
Abstract
The water-gas shift reaction (WGS, CO + H2O → H2 + CO2) was studied over CuO/CeO2 catalysts with two different ceria particle morphologies, in the form of nanospheres (ns) and nanocubes (nc). To understand the strong dependence of the WGS reaction activity on the ceria nanoshapes, pulses of CO (without and with water vapor) were employed during in situ X-ray diffraction and X-ray absorption near edge structure measurements done to characterize the catalysts. The results showed that CuO/CeO2 (ns) exhibited a substantially better activity than CuO/CeO2 (nc). The higher activity was associated with the unique properties of CuO/CeO2 (ns), such as the easier reduction of highly dispersed CuO to metallic Cu, the stability of metallic Cu and a larger concentration of Ce3+ in CeO2 (ns).
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The water-gas shift reaction (WGS, CO + H2O → H2 + CO2) has become one of the most important processes in the chemical industry for the production of clean molecular hydrogen for proton exchange membrane fuel cells (PEMFCs) [1]. As the reaction is exothermic (△H = −41.1 kJ/mol) [2] and equilibrium limited, a high conversion of CO can be obtained with a two-stage process: a high temperature shift (350–500 °C) using mixtures of Fe–Cr oxides as catalysts and a low temperature shift (200–250 °C) with Zn–Al–Cu oxides as catalysts [3]. However, the current industrial catalysts for the WGS reaction are not appropriate for use with fuel cells, particularly in mobile systems, due to the problems related to their narrow temperature regime of operation and pyrophoric characteristics, as well as the requirement of lengthy and complex activation steps before usage [4]. Great attempts have been made in recent years to seek WGS catalysts with high activity, stability, free of pretreatment requirements, that operate at lower temperatures.
Precious metal catalysts, such as Pt, Pd, Ru and Au, have been widely studied for the WGS reaction and have showed high catalytic activity and stability at low temperatures [5]. However, the high cost and the limited abundance of the precious metals inhibits their application and has encouraged researchers to look for base metal catalysts.
In recent years, cerium oxide has received considerable attention due to its unique reducibility properties and oxygen storage capability (OSC) [6]. This reducibility of CeO2 can be significantly enhanced by the presence of a small amount of a transition metal, for instance the CuO/CeO2 system. Because of its high oxygen storage capacity, strong metal-support interactions and facility to generate oxygen vacancies [7], CuO/CeO2 catalyst has been widely applied in many catalytic processes including: selective catalytic reduction of NO with NH3 [8] or C3H6 [9], selective oxidation of CO in excess hydrogen [10] and the WGS reaction [11]. Recently, it has been recognized that the morphology of CeO2 strongly influence the surface atom densities, electronic structure, and chemical reactivity of this oxide [12]. Therefore, many studies of CeO2 systems have focused on the shape-dependent properties. Meher and Rao [12] synthetized highly uniform CeO2 nanorods and nanoflowers and tested for their soot oxidation activity. They found that the CeO2 sample with nanorod morphology was more active due to larger CeO2/soot interface than the CeO2 sample with nanoflower morphology. Shen and co-workers [13] studied the catalytic activities of ceria nanowires, nanorods and nanoparticles for CO oxidation. It was found that nanowires expose a large proportion of active planes on the surface, resulting in a much higher activity than the others. Previously, we reported three CuO/CeO2 catalysts with different morphologies of ceria, namely nanospheres, nanorods and nanocubes, and investigated their catalytic performance for the WGS reaction [14]. The results showed that nanosphere ceria catalysts were more preferable than the other two types, due to their excellent water dissociation capability, the best dispersion of Cu and a strong Cu–CeOx interaction [14].
Pulse or transient experiments can provide more dynamic details than steady-state measurements. They have been widely used in many catalytic studies to reveal details of the reaction mechanism [15, 16]. In this study, we use the pulse method to investigate the relationship between ceria morphology (nanospheres and nanocubes) and catalytic activity, hoping to gain further insight to understand the performance of the CuO/CeO2 catalysts and how their components respond to CO and water.
2 Experimental
2.1 Catalysts Preparation
The ceria nanospheres were synthesized by a reverse microemulsion method containing n-Heptane, Triton-X-100 and n-Hexanol in amounts similar to those reported previously [3]. The required amount of ceria nitrate was dissolved in distilled water and dispersed within the previously mentioned organic components to form a reverse microemulsion. After 1 h stirring of the microemulsion, a (tetramethyl ammonium hydroxide) TMAH-containing solution was added to the cerium-containing solution and it was left for the period of 24 h in order to complete the precipitation reaction. The resulting solid was then separated by centrifugation and decantation; rinsed with methanol; dried overnight at 100 °C and finally calcined at 500 °C for 2 h in air. Ceria nanocubes were synthesized by a hydrothermal method. The cerium nitrate precursor was dissolved in a NaOH aqueous solution with stirring. The mixture was introduced into a stainless steel autoclave and kept at 180 °C for 24 h. The precipitants were further separated, washed and dried at 60 °C overnight and the resulting powder was calcined at 500 °C for 2 h in air. Copper based catalysts, employing the as-prepared CeO2 supports, were synthesized by incipient wetness impregnation using copper nitrate as the precursor (5 wt% Cu loading). The samples were dried overnight at 100 °C and then calcined in air at 500 °C for 2 h. The above as-prepared catalysts are labeled as CuO/CeO2 (ns) and (nc) for the nanosphere and nanocube ceria supports, respectively. Specific surface areas (SBET), as determined from N2 adsorption isotherms, were of 72 and 29 m2/g for CuO/CeO2 (ns) and (nc) catalysts, respectively. Annular dark field STEM (Fig. 1) showed that ceria nanocubes primarily expose (100) faces, with (110) terminations at the corners, and ceria nanospheres were dominated by (111) faces but in some cases the particles were irregular without well-defined facets [14]. CuO particles were not resolvable from TEM and STEM images mainly due to the low contrast between Cu and Ce, and the high dispersion of very small particles [14].

STEM and high-resolution STEM images of CuO/CeO2 (ns) (a) and CuO/CeO2 (nc) (b) catalysts
2.2 XRD
XRD was performed at beamline X7B (λ = 0.3196 Å) of the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory (BNL). The flow cell system was the same as in the reaction activity experiments as described in Sect. 2.4. Two-dimensional transmission diffraction data were collected with a PerkinElmer amorphous silicon detector, and the powder diffraction rings were subsequently integrated with the program Fit2D [17] to obtain XRD profiles (intensity vs. 2θ). The instrument parameters were derived from the fit of a LaB6 reference pattern. Lattice parameters and phase fractions were determined by Rietveld refinement using the program GSAS (General Structure Analysis System).
2.3 XANES
Cu K edge in situ XANES were collected at beamline X19A of the NSLS at BNL. The flow cell system was the same as in the reaction activity experiments, except that the sample was loaded into a Kapton capillary. The X-ray absorption spectra were taken repeatedly in the “fluorescence yield mode” using a passivated implanted planar silicon (PIPS) detector cooled with circulating water. The XANES data were then analyzed using the Athena program.
2.4 WGS Activity Experiment
WGS reaction activity experiments were performed in a quartz capillary (0.9 mm i.d.) mounted in a flow cell system described previously [15, 18]. 2–3 mg of the sample were loaded between two quartz wool beds at atmospheric pressure. A resistance heating coil was used to heat the sample capillary, and the catalyst temperature was monitored with a K-type thermocouple placed inside the capillary in the vicinity of the catalyst bed. In a set of initial experiments, the samples were exposed to CO or water alone. This was followed by a study of the behavior of the catalysts under a mixture of CO and water. CO was pulsed for periods of 25 min, while the flow of H2O was kept constant. The total feed gas mixture typically contained 1 % CO and/or 3 % H2O in helium with a flow rate of 10 mL/min. The outlet products were measured with a calibrated quadrupole mass spectrometer. During the studies with pulses, the temperature of the sample was maintained at 250 or 400 °C.
3 Results and Discussion
3.1 CO Reduction Experiments on CuO/CeO2 (nc) and CuO/CeO2 (nc) Catalysts
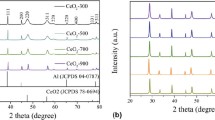
Figure 2 displays XRD patterns for the as-prepared CuO/CeO2 (nc) and CuO/CeO2 (ns) catalysts at room temperature. One can see strong diffraction lines for CeO2 and small features for CuO. The loading of Cu was similar in both cases (~5 wt%). From Rietveld refinement of these XRD data one can get CuO/CeO2 molar ratios of 0.16 for CuO/CeO2 (nc) and 0.08 for CuO/CeO2 (ns). In the case of the CuO/CeO2 (ns) sample, it is known that a significant part of the copper oxide is in an amorphous phase not seen by XRD but observed in measurements of X-ray absorption spectroscopy at the Cu K-edge [14]. On the other hand, almost all the CuO present in the CuO/CeO2 (nc) sample is in a crystalline phase [14].

Diffraction patterns for the CuO/CeO2 (ns) and CuO/CeO2 (nc) catalysts
Reduction experiments with a pulse of CO (T = 250 °C) were performed to evaluate the redox properties of CuO and CeO2 in the two catalysts. Results are shown for the CuO/CeO2 (nc) sample in Fig. 3 combining measurements of mass spectrometry at the outlet of the reactor (RGA intensities for CO and CO2) and the analysis of in situ time-resolved XRD data. As CO was pulsed into the reactor, the CuO phase transformed to metallic Cu very quickly and only a little Cu+ was detected by the XRD measurement.

In situ XRD: reduction of a CuO/CeO2 (nc) catalyst under a pulse of CO at 250 °C
Most of the transformation occurred within 5 min of the introduction of CO into the reactor. At the same time that there was an almost complete reduction of CuO, and we found a partial reduction of the ceria with a small expansion of the ceria lattice parameter (Ce3+ is bigger than Ce4+) [19].
The ceria lattice parameter changed from 5.424 Å (when the flow did not contain CO) to 5.427 Å (when CO was introduced). In Fig. 3, the consumption of CO stops almost at the same time that the signals for metallic copper and the ceria lattice parameter reach a constant value. This indicates that in the transient period, there is a strong link between the reduction of CuOy and CeOx (i.e. the transformations in Eqs. (1)–(4) take place simultaneously). This is somewhat surprising, since bulk ceria has a larger stability than copper oxides. Thus, it is likely that the Ce4+ sites reduced upon exposure of CuO/CeO2 (nc) to CO were probably associated with a CuO–CeO2 interface.
Figures 4 and 5 show data of in situ XRD and XANES for the exposure of a CuO/CeO2 (ns) catalyst to a pulse of CO at 250 °C, respectively. As mentioned above, this system contained almost equal fractions of crystalline and amorphous CuO [14] and for that reason was also studied with XANES. Both sets of results indicate that Eqs. (1)–(4) occur during the reduction of the CuO/CeO2 (ns) sample. Most of the transformations take place in the first 5 min of reaction. An important difference with respect to CuO/CeO2 (nc) is in the stability of the formed Cu+. In the case of CuO/CeO2 (ns), a substantial fraction of the Cu+ generated is not a transient species and remains in the sample after exposure to CO for a period of 25 min. This is interesting because bulk Cu2O is not stable in an atmosphere of CO at 250 °C [14, 18, 20]. The extra stability of Cu+ is probably a consequence of the existence of strong Cu2O ↔ CeOy interactions on the ceria (ns) support. The similar trends seen in Fig. 4 for the increase in the amount of metallic Cu formed and the expansion of the ceria lattice indicate that CO reduces Cu2+ and Ce4+ cations present in a CuO–CeO2 interface, as was also seen for the CuO/CeO2 (nc) sample. Furthermore, a comparison of the results in Figs. 3 and 4 shows a larger CeO2 cell expansion for CuO/CeO2 (ns) than for CuO/CeO2 (nc), which implies the formation of more oxygen vacancies in the CuO/CeO2 (ns) catalysts.

In situ XRD: reduction of a CuO/CeO2 (ns) catalyst under a pulse of CO at 250 °C

In situ XANES: reduction of a CuO/CeO2 (ns) catalyst under a pulse of CO at 250 °C
3.2 Exposure of CuO/CeO2 (nc) and CuO/CeO2 (ns) Catalysts to Water
When the CuO/CeO2 (nc) and CuO/CeO2 (ns) samples were exposed to pulses of water at 250 °C, we observed no changes in the XRD patterns.
3.3 WGS Activity of CuO/CeO2 (nc) and CuO/CeO2 (ns) Catalysts and the Pulsing of CO Under a Water Environment
Figure 6 shows data for the production of hydrogen over CuO/CeO2 (nc) and CuO/CeO2 (ns) catalysts through the WGS at temperatures between 200 and 400 °C. Both materials become catalytically active at temperatures above 200 °C [14]. CuO/CeO2 (ns) is always a better catalyst than CuO/CeO2 (nc), and the gap in catalytic activity increases when the temperature is raised. Previous experiments of in situ DRIFTS showed that these two catalysts exhibited infrared spectra with different line-shapes in the region between 1,700 and 800 cm−1 [14]. The different morphologies of the catalysts (Fig. 1) led to different types of adsorbed carbonates. CuO/CeO2 (nc) showed two strong bands centered at 1,466 and 1,394 cm−1 associated with carbonates adsorbed on the (100) face [14]. These bands were not seen on the CuO/CeO2 (ns) catalyst and were present on CuO/CeO2 (nc) at a wide range of temperature. These carbonate species were strongly bound to the nanocubes and do not participate in the catalytic process [14]. They may poison the active sites on the surface of CuO/CeO2 (nc) [14]. In this article, we examine how CuO/CeO2 (nc) and CuO/CeO2 (ns) respond to the pulsing of CO under a water environment. The sample was kept at a temperature of 250 °C when the onset of H2 production appears in Fig. 6.

Production of hydrogen during the WGS over CuO/CeO2 (ns) and CuO/CeO2 (nc) catalysts. The feed gas mixture contained 1 % CO and 3 % H2O in helium with a flow rate of 10 mL/min
Figure 7 displays in situ XRD results collected after exposing CuO/CeO2 (nc) to pulses of CO under a constant flow of water at 250 °C. As soon as CO was added to the water, there was a fast reduction of Cu2+ to Cu and an expansion of the ceria lattice due to the formation of some Ce3+. The amount of Cu+ formed as an intermediate was small and disappeared fast. After 2–3 min of the CO pulse, the system reached a steady state with Cu/CeO2−x as the active phase of the catalyst and H2 and CO2 evolving into gas phase. The magnitude of the change in the ceria lattice after exposure to the CO/H2O mixture (Fig. 7) was comparable to the magnitude seen upon exposure to CO alone (Fig. 3). The removal of the CO gas from the feed to the reactor (t = 50–60 min, Fig. 7) produced drastic changes in the chemical state of the catalyst. As soon as the amount of CO present in the reactor was reduced, the water started to reoxidize the Ce3+ sites to Ce4+ with a contraction in the lattice of ceria

In situ XRD results for a CuO/CeO2 (nc) catalyst under WGS reaction conditions at 250 °C. The sample was under a constant flow of water and the CO was pulsed as indicated. Under WGS reaction conditions, the feed gas mixture typically contained 1 % CO and/or 3 % H2O in helium with a flow rate of 10 mL/min
Eventually, the metallic Cu reacts with water to form Cu1+ and Cu2+ cations in a sequential way
These results clearly show that the active Cu/CeO2−x (nc) phase under WGS conditions is not stable under oxidizing conditions. However, upon a second pulse of CO (t > 80 min), there is a very fast reduction of CuO, Cu2O and CeO2 to yield the active Cu/CeO2−x (nc) phase under WGS conditions.
Figure 8 shows the data for the pulsing of CO, under a constant flow of water, over a CuO/CeO2 (ns) catalyst at 250 °C. As soon as CO is introduced into the reactor (t > 28 min), one sees a fast reduction of Cu2+ to eventually produce metallic Cu and an expansion of the ceria lattice as a consequence of the formation of some Ce3+. Interestingly, the amount of Cu+1 formed is smaller and less stable than seen after exposing the catalyst to pure CO (Figs. 4 and 5). A similar phenomenon has been observed for an inverse CeOx/CuO catalyst [7] and seems to go against the changes in oxidation state predicted by Eqs. (6) and (7). In principle, the water could modify the Cu2O–CeOy interactions to facilitate the reduction of Cu1+ by CO [7]. It could also help to remove carbonates formed on CuO/CeO2 (ns) upon exposure to CO that prevent a full reduction of copper [7]. In any case, the active phase for the WGS on CuO/CeO2 (ns) and CuO/CeO2 (nc) involves metallic copper on partially reduced ceria. Comparing the expansion of the ceria lattice in Figs. 7 and 8, it appears that the amount of Ce3+ formed is larger on the CuO/CeO2 (ns) catalyst.

In situ XRD results for a CuO/CeO2 (ns) catalyst under WGS reaction conditions at 250 °C. The sample was under a constant flow of water and the CO was pulsed as indicated. Under WGS reaction conditions, the feed gas mixture typically contained 1 % CO and/or 3 % H2O in helium with a flow rate of 10 mL/min
In Fig. 8, the removal of CO from the feed to the reactor (t > 53 min) starts a slow contraction of the ceria lattice produced by the disappearance of Ce3+ centers as a consequence of the reoxidation of ceria by water, Eq. (5). For a while (~20 min), there is no reoxidation of metallic Cu into Cu+1 [i.e. no reaction (6)]. A comparison of the results in Figs. 7 and 8 indicates that the active phase of the CuO/CeO2 (ns) catalyst is less sensitive to reoxidation by water than that of the CuO/CeO2 (nc) catalyst. This is probably a consequence of stronger Cu ↔ CeOx interactions in the CuO/CeO2 (ns) system and correlates with its higher activity for the WGS (Fig. 6). The introduction of a second pulse of CO in Fig. 8 (t > 78 min) produces a fast reduction of Cu+1 and the reoxidized ceria; in less than 2 min the catalyst adopts its active Cu/CeO2−x configuration.
As mentioned above, a comparison of the results in Figs. 7 and 8 indicates that the expansion of the ceria lattice in CuO/CeO2 (ns) is larger than in CuO/CeO2 (nc). The same trend was found in experiments done at 400 °C, see Fig. 9. An increase in temperature leads to a larger reduction of both ceria supports but the concentration of Ce3+ sites is always larger for the CuO/CeO2 (ns) system. This is important since the Ce3+ centers are associated with the dissociation of water and the production of H2 [19, 20]. Clearly, the higher the concentration of Ce3+, the greater the catalytic activity for H2 production as indicated by a comparison of Figs. 6 and 9.

Expansion in the ceria lattice at 250 and 400 °C for CuO/CeO2 (ns) and CuO/CeO2 (nc) catalysts under WGS reaction conditions. The feed gas mixture contained 1 % CO and 3 % H2O in helium with a flow rate of 10 mL/min
The reasons for the good performance on CuO/CeO2 catalysts have been studied before. Ozkan and co-workers [21] investigated the effect of support morphology and impregnation medium on the activity of Cu/CeO2 catalysts in the WGS reaction. They concluded that the finely dispersed copper species constitute the active sites for the WGS reaction. Yoon and co-workers [11] reported Cu–CeO2 catalyst prepared by a co-precipitation method and mainly ascribed the outstanding catalytic performance of 80 wt%Cu–CeO2 catalyst to Cu sintering resistance, resulting from a strong interaction between Cu and CeO2. According to the results of our experiments, the better activity of CuO/CeO2 (ns) may be associated with the following three factors.
In the first place, the easy reduction of highly dispersed CuO to metallic Cu is a very important factor for the high activity. In the CuO/CeO2 (ns) sample, about half of the CuO particles are in the highly dispersed state, while almost all the CuO is in the crystalline form on CuO/CeO2 (nc) [14]. It has been well documented that the finely dispersed CuO has a reduction temperature much lower than that of the bulk CuO [22, 23]. Since metallic Cu is commonly accepted as the active phase for the WGS reaction [24], it is no strange that CuO/CeO2 (nc) sample has a lower activity than CuO/CeO2 (ns).
Second, the stability of metallic Cu is another key factor. The metallic Cu seems very easy to be oxidized on CuO/CeO2 (nc), see Fig. 7. H2O can oxidize Cu to Cu+ and further to Cu2+ (Fig. 7). On the contrary, the metallic Cu is rather stable on CuO/CeO2 (ns), only a small extent to reoxidation to Cu+ can be observed (Fig. 8). The different stability of metallic Cu can be ascribed to the mobility of oxygen in ceria. Overbury and co-workers [25] found that the oxygen mobility on CeO2 nanocubes is much higher than that on CeO2 octahedra (nanosphere in our experiment since they have predominantly the same (111) face). The relative freedom for the movement of oxygen facilitated the oxidation of the metallic Cu on CeO2 nanocubes. Therefore, Cu on CuO/CeO2 (nc) exhibits a poorer stability than on CuO/CeO2 (ns), leading to a lower activity.
Third, CeO2 (ns) has a larger concentration of O vacancies and Ce3+, Fig. 9. These oxygen vacancies can act as sites for the dissociation of H2O molecules to form OH groups or H2 and O2− anions, a rate-limiting step independent of the reaction mechanism [26].
4 Conclusions
The effect of support morphology on the activity for the WGS reaction over copper supported on ceria nanospheres and nanocubes was investigated by using in situ XRD and pulsing one of the reactants (CO). In the presence of CO, CuO is rapidly reduced to Cu0 and Ce3+ sites are formed in ceria. The reduction of the copper and cerium cations in the CuO–CeO2 interface is strongly linked, revealing a synergistic redox partnership between copper and ceria. The results showed that the activity and stability of the catalyst active phase had a significant dependence on the ceria support morphology, with CuO/CeO2 (ns) showing the better activity and stability. The differences in the activity were associated with the unique properties of CuO/CeO2 (ns), such as the easier reduction of highly dispersed CuO to metallic Cu, the higher stability of the formed metallic Cu, and the larger concentration of Ce3+ in CeO2 (ns).
References
Shinde VM, Madras G (2013) Appl Catal B Environ 132–133:28–38
Zhang Y, Chen C, Lin X, Li D, Chen X, Zhan Y, Zheng Q (2014) Int J Hydrogen Energy 39:3746–3754
Zhao F, Liu Z, Xu W, Yao S, Kubacka A, Johnston-Peck AC, Senanayake SD, Zhang A-Q, Stach EA, Fernández-García M, Rodriguez JA (2014) J Phys Chem C 118:2528–2538
Wang T, Porosoff MD, Chen JG (2014) Catal Today 233:61–69
Roh H-S, Potdar HS, Jeong D-W, Kim K-S, Shim J-O, Jang W-J, Koo KY, Yoon WL (2012) Catal Today 185:113–118
Gayen A, Boaro M, Leitenburg C, Llorca J, Trovarelli A (2010) J Catal 270:285–298
López Cámara A, Monte M, Martínez-Arias A, Conesa JC (2012) Catal Sci Technol 2:2436–2439
Guo R-T, Zhen W, Pan W, Zhou Y, Hong J, Xu H, Jin Q, Ding C, Guo S (2014) J Ind Eng Chem 20:1577–1580
Amin NAS, Tan EF, Manan ZA (2003) Appl Catal B Environ 43:57–69
Laguna OH, Hernandez WY, Arzamendi G, Gandia LM, Centeno MA, Odriozola JA (2014) Fuel 118:176–185
Jeong D-W, Na H-S, Shim J-O, Jang W-J, Roh H-S, Jung UH, Yoon WL (2014) Int J Hydrogen Energy 39:9135–9142
Meher SK, Rao GR (2012) J Colloid Interface Sci 373:46–56
Tana ZM, Li J, Li H, Li Y, Shen W (2009) Catal Today 148:179–183
Yao SY, Xu WQ, Johnston-Peck AC, Zhao FZ, Liu ZY, Luo S, Senanayake SD, Martinez-Arias A, Liu WJ, Rodriguez JA (2014) Phys Chem Chem Phys 16:17183–17195
Hanson JC, Si R, Xu W, Senanayake SD, Mudiyanselage K, Stacchiola D, Rodriguez JA, Zhao H, Beyer KA, Jennings G, Chapman KW, Chupas PJ (2014) Martinez Arias A 229:64–71
Lin J, Huang Y, Li L, Wang A, Zhang W, Wang X, Zhang T (2012) Catal Today 180:155–160
Xu W, Si R, Senanayake SD, Llorca J, Idriss H, Stacchiola D, Hanson JC, Rodriguez JA (2012) J Catal 291:117–126
Rodriguez JA, Hanson JC, Wen W, Wang X, Brito JL, Martínez-Arias A, Fernández-García M (2009) Catal Today 145:188–194
Barrio L, Estrella M, Zhou G, Wen W, Hanson JC, Hungria AB, Hornes A, Fernández-García M, Martínez-Arias A, Rodriguez JA (2010) J Phys Chem C 114:3580–3587
Wang X, Rodriguez JA, Hanson JC, Gamarra D, Martinez-Arias A, Fernandez-Garcia M (2005) J Phys Chem B 109:19595–19603
Gawade P, Mirkelamoglu B, Ozkan US (2010) J Phys Chem C 114:18173–18181
Luo M, Ma J, Lu J, Song Y, Wang Y (2007) J Catal 246:52–59
Rao KN, Bharali P, Thrimurthulu G, Reddy BM (2010) Catal Commun 11:863–866
Pati RK, Lee IC, Hou S, Akhuemonkhan O, Gaskell KJ, Wang Q, Frenkel AI, Chu D, Salamanca-Riba LG, Ehrman SH (2009) ACS Appl Mater Interfaces 1:2624–2635
Wu Z, Li M, Overbury SH (2012) J Catal 285:61–73
Liu P, Rodriguez JA (2007) J Chem Phys 126:164705
Acknowledgments
The work performed at Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, and Catalysis Science Program under contract DE-AC02-98CH10886 contract. This work used resources of the National Synchrotron Light Source, which is a DOE Office of Science User Facility. The financial support from the National Natural Science Foundation of China (Grant 21303272) is gratefully acknowledged. Financial support from MINECO (CTQ2012-32928 project) and EU COST CM1104 action is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhao, F., Liu, Z., Xu, W. et al. Pulse Studies to Decipher the Role of Surface Morphology in CuO/CeO2 Nanocatalysts for the Water Gas Shift Reaction. Catal Lett 145, 808–815 (2015). https://doi.org/10.1007/s10562-015-1482-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-015-1482-y