehydrogenative coupling reaction. The 3.3 wt% Nb2O5/Co3O4 exhibited excellent catalytic activity, with the conversions reached as high as > 99% at 140 °C. The Nb5+ played a vital role by generating the Lewis acid species, oxygen vacancy, and defects on the Co3O4 could be responsible for excellent selective activation of alcohol in the presence of a reactive amine. Oxidant-free, easy product separation, and significant reusability makes our catalyst versatile for the synthesis of imine derivatives.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Dehydrogenative transformations with the concomitant release of hydrogen molecules have become an important thrust in synthetic organic chemistry. Imine derivatives are potential synthons of pharmaceuticals, fine chemicals, and biologically active heterocycles [1, 2]. Due to their huge demand, many synthetic and catalytic protocols have been developed to achieve higher conversion and selectivity [3]. The various synthetic methods for imine synthesis have been reported (Scheme 1). In this context, the dehydrogenative coupling reactions of alcohols with amines are oxidant and additives free to yield imines and generate merely water and hydrogen gas as a non-toxic byproduct [4, 5]. For this transformation, homogeneous metal complexes based on ruthenium, cobalt, iron, and iridium-containing catalysts have been developed in recent years [6, 7]. Also, Pincer-type complexes showed excellent catalytic activity for the dehydrogenative reactions [8]. For instance, Eizawa et al. synthesized [RuHCl(CO)(PPh3)3] containing PCP ligands in situ from the corresponding imidazolium salts with KN(SiMe3)2, in toluene under constant heating allows the formation of ruthenium complexes bearing a PCP ligand [RuHCl(CO)(PCP)] is used for the imine synthesis from amine and benzyl alcohol in the presence of bases and solvent under reflux conditions [9].

Imine formation via (1) The traditional method, (2) Previous approaches, and (3) The current approach
Also, Maggi et al. have synthesized imines through dehydrogenative reaction using homogeneous ruthenium N-heterocyclic carbine complex within 24 h. However, it required a stoichiometric amount of DABCO to obtain an excellent yield [10]. In another report, Han et al. used Ag–N heterocyclic carbine complexes for aerobic oxidation of alcohols followed by condensation at room temperature with an excellent yield of imines (98%) [11]. Peng et al. reported the direct self-coupling of amines to form imines using ruthenium containing pyridine-based (PNP = 2,6-bis(di-tert-butylphosphinomethyl)pyridine) and PNN (PNN = 2-(di-tert-butylphosphinomethyl)-6-(diethylaminomethyl)pyridine), pincer complex under argon atmosphere at 160 °C [12]. The majority of studies on the dehydrogenative coupling of alcohols and amines use metal-based pincer and carbine complexes to synthesize imines. Apart from these, inert conditions are required for the reaction to avoid the decomposition/oxidation of precious metal sources/complexes. There have some limitations as they cannot be reused, which raises concern of their applicability on an industrial scale. These considerations lead to a need for a bench-stable heterogeneous industrial viable catalyst that could promote the transformation with the advantage of easy separation and recyclability.
Due to environmental concerns, there is a constant search for an efficient and greener approach in catalytic dehydrogenative transformations [13]. In this context, metal oxides have been extensively explored and applied in various branches of chemistry. In particular, as heterogeneous catalysis, due to their outstanding redox properties, chemical and thermal stability, excellent catalytic activity, recyclability, cost-effectiveness, and applicability for various organic transformations [14]. These properties of metal oxides make them suitable for both industrial and academic research [15]. Typically, various metal oxides like CeO2, ZrO2, ZnO, Al2O3, SiO2, Fe2O3, and TiO2 containing metals have been used as catalysts [16] for various organic transformations such as oxidative, reductive, and dehydrogenative coupling reactions [17]. In most of these reactions, cobalt oxide (Co3O4) has been widely studied as a supporting material due to its unique chemical, mechanical, thermal, and magnetic properties [18]. Co3O4 is an excellent supporting material as it provides active promoting sites with other metals, and its mild Lewis acidic sites could increase the intrinsic activity [19]. Pure Co is very unstable at room temperature, and it converts easily into oxide forms such as CoO, Co2O3, and Co3O4. Among all the oxides of Co, Co3O4 is the most stable phase and shows variable oxidation states. The crystal structure of Co3O4 oxide ion belongs to form spinel based on a cubic close packing arrangement and p-type of semiconductor with potential applications in diverse fields [20]. This broad spectrum of properties makes it attractive for catalytic applications. Different transition metals, such as Pt/Co3O4 [21], Ni/Co3O4 [22], Fe/Co3O4 [23] are supported and exhibited efficient catalytic activity for imine synthesis. Interestingly, niobium (Nb) containing heterogeneous catalysts are widely used for variety of reactions [24]. The electronegativity and ionic radius of Nb metals provide strong surface acidity and stability, enhancing catalytic activity compared to other neighboring metals (V, Zr, Mo). Even a small amount of Nb had a significant effect in enhancing the lifetime of catalytic activity [25].
Solid Nb-containing catalysts are widely used for organic transformation, such as dehydration of alcohol, hydration of alkenes, condensation, alkylation, and esterification reactions [26]. Many solid-acid catalysts have been reported for the dehydrogenation of alcohol, in this context, niobic acid is known for its dehydrogenating catalytic activity. Armaroli et al. used an acidic niobium phosphate catalyst to dehydrate fructose to 5-hydroxymethyl-2-furaldehyde [27]. Also, Lizuka et al. used the Nb2O5 as a catalyst at high temperature for dehydration of 2-butanol to butene product [28]. Further, Can-Xiong et al. explain the acidic properties of niobium oxide, and it is utilized for dehydration of alcohol [29]. The binary system Nb2O5/Co3O4 almost compliments each other in valence electrons and is expected to show a cooperative effect. However, electronic and magnetic properties are still unclear due to their heteronuclear cluster of Nb2O5/Co3O4 challenging and giving structural variety [30]. Considering the literature reports, we investigated the catalyst activity of Nb2O5/Co3O4 oxide catalyst for imine synthesis by one-pot selective dehydrogenative coupling of alcohol and amine. The aforementioned catalyst was synthesized using co-precipitation, followed by the hydrothermal method with a series of Nb oxide loading amounts. The synthesized catalysts were characterized by various physicochemical methods and are examined for their catalytic activity.
2 Experimental Section
2.1 Materials
Co(NO3)2·6H2O (SRL), NbCl5 (V) (Aldrich), NaOH (S D Fine), aniline (99.5%) (AR) (Spectrochem), benzyl alcohol (S. D. Fine), p-nitroaniline (99.5%) (SRL), o-nitroanilne (SRL), 4-nitrobenzyl alcohol (Spectrochem) (99%), 4-hydroxy benzyl alcohol (TCI), benzylamine (SRL), 4-(hydroxymethyl)-2-methoxyphenol (TCI), 4-ethylaniline (TCI), 4-butylaniline (TCI).
2.2 Catalysts Synthesis
The Nb2O5/Co3O4 catalyst was synthesized via co-precipitation followed by hydrothermal method, with varying wt% (1.6, 3.3, and 6.7) of Nb loaded with Co3O4. In a typical synthesis, 5 g of cobalt nitrate was dissolved in 40 mL distilled water by moderately stirring at room temperature. The specified amount of NbCl5 was added and stirred for an additional 30 min. After the reaction mixture becomes homogeneous, NaOH, as a precipitating agent, was added dropwise until the pH comes in between 9 to 10 (6.2 M NaOH solution = 14 mL). Then the reaction was aged for 24 h at room temperature with moderately stirring. The obtained material was transferred to stainless steel autoclave and packed tightly. Then this autoclave was heated in an electrically heated oven at 120 °C for 5 h. The autoclave was cooled to room temperature, and solid was collected by centrifuge and washed with a copious amount of water till filtrate attendance neutral pH and finally with methanol for removing the adsorbed organic impurity. The obtained solid was dried at 70 °C for overnight in a hot air oven. Finally, the solid powder was calcined in the muffle furnace at 500 °C and hold for 5 h by a heating rate of 5 °C/min.
2.3 Catalytic Reactions
A dehydrogenative coupling reaction was carried out in a 35 mL screw-cap Borosil glass test tube. Typically, 1 mmol of alcohol, 3 mmol of aniline, and 25 mg freshly prepared catalyst were added and tightly screw-capped. The reaction tube was heated to the desired reaction temperature through the oil bath. The reaction mixture was stirred by using a magnetic stirrer. The progress of the reaction was monitored periodically by withdrawing samples and quantified by Gas Chromatography (Agilent-7890B) and GC–MS (Shimadzu QP-2010, Japan) with HP-5 column, which consists of 5% diphenyl and 95% dimethyl polysiloxane capillary column and FID as a detector). After the reaction, the solid catalyst was collected by centrifuge, washed with methanol and dried in an oven at 70 °C overnight and reuse for the further cycle.
3 Results and Discussion
The catalyst was synthesized by varying weight percent (wt%) of Nb2O5 on Co3O4 support by co-precipitation, followed by the hydrothermal method (Scheme 2). The hydration complex of both metals and metal salt was obtained by dissolving it in an aqueous solution under basic media (Scheme 3). Thus, the obtained aqua complex to crystals oxide was grown under the hydrothermal condition. The hydrothermal process builds the autogenous pressure to produce monomer, which is then nucleated and crystallized, subsequently calcined at a higher temperature [31].

Schematic diagram of Nb2O5/Co3O4 catalyst by a co-precipitation followed by hydrothermal method

Reactions involved during the synthesis of the Nb2O5/Co3O4 catalyst
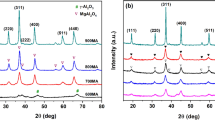
The phase purity of the synthesized material was identified by using the X-ray diffractions method (Fig. 1). The Nb2O5/Co3O4 catalyst exhibited diffraction at 2θ = 19°, 31.3°, 36.9°, 38.5°, 44.9°, 59.4°, 65.3° and 77.4° corresponding to the planes of (1 1 1), (2 2 0), (3 1 1), (2 2 2), (4 0 0), (5 1 1), (4 4 0), (5 3 3), and (6 2 2), indicating the cubic phase of spinel cobalt oxide with the average crystallite size 18.2 nm calculated by Scherrer’s equation [JCPDS file No. 04-003-0984] and confirm the formation of the spinel cubic crystalline structure of synthesized material [32]. Then, different wt% loading of Nb2O5 on Co3O4 oxide surface exhibit four sharp diffraction peaks at 2θ = 16 to 57°, corresponding to the planes 16.1° = (1 3 0), 22.8 = (0 0 1), 32.6 = (2 5 0), and 46.5° = (0 0 2), which confirms the orthorhombic crystal structure of Nb2O5 matching with (JCPDS No: 00-030-0873) [33]. Upon increasing the loading % of Nb2O5 on the Co oxide surface, observed the mixed phase of CoNb2O5 at 2θ = 57.8 with (1 1 3) plane are confirmed with JCPDS No. 00-032-0304.

XRD pattern of a Co3O4, b 1.6 wt% Nb2O5/Co3O4, c 3.3 wt% Nb2O5/Co3O4, d 6.7 wt% Nb2O5/Co3O4
The FESEM images of the 3.3 wt% Nb2O5/Co3O4 catalyst shows no distinct morphology (Fig. 2). The elemental mapping of the sample showed uniform distribution of Nb2O5 on the Co3O4 oxide surface (Fig. 2C–F). The FE-SEM–EDX of 3.3 wt% loading of Nb2O5 on Co3O4is shown in the ESI (See SI3, Fig. S2). The freshly prepared 3.3 wt% catalyst showed 2.3 atomic % of Nb2O5 content is present on the Co3O4 surface.

FESEM images and elemental mapping of 3.3 wt% Nb2O5/Co3O4 catalyst A 100 nm image, B 1 nm image, C Mixed elemental mapping; D Nb, E Co, F O distribution on the sample surface
Figure 3 displays the high-resolution transmission electron microscope (HRTEM) and SEAD image of 3.3 wt% Nb2O5/Co3O4 catalyst. Typically, the bimetallic Nb2O5/Co3O4 composited crystalline sample revealed lattice fringes seen from the different selected zones, which revealed the existence of single-crystalline phases of both the oxides in the sample. Figure 3A and B shows the lattice fringes regularly separated 0.23 nm, 0.25 nm, and 0.45 nm with well match with the XRD d-spacing of Co3O4 and which resembles with (2 2 2), (3 1 1), and (1 1 1) lattice index of cubic Co3O4 respectively (JCPDS No:04-003-0984).

HR-TEM images of 3.3 wt% Nb2O5/Co3O4 is showing different fringes in A A.1. Co3O4; A.2. Nb; A.3.Co; B.4.Nb; B.5. Co; C SEAD pattern of Nb2O5/Co3O4 catalyst
The prominent fringes that are displayed in Fig. 3A and B 0.27 nm, 0.39 nm well match with the d-spacing and co-related (2 5 0) (0 0 1) lattice index of crystalline Nb2O5, respectively (JCPDS No: 00-030-0873). The SEAD pattern also confirmed the plane, or d-spacing value of Nb2O5/Co3O4 (See Fig. 3C).
The N2 gas adsorption/desorption isotherms and specific surface area were determined by the multipoint Brunauer–Emmett–Teller (BET) method. Initially, all samples were degassed at 300 °C for 4 h before the measurements. The specific surface areas of the catalysts were determined by N2 adsorption and desorption at liquid nitrogen temperature. All the samples showed H3 type hysteresis with monolayer adsorption (See SI5. Figure S7). The surface area of pure Co3O4 has a much higher 5 m2/g as compared to Nb2O5 loaded on the Co3O4 oxide surface, while the pore size of pure Co3O4 was less (357.6 Å) than the Nb2O5/Co3O4 catalytic system. The surface area of 1.6, 3.3, and 6.7 wt% Nb2O5/Co3O4 catalysts were measured to be 9, 13, and 23 m2/g, respectively (Table 1). Upon increasing the wt% of Nb2O5 loading, the surface area also increases, and it was as expected because it may be due to high wt% of Nb2O5 loading [34]. It was also observed that increasing the amount of Nb2O5 loading (0, 1.6, 3.3 and 6.7 wt%) on the Co3O4 oxide surface, increases the pore size to be 357.6, 406.8,458, and 554 Å respectively [35].
X-ray photoelectron spectroscopy (XPS) has been used to analyze the chemical state of the solid surface. The full survey spectrum has confirmed the presence of synthesized composite of Nb 3d, Co 2p, and O 1s species with specific binding energy is shown in the Fig. 4A. By deconvolution of the XPS spectrum; the asymmetric Co 2p peak can be differentiated into two peaks centered at Co 2p3/2 and Co 2p1/2 spin–orbit and binding energy at 779.2 and 794.2 eV respectively, which indicate the presence of divalent and trivalent Co species in the Co3O4 spinel structure. The fitting peak at 779.4 was attributed to Co (2p3/2) are deconvoluted in to two peaks at binding energy 779.2 (FWHM 2.09) and 780.8 eV (FWHM 3.4) are assigned to Co3+ and Co2+ respectively. While the shoulder peak at a binding energy of 794.2 eV can be fitted into two components with binding energies at 794.2 (FWHM 2.0) and 796.05 eV (FWHM 2.8) corresponding to the 2p1/2 spin-orbits of Co3+ and Co2+ respectively. The two small peaks at 789.1 (FWHM 2.3) and 804 eV (FWHM 4.6) are typical Co shakeup satellite peaks that are conformed to Co3+, and Co2+ species are present in the catalyst see (Fig. 4B) [36]. Typically, the Co3O4 structure contains Co3+ is octahedrally coordinated cations, and Co2+ is tetrahedrally cations [37]. The electronic state of the active metal Nb was loaded on the Co3O4 oxide surface found out by XPS. The different binding energy position of the Nb 3d peak indicates the existence of Nb in different chemical state (Fig. 4C). The spin–orbit splitting centered of Nb 3d5/2 and Nb 3d3/2 with corresponding to binding energy 206 (FWHM 1.2) eV and 208.7 (FWHM 1.4) eV respectively, deconvolute both peaks in the form of NbO2, NaNbO2 and Nb2O5 on Co3O4 surface, in which niobium metal showing vary the oxidation state due to the Nb2O5 at higher temperature decompose and form suboxides NbO2 and NaNbO2 species [38, 39]. The fitting peak at 206 eV is decomposed into two peaks, 206 and 206.9 eV is assigned to the NbO2 and NaNbO2 species are present, then 208.7 are split into two peaks 208.7 and 209.8 eV are indicating that Nb2O5 species are present on Co3O4.

XPS analysis of 3.3 wt% Nb2O5/Co3O4, A Survey spectra of Nb2O5/Co3O4, B Co, C Nb, D O
The O 1s spectrum of the Nb2O5/Co3O4 catalytic system is shown in Fig. 4D, where two peaks were located at 529.2 eV and 532 eV. The major peak of O 1s of Nb2O5/Co3O4 catalytic system about 529.2 eV was assigned to lattice oxygen O2−, the higher binding energy can be attributed to the different types of bonding combination of O2− with Co3+ and Co2+ cubic spinel of Co3O4 oxide surface of the catalyst [40,41,42,43]. In the Co3O4 system may be O three coordinated with three Co3+ and two coordinated O bonded with one Co2+ and Co3+, prominent to the binding energy of O2− attached with Co ions having a considerable difference. However, for spinel Co3O4 new intense peak at 532 eV shows up, which is attributed to the high binding energy peak from the surface oxygen defect species [44]. It suggests that the Co3O4 spinel structure has more O vacancies; these results are in excellent agreement in correlation with surface area and pore size.
Figure 5 shows the ex-situ pyridine adsorption spectra of Nb2O5/Co3O4 and confirms the presence of Lewis and Brønsted acid sites in the catalyst. Initially, the freshly prepared catalyst was dried at 150 °C overnight, then make a pallet with KBr at room temperature. Initial spectra recorded at room temperature without pyridine is shown in Fig. 5a. Then pyridine was added on the same pellet and keep 15 minutes at room temperature (~ 32 °C) and record the FTIR (Fig. 5b). Then this pyridine added pellet keep in the oven at 100 °C for 1 h and then record the FTIR spectra (Fig. 5c).

Shows the ex-situ pyridine adsorption FT-IR study, (a) 3.3 wt% Nb2O5/Co3O4, (b) 3.3 wt% Nb/Co3O4 + Py, (c) 3.3 wt. % Nb2O5/Co3O4 + Py + 100 °C, (Py Pyridine, L Lewis acid, B Brønsted acid)
The pyridine adsorbed Nb2O5/Co3O4 catalyst shows three characteristic bands assign to the chemisorption of pyridine on the Nb2O5/Co3O4 oxide surface. The band located at 1350 to 1380 cm−1 corresponds to hydroxyl groups on the catalyst surface, and then the chemisorption band of pyridine at 1440 and 1588 cm−1 indicates the Lewis acid site. Additionally, a small band located at 1484 cm−1 corresponds to the vibrational mode of pyridine adsorbed on the catalyst and is assigned to both Lewis and Brønsted acidic sites on the catalyst [45].
The surface acidity of catalyst and reagent adsorption and desorption plays a vital role in the catalytic reaction and is an important aspect of the heterogeneous catalyst. The surface acidity strength of the Nb2O5/Co3O4 catalysts was investigated by the temperature program desorption (TPD) of NH3, and the result is shown in (Fig. 6A). NH3-TPD was carried out at a heating rate of 10 °C/min in He (30 mL/min) from room temperature to 600 °C. The Nb2O5/Co3O4 catalyst's TPD result indicates the wide-ranging distribution of acidic sites from mild to strong acidic sites in the catalyst. Typically, acidic strength, which determines by the NH3 desorption temperature range, can be classified as weak (< 250 °C), moderate (250–400 °C) and strong (> 400 °C) acidic sites [43]. The NH3-TPD profile of the Nb2O5/Co3O4 catalyst is shown in Fig. 6A, the NH3 desorption showed two regions, around the peak at 208 °C indicates weak and moderate acidic sites peak was observed at 328.6 °C. The quantitative analysis of the TPD result for Nb2O5/Co3O4 catalyst is showed us the presence of 0.441 mmol/g acidic sites. The NH3 TPD results of the catalyst’s acidic site corroborated with the py-FT-IR adsorption profile [46].

A NH3 TPD profile of 3.3 wt% Nb2O5/Co3O4 catalyst, B CO2 TPD profile of 3.3 wt% Nb2O5/Co3O4 catalyst
The strength of surface basicity of the Nb2O5/Co3O4 catalyst was investigated using temperature-programmed desorption of CO2 and results are shown in Fig. 6B CO2-TPD was performed from room temperature to 550 °C at a heating rate of 10 °C/min in Helium (30 mL/min). The Nb2O5/Co3O4 catalyst's TPD result indicates the wide-ranging distribution of basic sites from mild to the strong basic site in the catalyst. The quantitative analysis of the TPD result demonstrated the presence of Nb2O5/Co3O4 catalyst is 0.268 mmol/g basic sites. In Fig. 6B represent the CO2 desorption curve for the 3.3 wt% Nb2O5/Co3O4 catalyst sample. The desorption range around 107 to 283 °C can be attributed to the Brønsted site; it could be inferred from the distinct desorption peak at 107 and 283 °C that Nb's concentration on Co3O4 surface and hydroxyl groups on the catalyst surface directly correlates with the XPS studies [47, 48].
3.1 Catalytic Reactions
The synthesized Nb2O5/Co3O4 oxide catalysts were used in dehydrogenative coupling reaction to imine synthesis using benzyl alcohol and aniline as substrates (Scheme 4). The reaction was carried out in an inert atmosphere (under N2 and Ar) sealed tube for 20 h at 140 °C without any additives. Imine formation was analyzed by GC, GC–MS, and NMR (See in SI, S10).

Catalytic reaction of imine synthesis
The series of catalysts optimized under standard reaction conditions is shown in Table 2. Initially, the reaction was carried without catalyst at 140 °C for 24 h; no conversion of aniline and alcohol was observed. Furthermore, when pure Nb2O5 and Co3O4 were used as a catalyst at 140 °C for 20 h, obtained 11 and 18% conversion of benzyl alcohol with 98% selectivity to imines was achieved (Table 2 entries 2, 3). Then 1.6 wt% Nb2O5 loaded on Co3O4 surface gave 84% conversion with 98% selectivity to imine product in 20 h. This result suggests that the loading of Nb increases the rate of reaction as compared to the pure Co3O4. Further increase in catalyst loading (3.3 wt. %) resulted in excellent (95%) conversion with 98% selectivity to imine (Table 2, entry 5). However, no significant change was observed when the reaction was performed at 6.7 wt. % Nb2O5 loading (Table 2, entry 6). Notably, it was found that the catalyst (Nb2O5/Co3O4) showed a high rate of reaction up to 12 h, and after that, it slowed down gradually.
Among various polar and non-polar solvents tested for imine synthesis, initially used non-polar solvents like o-xylene gave 59% conversion and 100% selective desired product (See SI6 Table S1, entry 1). Whereas polar aprotic solvents such as DMSO and DMF gave 44 and 30% conversions, respectively (See SI6 Table S1, entries 2 and 3). This could be associated with the solvent's polarity, which possibly decreases the alcohol dehydrogenation rate. From these optimization studies, it was found that solvent-free conditions gave higher activity (95%) and selectivity of imine product (98%) (See SI6 Table S1, entry 4). Hence, we have adopted the solvent-free condition for the dehydrogenative coupling reaction of alcohols and amines.
The temperature effect on the imine synthesis was also studied, ranging from 100 to 160 °C (See SI7 Table S2). The results indicate that the reaction rate was slow at the lower temperature, subsequently increasing the temperature up to 140 °C, the conversion (96%) increased without losing the selectivity. Furthermore, increasing the temperature from 140 to 160 °C did not enhance the conversion (SI7 Table S2, entries 3 and 4). The controlled experiment was performed at the optimized reaction condition (See Fig. 7). Initially rate of reaction was high (7.5 h gave 34.8% conversion) and it further increased to 59.7% in 13 h. Then reaction rate was slowly decreased and it reached 95% with 98% selectivity to imine product in 24 h. (See Fig. 7a). A controlled experiment was performed at the same reaction condition without aniline, only using benzyl alcohol to cross-check the dehydrogenation rate without aniline (See Scheme 5b). Here we observed 25% the formation of aldehyde product. The conclusion of this study without amine dehydrogenation rate was slow, it might be amine functional groups also help to dehydrogenate with Nb2O3/Co2O3 catalytic system.

Controlled experiment time vs conversion a Benzyl alcohol + Aniline to Imine synthesis, b Benzyl alcohol to aldehyde synthesis

A plausible reaction mechanism for imine using alcohol and amine over Nb2O5/Co3O4 catalyst
Further, the catalytic activity was tested for hetero cross-coupling of anilines and benzyl alcohols under the optimized condition with a 1:1 ratio of starting material resulted in 88% conversion with 98% selectivity. Then, we changed the mole ratio of substrates to 3:1 (aniline and benzyl alcohol, respectively). It was observed that the increased conversion (98%) and selectivity (98%) while altering the mole ratio 1:3 of aniline and alcohol decreased conversion (25%) and selectivity (98%) (See SI8 Table S3). Furthermore, the obtained protocol was extended for a broader substrate scope. The optimized (3.3 wt% Nb2O5/Co3O4) catalyst was used for the hetero-coupling of alcohol and amine derivatives.
When simple benzyl alcohol was used as a substrate, it gave a 96% conversion with 98% selective to corresponding imine (Table 3, entry 1). Furthermore, when the electron-donating substrate, such as 4-hydroxy benzyl alcohol was used, the reaction rate was very fast and successfully converted (100%) all substrates at a lower temperature with 80% selectivity to imine in just 9 h reaction time. Because it is a very reactive substrate and easily converts into amine due to in situ generated H2 and other substituted product formation. In addition, the reaction was allowed to run for extended time (20 h). The formed product was further converted to an amine (Table 3, entries 2 and 3).
Notably, the electron-withdrawing groups such as –Cl and –NO2 on alcohol substrates were favourable to react slowly with the amine to give corresponding imines. In contrast, electron-donating alcohols tend to give less conversion but higher selectivity of corresponding imine products. For instance, 4-chloro benzyl alcohol gave only 18% conversion but 100% selectivity to the corresponding imine product (Table 3, entry 4). Furthermore, the strong electron-withdrawing effect on alcohol substrate on 2-nitro benzyl alcohol, 3-nitro benzyl alcohol, and 4-nitro benzyl alcohols achieved 38, 38, 70% conversion, respectively along with 100% selectivity (Table 3, entries 5, 6, and 7).
Moreover, the same protocol was extended for different types of amine derivatives with benzyl alcohol to synthesize imine (Table 4). This catalyst is also effective for the self-coupling of phenylmethylamine to provide the corresponding imine (N-benzyl-1-phenylmethylimine) product 48% yield with 100% selectivity (Table 4, entry 1). The same type of phenylmethylamine substrates was used for the cross-coupling with benzyl alcohol, resulting in the corresponding cross-coupled imine product with 100% conversion (Table 4, entry 2). Furthermore, benzene-1, 2-diamine was also converted to the corresponding cyclic 2-phenyl-1H-benzimidazole imine product with excellent conversion and selectivity under the same reaction conditions (Table 4, entry 3). The steric and electron-withdrawing groups (F, Br, and NO2) are favourable to react with benzyl alcohol to transform into resultant imines products such as N-(4-fluorophenyl)-1-phenylmethanimine, N-(4-bromophenyl)-1-phenylmethanimin and N-(4-nitrophenyl)-1-phenylmethanimine achieving 100, 50 and 25% conversions respectively with 100% selectivity to corresponding imines (Table 4, entries 4, 5, and 6). In addition, when slightly weak electron-donating alkyl group substituted aromatic amine was used for imine synthesis, 4-ethyl aniline gave 76% conversion with 97% selectivity 4-butyl aniline gave 45% conversion with 98% selectivity of corresponding imine products. (Table 4, entries 7 and 8). Compared to those electron-withdrawing functional amines, donating group substituted amine derivatives resulted in more conversion except fluorine substituted amine.
3.2 NMR
3.2.1 N, 1-Diphenylmethanimine
1H NMR (500 MHz, DMSO) δ 8.44, 7.89, 7.47, 7.37, 7.20.
13C NMR (126 MHz, CDCl3) δ 161.88, 153.52, 137.65, 132.84, 130.23, 127.39, 122.32.
3.2.2 1-(4-Nitrophenyl)-N-Phenylmethanimine
1H NMR (500 MHz, DMSO) δ 8.85, 8.17, 8.09, 7.86, 7.77, 7.44, 7.27. 13C NMR (126 MHz,
DMSO) δ 158.77, 152.67, 151.13, 135.44, 133.75, 131.42, 128.29, 126.40, 123.01.
3.2.3 N-(4-bromophenyl)-1-phenylmethanimine
1H NMR (500 MHz, CDCl3) δ 8.49, 7.96, 7.56, 7.32, 7.14. 13C NMR (126 MHz, CDCl3) δ 160.78, 151.02, 135.94, 132.20, 131.66, 128.84, 122.60, 119.32.
3.2.4 N-(4-butylphenyl)-1-phenylmethanimine
1H NMR (500 MHz, CDCl3) δ 8.47, 7.88, 7.46, 7.19, 2.63, 1.60, 1.38, 0.94. 13C NMR (126 MHz, CDCl3) δ 159.62, 149.60, 140.92, 136.38, 131.19, 128.56, 120.81, 35.20, 33.71, 22.36, 13.98.
3.2.5 N-(4-ethylphenyl)-1-phenylmethanimine
1H NMR (500 MHz, CDCl3) δ 8.45, 7.89, 7.45, 7.20, 2.68, 1.26. 13C NMR (126 MHz, CDCl3) δ 158.64, 148.66, 141.23, 135.38, 130.20, 127.74, 119.90, 27.67, 14.68.
3.2.6 Benzenamine, 4-fluoro-N-(phenylmethylene)
1H NMR (500 MHz, CDCl3) δ 8.51, 7.96, 7.55, 7.25, 7.14. 13C NMR (126 MHz, CDCl3) δ 162.23, 160.19, 148.05, 136.07, 131.46, 128.79, 122.27, 115.55.
3.2.7 (E)-N-benzyl-1-phenylmethanimine
1H NMR (500 MHz, CDCl3) δ 8.55, 7.95, 7.57, 7.50, 7.43, 4.98. 13C NMR (126 MHz, CDCl3) δ 162.19, 139.26, 136.13, 130.86, 128.66, 126.97, 65.04.
3.2.8 1-(4-Nitrophenyl)-N-phenylmethanimine
1H NMR (500 MHz, CDCl3) δ 8.56, 8.33, 8.07, 7.43, 7.28. 13C NMR (126 MHz, CDCl3) δ 158.54, 151.98, 150.95, 143.00, 130.76, 128.51, 125.11, 122.39.
3.3 Detection of H2 Gas Evolved During Dehydrogenation Reaction
An additional experiment was performed to demonstrate the evolution of H2 in the reaction. A dehydrogenative coupling experiment was carried out in a 15 mL screw-cap Borosil glass test tube. Typically, 2 mmol of alcohol, 6 mmol of aniline, and 50 mg freshly prepared catalyst were added and tightly screw-capped. The reaction tube was heated to the desired reaction temperature through the oil bath. The reaction mixture was stirred by using a magnetic stirrer. The formation of H2 was monitored by withdrawing gas by gas syringe and analyzed sample in GC (Thermo Scientific with Porapak Q column, connect TCD detector, He as a carrier gas, 120 °C temperature,). When the sample was withdrawn after 4 h H2 gas was produced in the dehydrogenation step was detected. In Fig. 8, (or SI8) initially confirmed a sample before reaction Fig. 8A there was observed only atmospheric gas mixture peak position range at 1.4 to 2.3 retention time, there was not observed any H2 gas peak. Then after 4 h withdraw the gas sample from the reaction tube and analyzed it by GC in which observed the H2 gas peak position at 0.54 min see Fig. 8B. Confirmed this H2 gas by injecting pure H2 gas in GC see in Fig. 8C graph. The conclusion of this study is H2 gas is generated during the dehydrogenation of alcohol.

Detection of H2 gas evolved during the reaction by gas chromatography, the sample analysed. A before reaction, B during reaction, and C standard hydrogen Gas
3.4 Mechanism
The synthetic parameters and types of the method adopted for the preparation affect the catalytic activity of mixed metal oxide (MMO) catalyst. The catalytic activity of MMOs depends on the particle size, oxidation state, surface properties, chemical composition of metals, and metal vicinity. The possible reaction mechanism of dehydrogenative coupling of amine and alcohol reaction over Nb2O5/Co3O4 for imine synthesis is shown in Scheme 5. The dehydrogenative reaction pathways for imine formation without any additives and the oxidant-free experiment were confirmed by a controlled experiment. In this regard, we perform the acceptor less dehydrogenation of alcohol under the same reaction condition, and alcohol conversion to aldehyde was obtained within 20 h. N2 and Ar, inert atmosphere also does not affect the reaction rate and product yields and gave the same percentile yield. Co3O4 in the MMO Nb2O5/Co3O4 catalyst has a fair amount of acidic sites which are of a weak acidic strength as compared to Nb2O5 [49] active metal Nb2O5 oxide has strong Lewis acid sites, in which Nb=O are associated, with highly polarised Nb–O which generate hydroxyl group on the surface, that functions as Brønsted acid sites [50]. Many solid acid catalysts and bases are promoted to dehydrogenation reactions. In which catalytic activity of solid acids is higher than those of solid bases. Among the solid acids, niobic acid seems to be most suitable for dehydration.25 Herein, it may be Lewis acidic properties of Nb2O5/Co3O4 catalyst to promote initial step dehydrogenation of alcohol as alkoxide species easily diffuse over Nb (1 1 1) plane, owing to the low energy. On the other side, it is usually convinced that additional basic site of Nb2O5/Co3O4 could promote the activation of the O–H bond in alcohol [51]. Thus, it can be stated that the basic redox site from the Nb2O5/Co3O4 support would be helpful in the activation of the O–H bond from the alkoxide intermediate. Then alkoxide -O to promote the release of benzylic –H and successive formation of benzaldehyde and H-Nb2O5/Co3O4 species via β-H elimination as the rate-determining step, and it may require Lewis acidic Nb2O5 (+ 5) with Co2+ and Co3+ states. Hence, the reactive Lewis acidic surface from the metal may participate in the reaction by abstracting the benzylic proton from the alkoxide intermediate and then forming benzaldehyde. Also, the strong interaction between Nb5+ and Co3O4 formation energy of oxygen vacancy which could further promote catalytic reaction at a lower temperature. Moreover, the Nb/Co3O4 catalyst's acidic and basic site might be releasing the H2 from the surface. The final step is the rapid and uncatalyzed condensation of benzaldehyde with aniline to give benzylidenealine and water as a side product. In this scheme, the prime Nb2O5/Co3O4 catalyst appears as the specific active site having bifunctionality, and also strong interaction with highly dispersed metallic Nb2O5 on the Co3O4 surface. These unique properties could be responsible for the outstanding catalytic performance without additives direct dehydrogenative coupling of alcohol and amine.
4 Conclusion
The Niobium oxide supported on spinel Co3O4 oxide was synthesized and examined for its catalytic activity over one-pot imine formation by dehydrogenative coupling of alcohol with an amine under solvent, oxidant and additives free condition is reported for the first time. The reaction conditions are very mild and environmentally benign, with the liberation of water and H2 as by-products. The present reaction approach has advantages such as solvent, oxidant and additive-free conditions. Significantly lower weight percent niobium containing spinal cubic phase cobalt oxide-containing catalyst demonstrated excellent (up to 100%) conversion and (up to 100%) selectivity to corresponding imine product. The rate of reaction was slow in electron-withdrawing substrates. The catalyst was tested for its recyclability studies and found to have no significant loss in its activity after three cycles. A plausible mechanism was proposed for the formation of imines, and this straightforward catalytic pathway could pave a path for accessing feedstock chemicals with high yields for a range of substrates.
References
Murahashi SI (1995) Synthetic aspects of metal-catalyzed oxidations of amines and related reactions. Angew Chem Int Ed 34:2443–2465
Adams JP (2000) Imines, enamines and oximes. J Chem Soc Perkin Trans 1:125–139
Patil RD, Adimurthy S (2013) Catalytic methods for imine synthesis. Asian J Org Chem 2:726–744
Choi J, MacArthur AH, Brookhart M, Goldman AS (2011) Dehydrogenation and related reactions catalyzed by iridium pincer complexes. Chem Rev 111:1761–1779
Corma A, Navas J, Sabater MJ (2018) Advances in one-pot synthesis through borrowing hydrogen catalysis. Chem rev 118:1410–1459
Bähn S, Imm S, Neubert L, Zhang M, Neumann H, Beller M (2011) The catalytic amination of alcohols. ChemCatChem 3:1853–1864
Dobereiner GE, Crabtree RH (2010) Dehydrogenation as a substrate-activating strategy in homogeneous transition-metal catalysis. Chem Rev 110:681–703
Gnanaprakasam B, Zhang J, Milstein D (2010) Direct synthesis of imines from alcohols and amines with liberation of H2. Angew Chem Int Edi 49:1468–1471
Eizawa A, Nishimura S, Arashiba K, Nakajima K, Nishibayashi Y (2018) Synthesis of ruthenium complexes bearing PCP-type pincer ligands and their application to direct synthesis of imines from amines and benzyl alcohol. Organometallics 37:3086–3092
Maggi A, Madsen R (2012) Dehydrogenative synthesis of imines from alcohols and amines catalyzed by a ruthenium N-heterocyclic carbene complex. Organometallics 31:451–455
Han L, Xing P, Jiang B (2014) Selective aerobic oxidation of alcohols to aldehydes, carboxylic acids, and imines catalyzed by a Ag-NHC complex. Org Lett 16:3428–3431
He LP, Chen T, Gong D, Lai Z, Huang KW (2012) Enhanced reactivities toward amines by introducing an imine arm to the pincer ligand: direct coupling of two amines to form an imine without oxidant. Organometallics 31:5208–5211
Shiraishi Y, Ikeda M, Tsukamoto D, Tanaka S, Hirai T (2011) One-pot synthesis of imines from alcohols and amines with TiO2 loading Pt nanoparticles under UV irradiation. ChemComm 47:4811–4813
Tamura M, Tomishige K (2015) Redox properties of CeO2 at low temperature: the direct synthesis of imines from alcohol and amine. Angew Chem 127:878–881
Qin J, Long Y, Wu W, Zhang W, Gao Z, Ma J (2019) Amorphous Fe2O3 improved [O] transfer cycle of Ce4+/Ce3+ in CeO2 for atom economy synthesis of imines at low temperature. J Catal 371:161–174
Ruiz PA, Schlexer P, Tosoni S, Pacchioni G (2017) Increasing oxide reducibility: the role of metal/oxide interfaces in the formation of oxygen vacancies. ACS Catal 7:6493–6513
Gawande MB, Pandey RK, Jayaram RV (2012) Role of mixed metal oxides in catalysis science versatile applications in organic synthesis. Catal Sci Technol 2:1113–1125
Al-Senani GM, Deraz NM, Abd-Elkader OH (2020) Magnetic and Characterization Studies of CoO/Co3O4 Nanocomposite. Process 8:844
Jacobs G, Das TK, Zhang Y, Li J, Racoillet G, Davis BH (2002) Fischer–Tropsch synthesis: support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl Catal A 233:263–281
Pal J, Chauhan P (2010) Study of physical properties of cobalt oxide (Co3O4) nanocrystals. Mater Charact 61:575–579
Amin RS, Elzatahry AA, El-Khatib KM, Youssef ME (2011) Nanocatalysts prepared by microwave and impregnation methods for fuel cell application. Int J Electrochem Sci 6:4572–4580
Broicher C, Zeng F, Artz J, Hartmann H, Besmehn A, Palkovits S, Palkovits R (2019) Facile synthesis of mesoporous nickel cobalt oxide for OER–insight into intrinsic electrocatalytic activity. ChemCatChem 11:412–416
Laouini E, Hamdani M, Pereira MI, Douch J, Mendonça MH, Berghoute Y, Singh RN (2008) Preparation and electrochemical characterization of spinel type Fe–Co3O4 thin film electrodes in alkaline medium. Int J Hydrog Energy 33:4936–4944
Ziolek M, Sobczak I (2017) The role of niobium component in heterogeneous catalysts. Catal Today 285:211–225
Tanabe K, Okazaki S (1995) Various reactions catalyzed by niobium compounds and materials. Appl Catal A 133:191–218
Li B, Gu M, Nie Z, Wei X, Wang C, Sprenkle V, Wang W (2014) Nanorod niobium oxide as powerful catalysts for an all vanadium redox flow battery. Nano Lett 14:158–165
Armaroli T, Busca G, Carlini C, Giuttari M, Raspolli Galletti AM, Sbrana G (2000) Acid sites characterization of niobium phosphate catalysts and their activity in fructose dehydration to 5-hydroxymethyl-2-furaldehyde. J Mol Catal A 151:233–243
Iizuka T, Ogasawara K, Tanabe K (1983) Acidic and catalytic properties of niobium pentaoxide. Bull Chem Soc Jpn 56:2927–2931
Zhao Y, Zhou X, Ye L, Tsang SCE (2012) Nanostructured Nb2O5 catalysts. Nano Rev 3:17631
Wang X, Cao Z, Lu X, Lin M, Zhang Q (2005) Structure and stability of binary transition-metal clusters (NbCo) (n⩽5): A relativistic density-functional study. J Chem Phys 123:064315
Sharma RK, Ghose R (2016) Synthesis of Co3O4–ZnO mixed metal oxide nanoparticles by homogeneous precipitation method. J Alloys Compd 686:64–73
Wang C, Hua W, Chai G, Zhang C, Guo Y (2019) Insights into the morphological effect of Co3O4 crystallite on catalytic oxidation of vinyl chloride. Catal 9:1–10
Tamura S, Kato K, Goto M (1974) Single crystals of T-Nb2O5 obtained by slow cooling method under high pressures. ZAAC J Inorg Gen Chem 410:313–315
Mejía CH, De-Otter JH, Weber JL, de Jong KP (2017) Crystalline niobia with tailored porosity as support for cobalt catalysts for the Fischer Tropsch synthesis. Appl Catal A 548:143–149
Atribak I, Bueno-López A, Garcia-Garcia A (2008) Combined removal of diesel soot particulates and NOx over CeO2–ZrO2 mixed oxides. J Catal 259:123–132
Cai Y, Xu J, Guo Y, Liu J (2019) Ultrathin, polycrystalline, two-dimensional Co3O4 for low-temperature CO oxidation. ACS Catal 9:2558–2567
Raveau B, Seikh M (2012) Cobalt oxides: from crystal chemistry to physics. Wiley, Hoboken
Usha N, Sivakumar R, Sanjeeviraja C, Arivanandhan M (2015) Niobium pentoxide (Nb2O5) thin films: rf Power and substrate temperature induced changes in physical properties. Optik 126:1945–1950
. Ma Q, Rosenberg RA (2001) Surface study of niobium samples used in superconducting RF cavity production. Proceedings of the 2001 Particle Accelerator Conference 2: 1050–1052.
Aufray M, Menuel S, Fort Y, Eschbach J, Rouxel D, Vincent B (2009) New synthesis of nanosized niobium oxides and lithium niobate particles and their characterization by XPS analysis. J Nanosci Nanotechnol 9:4780–4785
Jiang DE, Dai S (2011) The role of low-coordinate oxygen on Co3O4 (110) in catalyticCO oxidation. Phys Chem Chem Phys 13:978–984
Wang K, Cao Y, Hu J, Li Y, Xie J, Jia D (2017) Solvent-free chemical approach to synthesize various morphological Co3O4 for CO oxidation. ACS Appl Mater Inter 9:16128–16137
Zhang X, Zhang H, Zhu H, Li C, Zhang N, Bao J, He G (2019) Co3O4 nanorods with a great amount of oxygen vacancies for highly efficient Hg0 oxidation from coal combustion flue gas. Energy Fuels 33:6552–6561
Srivastava S, Jadeja GC, Parikh J (2016) A versatile bi-metallic copper–cobalt catalyst for liquid phase hydrogenation of furfural to 2-methylfuran. RSC Adv 6(2):1649–1658
Hu H, Cai S, Li H, Huang L, Shi L, Zhang D (2015) In situ DRIFTs investigation of the low-temperature reaction mechanism over Mn-Doped Co3O4 for the selective catalytic reduction of NOx with NH3. J Phys Chem C 119(40):22924–22933
Zhang Y, Wang J, Ren J, Liu X, Li X, Xia Y, Lu G, Wang Y (2012) Mesoporous niobium phosphate: an excellent solid acid for the dehydration of fructose to 5-hydroxymethylfurfural in water. Catal Sci Technol 2:2485–2491
Díez VK, Apesteguıa CR, Di Cosimo JI (2003) Effect of the chemical composition on the catalytic performance of MgyAlOx catalysts for alcohol elimination reactions. J Catal 215:220–233
Fan Z, Fang W, Zhang Z, Chen M, Shangguan W (2018) Highly active rod-like Co3O4 catalyst for the formaldehyde oxidation reaction. Catal Commun 103:10–14
Ai M (1979) The oxidation activity and acid–base properties of CO3O4–V2O5 and CO3O4–MoO3 systems. Bull Chem Soc Jpn 52:2766–2770
Jouve A, Cattaneo S, Delgado D, Scotti N, Evangelisti C, López Nieto JM, Prati L (2019) Furfural hydrogenation on modified niobia. Appl Sci 9(11):2287
Wu S, Sun W, Chen J, Zhao J, Cao Q, Fang W, Zhao Q (2019) Efficient imine synthesis from oxidative coupling of alcohols and amines under air atmosphere catalysed by Zn-doped Al2O3 supported Au nanoparticles. J Catal 377:110–121
Acknowledgements
BDB acknowledges to UGC government of India for the senior research fellowship. CSMCRI communication No. 89/2020. AVB acknowledges MLP 0028, India, for financial support. The analytical division provides the centralized instrumentation facility with all requisite instrumental analysis of CSIR- Central Salt and Marine Chemicals and Research Institute, Bhavnagar.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bankar, B.D., Ravi, K., Subramanian, S. et al. Niobium Oxide Supported on Cubic Spinel Cobalt Oxide as an Efficient Heterogeneous Catalyst for the Synthesis of Imines via Dehydrogenative Coupling of Amines and Alcohols. Catal Lett 152, 3733–3746 (2022). https://doi.org/10.1007/s10562-022-03943-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-022-03943-2