
Abstract
Pd–Cu/APT catalysts were prepared by an impregnation method (PC-IM) and an evaporation ammonia method (PC-AE), respectively. Their catalytic activities for low-temperature CO oxidation were studied. It is found that PC-AE shows much higher catalytic activity. Characterizations results suggest that Cu species exist as the Cu2(OH)3Cl phase with nanoplatelet morphology in PC-AE. In addition, PC-AE possesses high specific surface area and enhanced reducibility, which may contribute to the excellent catalytic ability.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The low-temperature CO catalytic oxidation is of great significance owing to its practical application such as air purification, CO gas sensors, gas mask, lowering automotive emission and purification of hydrogen for proton exchange membrane fuel cells (PEMFCs), etc. [1–5].
Among various catalysts, the hopcalite catalyst (a mixed manganese and copper oxides) and Co3O4 have high catalytic activity at room temperature, especially Co3O4 catalyst that is active even at −77 °C, however, both of them severely deactivate in the presence of trace amount of moisture due to the blocking of active sites by water adsorption [6–8]. Also, noble metal catalysts (Pt, Pd and Au) show excellent catalytic performance for CO oxidation at low temperature [9–11]. In particular, Au-based catalysts could be superior to Pd and Pt catalysts. But Au-based catalysts also have several unsolved problems, for example, they are sensitive to halogen-including compounds and indoor light (light from fluorescent lamps), resulting in deactivation during reaction or storage [12, 13]. The supported Wacker catalyst, in which an aqueous solution of PdCl2–CuCl2 is loaded into the pores of the high surface area supports, is known to accelerate the oxidation of CO near room temperature and is stable even in the presence of organic halogen compounds and large amounts of moisture in the feed gas [3, 14, 15]. The catalytic performance of supported Wacker catalyst is highly dependent on several parameters, such as support, addition agent, preparation method and reaction conditions. In order to improve the catalytic activity, previous works mainly focused on searching for a proper support or adding different addition agents. Lee et al. [16]. investigated the effect of supports on the catalytic performance of supported Pd–Cu catalysts, and found that the catalytic activity decreased approximately in the order of active carbon > (active carbon + ZSM-5) > ZSM-5 > molecular sieve-13X > molecular sieve-5A > alumina. They thought that the poor activity for zeolite-based catalyst is largely due to the pore-filling with water, which decreased the gas–liquid interfacial surface area. Wang et al. [17] claimed that Pd–Cu catalyst supported on Al2O3 possessed better catalytic performance than that on TiO2, because the higher BET surface of Al2O3 support facilitates the dispersion of Pd2+ species and the presence of Cu2Cl(OH)3 over Pd–Cu/Al2O3 catalyst accelerates the re-oxidation of Pd0–Pd2+. Park et al. [18]. tested other Cu precursors as an addition agent for CuCl2, such as Cu(OH)2, Cu(CH3COO)2 and Cu(NO3)2, and found that Cu(NO3)2 was the best Cu addition agent precursor, because Cu(NO3)2 can promote the formation of Cu2(OH)3Cl in catalysts. In recent years, some researchers paid attention to developing a suitable preparation method to promote the catalytic performance [3, 19, 20]. Shen et al. reported the Pd–Cu–Clx/Al2O3 catalyst prepared by a NH3 coordination-impregnation method, which exhibited an excellent activity for low-temperature CO oxidation due to the higher dispersion of active copper species and the excellent low-temperature reduction characteristic [3].
The Pd–Cu/attapulgite clay (Pd–Cu/APT) catalyst prepared by an evaporation ammonia method has not been reported. In this paper, the evaporation ammonia method was employed to prepare an APT supported PdCl2–CuCl2 catalyst (PC-AE), which exhibits excellent catalytic performance for CO oxidation at low temperature in the presence of large amounts of moisture. For comparison, the Pd–Cu/APT catalyst was also prepared by using a traditional wet impregnation method (PC-IM). Herein, PC-AE and PC-IM were characterized by XRD, N2-physisorption, FTIR, TEM and TPR, and the relationships between the preparation method and the structure, catalytic performance were also discussed in detail.
2 Experimental
2.1 Preparation of Catalysts
Pd–Cu/APT catalyst was prepared by an evaporation ammonia method as follows: 0.02 g of PdCl2 and 1.82 g of CuCl2·2H2O were dissolved in 10 ml of H2O, and 20 ml of 28 % ammonia aqueous solution was added and stirred for 30 min. Then 5 g of APT was added into the above solution and the initial pH of the suspension was 11.5. The suspension was stirred for another 4 h. All the above operations were performed at room temperature. The suspension was transferred to an water bath preheated at 60 °C to allow for ammonia evaporation. When the pH value of the suspension decreased to 6.5, the suspension was removed from the water bath, and the evaporation process was terminated. After being filtered, the residue was dried in air at 80 °C for 3 h and at 120 °C for other 3 h, then calcined at 300 °C for 3 h in air. The sample prepared was designated as PC-AE. The average contents determined by ICP-AES were 0.34 wt% and 11.92 wt% for Pd and Cu, respectively, which were a little lower than the theoretical values (Pd 0.35 wt%, Cu 12 wt%).
Pd–Cu/APT catalyst was prepared by a wet impregnation method as follows: 0.02 g of PdCl2 and 1.82 g of CuCl2·2H2O were dissolved in 10 ml of H2O, and then 5 g of APT was added into the above solution. After being aged at room temperature for 3 h, the solid was dried in air at 80 °C for 3 h and at 120 °C for other 3 h, and then calcined at 300 °C for 3 h in air. The sample prepared was designated as PC-IM. The average contents determined by ICP-AES were 0.35 wt% and 11.98 wt% for Pd and Cu, respectively, which were almost the same as the theoretical values.
2.2 Characterization of Catalysts
The contents of Cu and Pd in the catalysts were determined by a Varian 710ES inductively coupled plasma-atomic emission spectroscopy (ICP-AES). All determinations were conducted at least in triplicate. Powder X-ray diffraction (XRD) pattern was performed on a Bruker D8 Advance diffraction spectrometer with CuKα radiation (λ = 0.154 nm) at a scanning rate of 2.4°/min in the 2θ range of 5–50°. N2-physisorption analysis was measured on a Micromeritics ASAP-2020 apparatus. Sample was degassed at 150 °C for 5 h before analysis. Specific surface area (SBET) was calculated following the multi-point BET procedure. Hydrogen temperature programmed reduction (H2-TPR) was performed with a Micromeritics AutoChemII 2920 instrument. 30 mg of sample was loaded. 5 vol% H2/N2 (30 mL/min) mixture was introduced and the temperature was raised from room temperature to 700 °C at a rate of 10 °C/min. FT-IR spectrum was recorded on a Bruker Tensor 27 Fourier transform infrared spectrometer at room temperature. One milligram of each powder sample was diluted with 100 mg of vacuum-dried IR-grade KBr. 16 scans were collected for each sample at a resolution of 4 cm−1. Transmission electron microscopy (TEM) measurement was carried out using JEM-2100 model TEM with accelerating voltage of 200 kV. Sample for TEM was dispersed by sonicator in ethanol followed by deposition of the suspension onto a standard Cu grid covered with a holey carbon film.
2.3 Catalytic Activity Test
The measurements of catalytic performance for low-temperature CO oxidation were carried out in a flow-through laboratory microreactor under atmospheric pressure. The microreactor was a 8 mm i.d. quartz u-tube, and a thermocouple was set into the catalyst bed to measure the temperature. The samples were sieved to 40–60 mesh so that temperature gradients and pressure drop over the catalyst bed were negligible. 300 mg of catalyst was used for each test. The feed gas adjusted by mass flow controllers consisted of 0.5 vol% CO, 3.3 vol% H2O and balance of air, passed through the catalyst bed with a total flow rate of 30 mL/min. No pretreatment was applied before catalytic activity test. The quantitative analysis of CO was performed with an on-line gas chromatograph equipped with a 3 m column packed with carbon molecular sieve, a methanator and a flame ionization detector (FID). In order to enhance the sensitivity of the detection, CO and CO2 were converted to CH4 by the methanator at 360 °C before entering into the FID. The activity was expressed by CO conversion, which was calculated according to:
where X is the CO conversion, COinlet presents the initial CO concentration in the inlet, and COoutlet presents the CO concentration in the outlet.
3 Results and Discussion
3.1 Catalytic Performance for Low-Temperature CO Oxidation
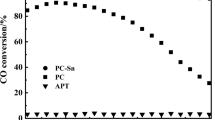
CO catalytic oxidation performances of APT, PC-IM and PC-AE as a function of reaction time are compared and given in Fig. 1. As shown in Fig. 1, both of the catalysts show catalytic activity for CO oxidation at room temperature except the support (APT). PC-AE exhibits much higher catalytic activity for CO oxidation than PC-IM. The initial CO conversion over PC-AE can reach 100 %, and has almost no change with continuously increased reaction time. However, the initial CO conversion over PC-IM is less than 10 %, but the CO conversion obviously increases with the extension of reaction time. Then, the CO conversion could reach a maximum of 60 % after 120 min. It needs further study whether the PC-IM catalyst has changed in this process.

Catalytic performance of support (APT) and catalysts prepared by the impregnation method (PC-IM) and evaporation ammonia method (PC-AE) Reaction Conditions: 0.5 vol% CO, GHSV 6000 h−1, 3.3 vol% H2O, room temperature Inset: The inset shows the effect of reaction temperature on the catalytic activity of PC-AE
The inset shows the effect of reaction temperature on the CO conversion over the two catalysts. From the inset it can be seen that PC-AE also shows higher catalytic activity for CO oxidation than PC-IM not only below room temperature, but also above room temperature. In order to readily compare with the literatures, the specific reaction rate for CO oxidation over PC-AE has been calculated. In general, the Pd2+ is regard as the active site of the supported Wacker catalyst [3, 14, 18], so the specific reaction rate is calculated based on the number of moles of converted CO on per gram of Pd per hour. PC-AE gives a specific reaction rate of 0.20 molCO/gPd/h at 0 °C, which is much higher than for 4.4 % Au/Fe2O3 (0.091 molCO/gAu/h) at 30 °C [21], and also higher than for Pd–Cu–Clx/Al2O3 (0.18 molCO/gPd/h) at 0 °C [3].
3.2 Characterization of Catalysts
3.2.1 XRD Analysis
X-ray diffraction patterns of support (APT) and the catalysts are shown in Fig. 2. The typical peak at 2θ = 8.4° (d110 = 1.045 nm) corresponds to (110) of APT, which is attributed to the basal plane of the APT structure. The intensity of the peak at 2θ = 8.4° indicates the degree of crystallinity of APT. Peaks at 2θ = 19.7° and 20.7° represent the Si–O-Si crystalline layers in APT. Additionally, the peak scanned at 2θ = 26.7° is attributed to quartz impurities.

XRD patterns of support (APT) and catalysts prepared by the impregnation method (PC-IM) and evaporation ammonia method (PC-AE) Inset: The inset shows a magnification of the XRD patterns from 7° to 10° for a clear comparison
Compared with the diffraction pattern of APT, the weak diffraction peaks at 2θ = 16.2°, 17.6°, 31.0°, 32.2° and 39.5°, characterization of Cu2(OH)3Cl structure, were observed in the pattern of PC-AE. However, CuCl2·2H2O (2θ = 16.2°, 21.9° and 33.9°) is a dominant solid copper phase in PC-IM. In addition, the XRD pattern of PC-IM after reaction showed that the CuCl2·2H2O species transformed to Cu3Cl4(OH)2·H2O with time on stream (see supplementary material, Figure S1). The diffraction peak of Pd species cannot be observed in both of the catalysts, which indicates Pd species exist as a high dispersed state or below the detection limit of XRD. Furthermore, it can be seen that the intensity of diffraction peak (2θ = 8.4°) in the catalysts are less than that of APT support, and the intensity order is: APT(793) > PC-IM(675) > PC-AE(579), which indicates the decrease of the degree of crystallinity of APT in catalysts. It has been reported that when CuO is loaded on the surface of APT, the fine dispersal of CuO particles and the strong interaction between the CuO and APT support could result in the decreased diffraction intensity of APT. In the present Pd–Cu/APT catalyst system, the decreased diffraction intensity of APT might also derive from the high dispersion of Cu, Pd species and the interaction between them and APT support. In addition, the position of the peak 2θ = 8.4° shifts to 2θ = 8.3° in PC-AE, indicating the basal plane distance of APT interlayers slightly increase to d110 = 1.057 nm. This might suggest that the changes of APT structure in PC-AE result from not only the more strong interaction between Cu, Pd species and APT, but also the effect of evaporation ammonia process.
3.3 N2-Physisorption Analysis
Figure 3 shows N2-physisorption isotherms of APT and PC-IM, PC-AE catalysts, and their texture properties are listed in Table 1. On the basis of the IUPAC classification, the isotherms of APT, PC-IM and PC-AE exhibit a typical type III class, with a H3-type hysteresis loop characteristic. All samples show a very strong increase of N2-adsorbed volume at a relative pressure higher than 0.85, which is the characteristic of the presence of an appreciable amount of macropores [22]. The H3-type hysteresis loop indicates the materials comprised of aggregates (loose assemblages) of platelike particles, which is the nature of APT. Specific surface area of APT is 137 m2 g−1, which decreases to 62 m2 g−1 when APT is used as the support of PdCl2–CuCl2 catalyst prepared by the wet impregnation method. This is mainly resulting from the distribution of copper and palladium species on the surface or in the channel of APT. SBET and pore volume of PC-AE are 107 m2 g−1 and 0.34 mL g−1, respectively, which are much larger than that of PC-IM. This suggests that catalyst prepared by the evaporation ammonia method possesses higher specific surface area and larger pore volumes, which are the advantage for the catalytic CO oxidation.

N2-physisorption isotherm of support (APT) and catalysts prepared by the impregnation method (PC-IM) and evaporation ammonia method (PC-AE)
3.4 FT-IR Analysis
The samples were characterized by FT-IR to understand the structure and the interaction between Cu, Pd species and APT. Figure 4 gives the FT-IR spectra between 4000 and 400 cm−1 of the PC-IM, PC-AE and the support APT. FT-IR spectrum of APT shows two bands at 3631 and 3552 cm−1, corresponding to the stretching vibrations of –OH units in the coordinated water and the bond water, respectively. A broad band between 3500 and 3200 cm−1 is observed in APT that possibly involved stretching vibrations of –OH in the zeolitic water and surface adsorbed water in APT. The band at 1639 cm−1 is ascribed to the –OH bending mode of all kinds of water. Two bands at approximately 1026 and 989 cm−1 are present in APT and they have been described as the Si–O vibrations and the in-layer Si–O bands. However, the two bands change into a broad band in catalysts, probably originating from the structure change of APT and the interaction between APT and copper, palladium species. The band at 798 cm−1 confirms the presence of quartz and the band at 473 cm−1 is assigned to the Si–O–Si deformation vibration. The relative degree of crystalline of APT is calculated by considering the transmittance of band at 473 cm−1 to the transmittance of band at 534 cm−1, which is termed as T 473 /T 534 . It is worthwhile to note that the T 473 /T 534 ratio only gives a qualitative estimation of the degree of crystalline of ATP. Inset in Fig. 4 clearly shows that the relative degree of crystalline of APT decreases in the order of APT > PC-IM > PC-AE, which are consistent with the XRD results.

FT-IR spectra of support (APT) and catalysts prepared by the impregnation (PC-IM) and evaporation ammonia method (PC-AE) Inset: The inset shows the T 473 /T 534 intensity ration representing the relative degree of crystalline of APT
Two bands at 3448 and 3359 cm−1 in PC-AE are attributed to the -OH stretching modes of Cu2(OH)3Cl, indicating the existence of Cu2(OH)3Cl in PC-AE catalyst. However, the positions of the two bands are not consistent with the two adsorption peaks at 3445 and 3341 cm−1 of the uniform Cu2(OH)3Cl [23], demonstrating the partially amorphous Cu2(OH)3Cl on the APT support. Furthermore, it can also reveal that the interaction between Cu species and Pd species or the support APT, which can be given in TPR results (will be present shortly).
3.5 TEM Analysis
TEM images of PC-AE (a), PC-IM (b) and the support APT (c) are shown in Fig. 5. APT exhibits the bundles of attenuated fibrous structures, in which some impurities of granular particles and their aggregates can also be observed from the TEM images (Fig. 5c). The fiber appearance of APT has no obvious changes after loading with Pd and Cu species (Fig. 5a, b). In PC-IM (Fig. 5b), there are numerous nanoparticles attached favorably on the surfaces of APT fibers, and the particle size is not uniform, which is the feature of catalysts made by the impregnation method. However, active component on PC-AE (Fig. 5a) is a kind of nanoplatelet with the size of 15–20 nm, and through which the fibrous structure of the support (APT) can be seen clearly. The nanoplatelet seems to present a hexagonal morphology. According to the XRD results, the hexagonal nanoplatelet may be the Cu2(OH)3Cl formed in the process of evaporation ammonia, which is essential for high activity of the supported PdCl2–CuCl2 catalysts [15, 17].

TEM images of support (APT) and catalysts prepared by the impregnation method (PC-IM) and evaporation ammonia method (PC-AE)
As to the formation process of the Cu2(OH)3Cl nanoplatelet, an analysis of the liquid-phase chemistry reaction may be as follows. The reaction solution initially contains Cu(NH3) 2+4 with excess dissolved ammonia. When heated in the water bath, with the evaporation of ammonia the hydrolysis of the Cu(NH3) 2+4 complex occurs, forming Cu2(OH)3Cl species, which attach to each other, extend along with the fiber surface of APT and form the nanoplatelet. The similar analysis had been reported by Wu et al., which was concerning the synthesis of metal hydroxide/oxide [24]. We also measured the decrease of the solution pH with reaction time, which reflected the decrease of the ammonia concentration during the reaction process. In order to further confirm the formation of Cu2(OH)3Cl in the liquid-phase chemistry reaction, the sample above mentioned before calcination was measured by XRD, and the result showed that the Cu species existed as the Cu2(OH)3Cl (not given here).
There are the following two major chemical equilibriums during the formation of Cu2(OH)3Cl:
3.6 H2-TPR Analysis
TPR profiles of the PC-IM, PC-AE and APT are shown in Fig. 6. According to the XRD results, CuCl2·2H2O is a dominant solid copper phase in PC-IM, while Cu2(OH)3Cl in PC-AE. In the TPR profile of PC-IM, the sharp peak at 311 °C and the peak at 352 °C are ascribed to the reduction of CuCl2 dispersed on APT and bulk CuCl2 species, respectively. The shoulder reduction peak at 263 °C in TPR profile of PC-IM attributes to the co-reduction of Pd and Cu species. It can be seen clearly that the TPR profile of PC-AE is distinctly different from that of PC-IM. There is a main sharp peak at 282 °C originating from reduction of crystal Cu2(OH)3Cl. Other peaks presented at 204 and 252 °C ascribe to co-reduction of Pd and Cu species, and Cu2(OH)3Cl dispersed on the APT, respectively. On the whole, the reduction peaks of PC-AE shift obviously to lower temperature compared with that of PC-IM, which indicates Cu2(OH)3Cl species are more easily reduced than CuCl2. In addition, based on the reduction temperature and area of the peak ascribed to co-reduction of Pd and Cu species, it can be demonstrated that the Cu2(OH)3Cl species has much stronger interaction with Pd species than CuCl2. It can be concluded that the preparation method has a marked influence on the reducible nature of the Pd–Cu/APT catalyst. Furthermore, the TPR profile of PC-IM after reaction indicates that the Cu3Cl4(OH)2·H2O phase formed during reaction is more reducible than the CuCl2·2H2O phase of PC-IM before reaction, but is still less reducible than the Cu2(OH)3Cl phase present on PC-AE (see supplementary material, Figure S2).

TPR profiles of support (APT) and catalysts prepared by the impregnation method (PC-IM) and evaporation ammonia method (PC-AE)
As seen clearly from the results of XRD and FT-IR, the Cu species exist as the Cu2(OH)3Cl form in the catalyst prepared by the evaporation ammonia method, which is entirely different from that in the catalyst prepared by the impregnation method. N2-physisorption and TEM results suggest that the PC-AE catalyst possesses high specific surface area, and the Cu2(OH)3Cl presents the nanoplatelet morphology. The TPR results indicate that the Cu2(OH)3Cl possesses much stronger interaction with Pd species than CuCl2. The appropriate interaction enhances the reduction degree of the active species. So it can be concluded that the excellent reduction property of Cu2(OH)3Cl nanoplatelet result in the higher catalytic activity of the PC-AE catalyst for CO oxidation. Further works related to the influences of evaporation time and initial ammonia concentration on the catalytic activity of the PC-AE catalyst for low-temperature CO oxidation are in progress.
4 Conclusions
Pd–Cu/APT catalysts were prepared by an impregnation method (PC-IM) and an evaporation ammonia method (PC-AE), respectively. Effect of preparation methods on the structure and catalytic performance of Pd–Cu/APT had been investigated. The tests for CO oxidation indicate that the PC-AE shows higher catalytic activity than PC-IM. The characterization results reveal that the Cu species exist as Cu2(OH)3Cl nanoplatelet on the PC-AE catalyst. The excellent reduction property of Cu2(OH)3Cl nanoplatelet obtained via the ammonia evaporation process contribute to the higher catalytic activity of PC-AE. Further works related to the optimization of the design and preparation for this catalyst system are in progress, since this work can lead to exploiting a catalyst with higher activity for low-temperature CO oxidation.
References
Xie XW, Li Y, Liu ZQ, Haruta M, Shen WJ (2009) Nature 458:746–749
Zhu HQ, Qin ZF, Shan WJ, Shen WJ, Wang JG (2007) Catal Today 126:382–386
Shen YX, Lu GZ, Guo Y, Wang YQ (2010) Chem Commun 46:8433–8435
Si RR, Liu JF, Yang K, Chen X, Dai WX, Fu XZ (2014) J Catal 311:71–79
Twigg MV (2007) Appl Catal B 70:2–15
Wang YZ, Zhao YX, Gao CG, Liu DS (2007) Catal Lett 116:136–142
Wang YZ, Zhao YX, Gao CG, Liu DS (2008) Catal Lett 125:134–138
Hou XD, Wang YZ, Zhao YX (2008) Catal Lett 123:321–326
Grünert WG, Großmann D, Noei HM, Pohl MM, Sinev L, Toni AD, Wang YM, Muhler M (2014) Angew Chem Int Ed 53:3245–3249
Qian K, Luo LF, Bao HZ, Hua Q, Jiang ZQ, Huang WX (2013) Catal Sci Technol 3:679–687
Carlsson PA, Skoglundh M (2011) Appl Catal B 101:669–675
Schumacher B, Plzak V, Kinne M, Behm RJ (2003) Catal Lett 89:109–114
Oxfor SM, Henao JD, Yang JH, Kung MC, Kung HH (2008) Appl Catal A 339:180–186
Wang FG, Zhao KF, Zhang HJ, Dong YM, Wang T, He DN (2014) Chem Eng J 242:10–18
Lee JS, Park ED, Song BJ (1999) Catal Today 54:57–64
Lee CW, Park SJ, Kim YS, Chong PJ (1995) Bull Korean Chem Soc 16:296–298
Wang FG, Zhang HJ, He DN (2014) Environ Technol 35:347–354
Park ED, Lee JS (2000) J Catal 193:5–15
Shen YX, Lu GZ, Guo Y, Wang YQ, Guo YL, Gong XQ (2011) Catal Today 175:558–567
Shen YX, Lu GZ, Guo Y, Wang YQ, Guo YL, Gong XQ (2011) Catal Sci Technol 1:1202–1207
Qiao BT, Liu LQ, Zhang J, Deng YQ (2009) J Catal 261:241–244
Cao JL, Shao GS, Wang Y, Liu YP, Yuan ZY (2008) Catal Commun 9:2555–2559
Wei W, Gao P, Xie JM, Zong SK, Cui HL, Yue XJ (2013) J Solid State Chem 204:305–313
Li YG, Tan B, Wu YY (2008) Chem Mater 20:567–576
Acknowledgments
Authors gratefully acknowledge financial support from International S&T Cooperation Program of China (2013DFA40460), the National Natural Science Foundation of China (21073114), and the Shanxi Youth S&T Foundation (2010021008-3).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, Y., Fan, L., Shi, J. et al. Effect of Preparation Method on the Catalytic Activities of Pd–Cu/APT Catalysts for Low-Temperature CO Oxidation. Catal Lett 145, 1429–1435 (2015). https://doi.org/10.1007/s10562-015-1536-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-015-1536-1