
Abstract
In this paper, fundamental issues of the NSR technology are addressed. The reaction paths and the mechanism involved in the storage of NO x and the relevance of the proximity between the noble metal and the alkali or alkaline-earth component of the catalyst are discussed first. Then the release of NO x , the reaction pathways of NO x reduction by different reducing agents, and the mechanisms involved in the formation of N2, NH3 and N2O are illustrated. The use of the combined technologies LNT + SCR and DPNR is addressed next and key aspects that highlight the synergies of the individual technologies are discussed. Finally, the perspectives of the NSR technology are outlined.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Environmental policy in industrialized countries forces a drastic reduction of NO x emissions from diesel engines that operate under lean conditions and have significantly better fuel economy compared to gasoline stoichiometric engines. The EU emission standards for passenger cars are summarized in Table 1 [1]. Since the Euro 2 stage, EU regulations introduce different emission limits for diesel and gasoline vehicles. Diesel have more stringent CO standards but are allowed higher NO x . However, significant reductions in NO x limits are introduced for diesel engines with Euro 6 (from 0.18 g/km in Euro 5 down to 0.08 g/km in Euro 6), which makes NO x emission standards essentially fuel neutral (0.08 g/km in diesel engines vs. 0.06 g/km in gasoline engines). Table 1 also shows that gasoline vehicles are exempted from particulate matter (PM) standards through the Euro 4 stage. Euro 5 and 6 regulations reduce the PM mass emission standard for diesel and introduce PM standards for gasoline cars with direct injection (DI) engines as well, numerically equal to those for diesel. The legislation also introduces a particulate number emission standard (P or PN) at the Euro 5/6 stage. At the time of adoption of the Euro 5/6 regulation, its mass-based PM emission limits could only be met by closed particulate filters (wall through filters). Number-based PM limits will prevent the possibility that in the future open filters are developed that meet the PM mass limit, yet enable a high number of ultrafine particles to pass.
The light-duty (LD) NO x regulations in Europe and US are compared in Fig. 1 [2]: the trend is always for very low NO x emissions. Tighter regulations are foreseen for developing markets like Brazil, China and India.

Euro V and Euro VI light-duty NOx regulatory limits compared to the U.S.: a About 55 to 60 % NOx control will be needed for a Euro V (2009) diesel to hit the U.S. Bin 8 maximum allowable emission (45 states). For Bin 5 (50 states) nominally 85 to 90 % NO x control is needed; b For Euro VI (2014), the requirement is 60 to 65 % additional NO x reduction (reprinted from [2])
While compliance with previous NO x regulatory limits under lean conditions has been largely achieved without resort to NO x after-treatment, either selective catalytic reduction (SCR) or NO x storage reduction (NSR) need to be applied to achieve the most severe NO x standards. Indeed, with the proven technologies of automotive catalysis, the three-way catalytic (TWC) converter used for stoichiometric gasoline engines requires the absence of oxygen or very little excess oxygen and can reduce some NO x under lean conditions. An additional driver for the use of de-NO x after-treatment systems is emerging now in Europe: real world driving emissions. Investigators have found that the NO x emissions from LD diesels can be 3–5 times higher than the laboratory certification level [3]. Along these lines new test cycles (e.g. worldwide harmonized Light duty driving test cycle, WLTC) are under development that are obtained from real world data and require advanced control for cold start, steeper transients accelerations, and higher speed and load conditions. These will make the removal of NO x extremely challenging. Finally, the strategy to improve the fuel economy of cars and to reduce CO2 emissions will motivate more extensive use of diesel vehicles and lean burn DI gasoline vehicles due to the more effective combustion realized under lean conditions.
The SCR approach is based on the reaction between NO and NH3 [4], which is produced by hydrolysis of an aqueous urea solution injected into the exhausts from an on-board tank, according to the standard SCR reaction:
The activity at low temperature, where most of NO x emissions are generated, can be improved by positioning a DOC unit upstream of the SCR converter to oxidize NO to NO2. This allows the occurrence of the fast SCR reaction, which is considerably faster than reaction (1) at low temperatures [5, 6]:
The first commercial use of SCR NO x control in heavy duty (HD) trucks employed traditional vanadia-tungsta-titania catalysts. Nowadays Fe- and Cu-zeolites are preferred, in view of better high-temperature durability, since the SCR catalyst receives hot gases during regeneration of the upstream diesel particulate filter (DPF), and in view of higher low temperature activity. Copper zeolite is the preferred catalyst, has the best low-T performance and shows very little sensitivity to NO2 concentration. Iron zeolite has the best high-T performance but requires NO2 management in the inlet gas to improve LT performance, as do vanadia catalysts. This in turn, implies a higher precious metal usage in the DOC unit.
The NH3/urea SCR is recognized as the most effective technology for the abatement of NO x emissions from HD diesel vehicles, and a growing interest in the same technology is coming also from LD vehicles and passenger cars, in order to combine low fuel consumption and low NO x emissions.
The NSR, also referred to as lean NO x traps (LNT), represents an alternative viable technology for the removal of NO x under lean conditions in vehicles [7–9]. The system is operated in a cyclic manner by alternating a long lean phase (30–90 s) and a short rich phase (3–5 s). The basic concept is that NO x are trapped onto the catalyst when the engine is running lean and are reduced to produce N2 when the engine is running rich. A typical NSR catalyst comprises a noble metal such as Pt to oxidize NO, CO and unburned hydrocarbons and to reduce trapped NO x , and an alkaline and/or alkaline-earth component such as barium and/or potassium to trap the NO x as nitrites/nitrates. Besides the catalysts typically contain Rh and Ce. These catalyst components are dispersed on a high surface area alumina support. The overall principle of operation of NSR has been described by the following five steps [9]: (i) oxidation of NO to NO2; (ii) NO and/or NO2 up-take in the form of nitrite and/or nitrates; (iii) evolution of the reducing agents when the exhaust is switched to the rich condition; (iv) NO x release from stored nitrites and nitrates; (v) reduction of released NO x to N2.
The NSR technology has been commercialized for passenger cars equipped with lean burn engines in the early 2000s. In spite of the cost of the noble metal catalysts, NSR is a favored approach for lighter vehicles (i.e. engines of less than about 2.0 to 2.5 l capacity), mainly due to the fact that big layout modifications are unnecessary and the relatively fixed cost of an on-board urea system. However, since mixed-mode engines greatly reduce low-load NO x , NSR systems could be operated at temperatures greater than 300 °C with a substantially lower loading of the platinum group metals (PGM). This could make NSR economically attractive for cars with mixed-mode engines of higher capacity as well (i.e. up to 5 or 6 l) [2].
The durability of LNTs to sulphur contamination has always been a major problem of NSR, based on the fact that sulphates are more stable than nitrites/nitrates and tend to poison the catalyst basic sites where NO x are adsorbed [10, 11]. Accordingly, the catalyst is typically oversized to compensate for degradation during a given period and must be periodically regenerated; this is accomplished by decomposing the sulphates at high temperature under reducing conditions to give SO2 and H2O for a total of few minutes every few thousands km. In view of the introduction of ultra-low sulphur fuels, improvements in the catalyst resistance to sulphur poisoning and new control strategy, which limit the desulfation temperature, LNTs are effective to about 80 % NO x efficiency in real world Light Duty systems [12]. Attention is also paid to limit the H2S peak amount released during desulfation.
In order to decrease the size and the weight of the after-treatment devices, integrated systems have been developed where existing complementary technologies are combined. In the LNT + SCR configuration, ammonia slipped from the LNT catalyst during the rich phase is adsorbed onto the SCR catalyst bed placed downstream. Such adsorbed NH3 reacts according to the SCR reactions with upcoming NO x present in the exhausts which have not been trapped onto the NSR catalyst bed during the lean phase and with NO x released during the fast lean-rich switch that escape from the NSR catalyst bed. Accordingly, over the LNT + SCR system the NO x removal efficiency is increased, the ammonia slip is reduced and the PGM usage is lower. This technology was introduced in the US and in Europe in the 2000s [13].
Besides, in order to control soot emissions, technologies like DPF and continuously regeneration trap (CRT) [14, 15] are used in combination with SCR and LNT catalysts. Although changes to engine design will improve engine emissions in order to meet both PM and NO x regulatory limits, combined systems including PM and NO x after-treatment can be utilized. Soot treatment, NO x reduction and CO and hydrocarbons oxidation functions are combined on a single monolith developed by Toyota and known as diesel particulate—NO x reduction (DPNR) [16]. The commercialized “4 way catalytic converters” are composed of a particulate wall-flow filter coated with an NSR catalyst layer and work under cyclic conditions, as LNT catalyst does. Particulate filter regeneration is supposed to be effective during the lean phase, due to the presence of NO x and the excess of oxygen in the exhaust gas, similarly to that occurring in the CRT technology (soot combustion by direct and cooperative reactions).
In this paper, fundamental issues of the NSR technology are addressed with the aim to provide a critical survey, and to identify open issues and space for future advancement. The reaction paths and the mechanism involved in the storage of NO x and the role of proximity between the noble metal and the alkali or alkaline-earth component of the catalyst are discussed in Sect. 2. The release of NO x , the reaction pathways of NO x reduction using different reducing agents (H2, CO, hydrocarbons), and the mechanisms involved in the formation of N2, NH3 and N2O are illustrated in Sect. 3. The use of LNT in combination with other technologies like LNT + SCR and DPNR is addressed next and key aspects that highlight the synergies of the individual technologies are discussed. Finally, the perspectives of the NSR technology are outlined.
2 Reaction Pathways and Mechanistic Features in the Storage of NO x
2.1 The NO x Storage Mechanism
There is ample evidence in the literature that suggests NO2 is a precursor in the storage of NO x ; the NO2 can be gas-phase, a surface-bound precursor, or an intermediate species.
Fridell et al. [17] suggested that NO2 is formed from NO oxidation and is stored onto the catalyst surface as nitrates:
In this pathway, hereafter quoted as the nitrate route, Pt catalyses the oxidation of NO to NO2 and the alkali- or alkali-earth metal compound participates in the NO2 storage reaction. It is noted that reaction (4) involves the disproportionation of NO2 to nitrite and nitrate intermediates, followed by the oxidation of nitrites to nitrates:
Indeed NO is effectively oxidized to NO2 over Pt, although Ba has a negative effect on the rate of NO oxidation, and NSR trapping materials sorb more effectively NO2 compared to NO [9]. Besides both nitrite and nitrate ad-species have been simultaneously observed by several authors by means of FTIR at the preliminary stage of storage from NO2. Nitrites eventually are oxidized to nitrates, that prevail after a sufficiently long storage period [17–20].
However spectroscopic data in the early LNT literature suggest that the formation of nitrites may precede that of nitrates during NO/O2 adsorption [21, 22]. Indeed some of us have indicated another pathway for the storage from NO/O2, on the basis of combined pulse reactor experiments and in situ FTIR measurements accomplished over Pt–Ba/Al2O3, Ba/Al2O3, Pt/Al2O3 catalysts and bare Al2O3 [23, 24]. This route, hereafter called the nitrite route, is based on the stepwise oxidation of NO at a Pt site followed by adsorption at a neighboring Ba site to form nitrite ad-species:
In this route, the cooperative action between Pt and nearby Ba sites prevents the over-oxidation of NO to NO2 due to the trapping of nitrite species; nitrites can be oxidized to nitrates. In addition to the formation of nitrites, the oxidation of NO to NO2 over Pt may also occur and NO2 can be stored on Ba sites directly in the form of nitrates via reaction (4). A sketch of the nitrite and nitrate pathways for NO x storage is given in Fig. 2.

Sketch of the nitrite and nitrate routes in the process of NO x storage
It is worth noting that the formation of nitrates during NO/O2 uptake followed by FTIR collected in situ under static conditions may be biased by the spurious oxidation of NO to NO2 in the gas phase followed by adsorption of NO2; this is prevented under operando conditions.
Recently it has been confirmed by FTIR under operando conditions [20] that NO/O2 adsorption on Pt–Ba/Al2O3 at low temperature results exclusively in the formation of nitrites. Indeed, as shown in Fig. 3a, the adsorption at 150 °C results in the formation of chelating nitrites with characteristic bands at 1,350 cm−1 [νsym (NO2)] and at 1,217 cm−1 [νasym (NO2)], that grow during storage. A small band at 1,544 cm−1 is seen only at high exposure times whose assignment is still under debate (monodentate nitrites/bidentate nitrates). Besides, as shown in Fig. 3d, immediately upon admission NO is detected at the reactor exit and the formation of NO2 at catalyst saturation is at most negligible. It was concluded that at low temperature (150 °C) NO x are stored only via the nitrite route in the form of nitrites.

Results of operando FT-IR analysis upon NO/O2 exposure (1,000 ppm NO in He + 3 % v/v O2) of the model Pt–Ba/Al2O3 sample: a at 150 °C; b at 250 °C; c 350 °C. FT-IR spectra are recorded every 30 s (increasing storage time indicated by the arrow in the Figures) and the subtrahend spectrum is the catalyst before NO/O2 admission. In d–f the gas phase analysis at the same temperature, respectively, in terms of NO, NO2 and NO x is reported
At higher T (250 °C), as shown in Fig. 3b, the band of nitrites at 1,217 cm−1 is observed as well, that increases with storage, goes through a maximum and then almost disappears, while the bands of nitrates at near 1,320 and 1,420 cm−1 increase markedly. The presence of the isosbestic point in the FTIR spectra confirms that nitrites are transformed into nitrates. Besides the oxidation of NO to NO2 increases and a dead time in the NO breakthrough is observed (Fig. 3e), that has been associated with the occurrence of the nitrite route and with the integral behavior of the reactor. It was concluded that in addition to the nitrite pathway, the nitrate route was also effective at this temperature (250 °C).
At 350 °C both nitrites and nitrates are formed initially (Fig. 3c). The band of nitrites at 1,217 cm−1 disappears soon while the bands of nitrates markedly increase and are the only bands present after 10 min of exposure. Besides, the dead time in NO breakthrough is greater and the oxidation of NO to NO2 is more significant at this temperature (Fig. 3f). It is noted that the storage of NO2 does not allow conversion of NO to NO2 to be monitored using NO2 evolution until a significant amount of sorption has occurred. It was concluded that at high temperature both the nitrite and nitrate pathways are rather effective and NO x are stored only in the form of nitrates after prolonged time of exposure.
By combining the results of operando FTIR spectroscopy with those of the synchronous quantitative analysis of the gaseous species, the concentrations of nitrites and nitrates ad-species as function of time were evaluated at the quantitative level, as shown in Fig. 4. Inspection of the Figure indicates that the storage of NO x at 150 °C from NO/O2 occurs exclusively through the nitrite route: in fact only nitrites are formed at this temperature, and NO2 and nitrates are not observed. The nitrite route is faster at higher temperatures as well. However at 200 °C and above the nitrites are oxidized to nitrates and nitrates are also formed directly from NO2 via the nitrate route. Because of this, a maximum in the concentration of nitrite species is observed. At 350 °C the maximum is smaller and is seen at shorter time of exposure. Still the nitrites prevail over the nitrates for a storage period of about 15 min at 250 °C and of a couple of minutes at 350 °C.

Evolution versus time of the main bands for different surface species detected by using FT-IR spectroscopy under operando conditions upon NO/O2 exposure (1,000 ppm NO in He + 3 % v/v O2) at 150, 200, 250 and 350 °C of the model Pt–Ba/Al2O3 sample (reprinted from [20])
The concentrations of the adsorbed species provided by operando FTIR and of the gaseous species provided by the simultaneous on line analysis of the gas phase upon exposure of Pt–Ba/Al2O3 to NO/O2 mixture have been used as direct experimental responses to estimate the kinetic parameters of the steps involved in the process of NO x storage. At variance, in traditional kinetics the concentration of adsorbed species is estimated in order to gain a satisfactory fitting of the time evolution of the gaseous species. Appropriate reaction scheme, kinetic models and reactor model of the IR cell were developed. Considering the fact that this approach is the result of the joint application of spectroscopic and kinetic methods, it has been termed as spectro-kinetics. The model was found to fit nicely with the time evolution of the concentration of NO and NO2 in the gas phase and the surface coverage of nitrites and nitrates in the investigated temperature range [25].
The effect of CO2 and H2O, that are present in the exhausts, on NO x storage over Pt–Ba/Al2O3 has been addressed recently in our labs [26], and among others by Weiss et al. [27], Epling et al. [28] and by Chaugule et al. [29]. We found that the nitrites pathway is not significantly affected by the presence of CO2 at low temperature, but is inhibited at higher temperatures. However, the nitrate pathway is inhibited as well, particularly at high temperature, but to a lower extent. The different inhibiting effect of CO2 has been ascribed to the different relative thermal stability of carbonates, nitrites and nitrates as function of temperature, which has been evaluated by means of dedicated adsorption–desorption experiments. The effect of water was minor, because BaCO3 is more stable than Ba(OH)2 [30, 31]. It is concluded that nitrites are formed preferentially during the early period of NO x storage from NO/O2 also in the presence of CO2 and H2O.
2.2 Relevance of the Proximity Between the Noble Metal and the Alkali or Alkaline-Earth Component
To investigate the importance of close contact between Pt and BaO during the storage, Cant et co-workers compared the NO x storage characteristics of four systems: BaO/Al2O3, a sequential system with Pt/SiO2 ahead of BaO/Al2O3, a combined system with Pt/SiO2 and BaO/Al2O3 physically mixed, and BaO/Al2O3 with Pt deposited on it [32]. They found that storage of NO x from NO/O2 by the Pt–BaO/Al2O3 sample differed from that of the sequential and combined systems. Indeed, the rate of storage on Pt–BaO/Al2O3 is faster, and the total storage capacity under the conditions used is attained more quickly. The implication is that Pt, in close proximity to BaO, either allows uptake of NO or NO2 in a way that is not feasible when Pt is absent or enables conversion of NO x to nitrites/nitrate as storage proceeds. Spillover of NO2 from Pt to BaO/Al2O3 is one way in which the latter might occur. These authors also investigated the isotopic exchange between 15NO and stored 14NO x . The rate of exchange was more than five times as fast for Pt/BaO/Al2O3 than for the combined Pt/SiO2 + Ba/Al2O3 system, demonstrating spillover of NO x species between Pt and Ba in close proximity.
Similar results have been reported by Nova et al. [33]. Indeed, comparing the results between the physical mixture made of Pt/γ-Al2O3 and Ba/γ-Al2O3 and the ternary Pt–Ba/γ-Al2O3 sample it clearly appeared that the two systems are characterized by a similar NO x breakthrough time but the physical mixture presents a higher oxidizing capacity, as revealed by the higher conversion of NO to NO2 measured at catalyst saturation. In addition, the physical mixture resulted able to adsorb significant amounts of NO x . It was assumed that the mechanism of NO x adsorption operating on the physical mixture, i.e. in the absence of interaction between Pt and Ba atoms, is the nitrate route: NO is oxidized to NO2 on Pt sites (reaction (3)) and then is adsorbed on Ba (and Al sites) via the disproportionation reaction (reaction (4)). NO, which is released during the same reaction is re-oxidized to NO2 and this results in a significant delay in the NO x breakthrough also in this case.
Hence, the data on the physical mixture confirm the feasibility of the NO x storage mechanism proposed in the literature implying the oxidation of NO to NO2 followed by NO2 adsorption on the catalyst surface to form nitrates. In this case, no direct interaction is necessary to have an effective NO x uptake. However, this is not the case of representative NSR catalysts, in which Pt and Ba are on the same support and which must be able to catalyze the reduction of the stored NOx, as indeed realized in the NSR technology. As a matter of fact, the physical mixtures made of Pt/SiO2 and Ba/Al2O3 or Pt/Al2O3 and Ba/Al2O3 are active towards the NO x reduction at considerably higher temperatures compared to the ternary system Pt–Ba/Al2O3 [32, 33]. This is consistent with the decomposition of stored NO x to gaseous NOx and subsequent reduction by a catalytic reaction on Pt particles located remotely from BaO.
To investigate this interaction further and to better understand its role in the storage mechanism, a study was undertaken by Castoldi et al. [34] using Pt–Ba/Al2O3 samples with different Ba loadings, in order to vary the number of Pt–Ba neighboring couples. The CO chemisorption measurements performed by means of in situ FTIR analysis showed the characteristic band of CO linearly adsorbed on Pt sites in all cases; however, upon increasing the Ba loading the band progressively shifts towards lower energy due to the increase of the catalyst basic character. Hence, the data indicate a strong interaction between Pt and the basic oxygen anions of the Ba phase, suggesting that the exposed Pt sites and the Ba component are in close proximity. In addition, it has been shown that when applying a rectangular step feed of NO in He + O2 (3 % v/v) both the NO x breakthrough and the storage capacity of the catalyst increase with the Ba content not only because of the amount of Ba, but also the number of Pt-neighboring Ba sites increase, the uptake of NO/O2 is faster over these sites and a large percentage of Ba participates to NSR [34]. The fraction of Ba that participates in the storage process (“active” Ba) increases linearly with the catalyst Ba content passing through a maximum that represents the best exploitation of the Ba component, while the Pt dispersion decreases from 80 to 40 % [34].
The relevance of the Pt–Ba proximity in the storage of NO x implies that Ba sites distant from Pt are less reactive in the storage. This eventually accounts for the slower uptake of NO x which is observed at the later stages of the trapping process, and that was explained invoking a nitrate spillover type mechanism [35, 36]. In fact the adsorption sites near Pt react and saturate first, then the trapping rate slows due to increasing resistance to nitrate surface diffusion as the N-species build around the Pt particles (two dimensional shrinking core model) and/or into the inner core of the Ba particles (three dimensional shrinking core model). The close proximity between Pt and BaO facilitate the removal of stored NO x as well by lowering the stability of adsorbed species.
3 Reaction Pathways and Mechanism in the Reduction of the Stored NO x
There are three methods for introduction/generation of the reductant in NSR [9]. The first strategy is the DI of the fuel into the exhausts upstream of the catalyst during the rich event. In the second strategy, the fuel is continuously processed over a reformer or other devices such as plasma or a partial oxidation catalyst to generate reductants, which are more effective in accomplishing the regeneration of the NO x trap. In the third strategy, the lean burn engine is operated to generate intermittent pulses of rich exhausts to the LNT catalyst that either directly contain the reductant or are processed by the NSR catalyst or by an upstream catalyst to form the reductant. In this case, the regeneration event involves a rich transient originating from the engine.
It is difficult to determine what exact reducing species participates in the NO x reduction process since the original reductant may undergo substantial changes over the NSR catalyst or the reformed devices added upstream. Besides, extensive heat is evolved due to catalytic combustion of the reductant during regeneration events in some modes of operation. Accordingly, the catalyst operation is highly non isothermal, resulting in complex dynamic temperature distribution profiles. Temperature swings, measured as gas phase temperature changes, can be as high as 50–100 °C at the front of the catalyst and only 2–3 °C at the back of the catalyst [9]. Keeping in mind these aspects, it is obvious how complex the mechanism of reduction is for the stored NO x .
3.1 The Release of NO x
It is a common belief that the reduction of stored NO x over LNT catalysts implies the release of NOx from the catalyst surface, followed by the reduction to N2 and other by-products such as NH3 and N2O. The process of NO x release is most commonly discussed in terms of the breakthrough of desorbed and unconverted NO x during the rich event as schematically shown in Fig. 5. However, the detailed mechanism of the release of NOx and of their further reduction remains under debate.

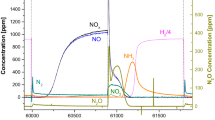
A representative NO x breakthrough and release profile. The catalyst was Pt/Ba/Al2O3,the reactant gas contained 250 ppm NO, 8 % H2O, 8 % CO2, 8 % O2, balance N2 in the lean/trapping phase and 2,000 ppm H2, 8 % H2O, 8 % CO2, and a balance of N2 in the regeneration/rich phase. The space velocity was 25,000 h−1. The outlet NO x concentration was monitored with a chemiluminescence detector (reprinted with permission from [9])
Some authors [37, 38] proposed that the NOx are released as a result of the heat generated by the exothermic reactions upon the lean/rich switch (thermal release). Indeed, as the rich front passes over the catalyst and the surface temperature increases, stored NOx can be released because their stability decreases with increasing temperature. However it has been shown that the reduction process, even when accomplished under nearly isothermal conditions (i.e. with small concentration of the reductant), occurs at temperatures well below that at which stored NO x thermally decompose. Accordingly, under these conditions, the process is not initiated by the thermal decomposition of stored nitrates, but rather by a catalytic pathway involving Pt [33]. Other authors [39, 40] have proposed that the release of NO x is caused by a decrease in the equilibrium stability of the stored nitrates due to the decrease in the partial pressure of NO and/or of oxygen upon the lean/rich switch. However this contribution is small during typical NSR operation since only a negligible desorption of weakly adsorbed NO x species was observed in the time range of few seconds characteristic of the rich period, when the NO feed and the O2 feed were shut down after the storage of NO x was completed [30, 33]. Finally, the release of NO x may be promoted by the establishment of a net reducing environment [41, 42].
To account for the reduction of NO x either the reverse spill-over of NO x from Ba to Pt is occurring or alternatively the reductant activates and then spills over from Pt sites to the storage component and reacts directly with nitrites and nitrates, so that they are eventually released as NO x [32, 43, 44]. The subsequent reduction of the released NO x may occur at the Pt metal sites following a TWC-like chemistry [45]. The transient product distribution displayed by three catalysts having varied dispersions (3.2, 8 and 50 %) was described by Harold and coworkers [35, 46] using a model based on global kinetics and detailed crystallite-scale catalyst features and was explained by the localized stored NO x gradients in the Ba phase. The rate determining process during the regeneration was found to be the diffusion of stored NO x within the Ba phase towards the Pt/Ba interface. Indeed the results reported by Coronado and Anderson [47], Cant et al. [32] and Nova et al. [33] clearly show that NO x decomposition is easier when Pt and Ba are in close proximity.
Even if the net NO x release at the lean/rich interface represents a small loss under many of the operating conditions, this release increases at higher temperatures and can result in not attaining the required emissions standards. Therefore, one of the most important issues is understanding the pure contributes that cause the NO x release and the selectivity to N2. Although the proposed pathways of NO x release are relatively well accepted, decoupling the release of NO x from their subsequent reduction to N2 or other products is not possible during typical NSR operation. Indeed, in the presence of a reducing agent the NO x release is immediately followed by reduction of the released NO x , which is a very fast reaction. However, recently the release of stored NO x has been decoupled from the reduction of the released NO x and measured by performing NO isotopic exchange experiments under temperature programming, using labelled gaseous 15NO and unlabelled 14NO x stored on the Pt–Ba/Al2O3 catalyst. The exchange between gaseous 15NO and stored 14NO x was previously used by Cant et al. [32] to probe the forward and reverse spillover of NO x species between Pt and BaO.
NO isotopic exchange experiments have shown that [48]: (i) Pt is necessary for the exchange because this is observed over Pt–Ba/Al2O3 but not over Ba/Al2O3; (ii) the NO isotopic exchange implies the release of stored NO x and is observed from a temperature well below that of thermal decomposition of stored NO x , thus pointing to a Pt-catalysed process. The exchange is observed from about 50 °C for nitrites stored from NO/O2 at 150 °C and from about 250 °C for nitrates stored from NO/O2 at 350 °C; (iii) all stored NO x have been exchanged after T programming up to 350 °C and subsequent hold at this temperature for half an hour so that both nitrites and nitrates migrate between Pt and Ba sites.
The mechanism of NO isotopic exchange is sketched in Fig. 6. Upon interaction of unlabelled stored nitrite or nitrate species with reduced Pt sites unlabelled NO is released in the gas phase and PtO species are formed. PtO species oxidize labelled NO present in the gas phase to give labelled nitrite and nitrate adsorbed species restoring the reduced Pt sites. The NO isotopic exchange is a redox process. The reduced Pt sites activate NO x stored species that are destabilized and eventually released as NO in the gas phase. It is worth to note that during typical NSR operation stored NO x upon activation could either react with the reducing agents present in the exhausts to give N2O/N2/NH3 or produce released but unconverted NO.

Sketch of the mechanism of NO isotopic exchange/NO x release
It is worth noting that in the NO isotopic exchange process labelled NO represents the reducing agent so that the released unlabelled NO cannot be reduced further. For this reasons, in NO isotopic exchange experiments the release of stored NO x could be decoupled from its further reduction and the process of NO x release could be characterized unambiguously.
3.2 Pathways of the Reduction of Stored NO x Using H2
The regeneration of the NO x storage component is a crucial phase of the NSR process. The regeneration has been studied by several authors using H2 as the reductant: the formation of NH3, N2O and N2 was observed depending on temperature, H2 concentration, time of exposure to the reductant.
Harold and coworkers [49] on the basis of measurements performed with different catalyst length sections proposed that there are two primary competing routes to the desired N2 product: a direct route from the reduction of stored NO x by H2 (H2 + NO x → N2) and a sequential route of stored NO x reacting with H2 forming NH3 and then NH3 reducing NO x stored downstream to give N2 (H2 + NO x → NH3; NH3 + NO x → N2). Evidence for the direct route and the sequential route with ammonia as intermediate has also been provided by means of in situ measurements for the transient intra catalyst species (H2, NH3, N2, NO x ) using spatially resolved capillary inlet mass spectrometry (SpaciMS), which provides a spatiotemporal resolution during NSR catalyst regeneration as shown in Fig. 7 [50]. While the data clearly prove that intermediate NH3 plays a significant role in LNT catalyst regeneration, it was further recognized that experimental and numerical work are needed to more clearly understand the partitioning between the two suggested regeneration pathways, the direct route and the sequential route with ammonia as intermediate.

Species transients at different locations through the catalyst for a H2, b N2 and c NH3 at 200 °C. The legend indicates the specific measurement location in millimeters relative to the catalyst inlet face. (reprinted with permission from [50])
Lindholm et al. [31] showed that N2O and NH3 can be formed when reducing NO x with H2 and that NO reduction, NH3 formation and N2O formation depend on the H2 concentration. A delay in the ammonia signal was observed and was explained by the SCR-type reaction between nitrites/nitrates adsorbed on the storage component and ammonia; when the coverage of nitrites/nitrates has decreased NH3 is observed in the gas phase.
Ribeiro et al. [51, 52] proposed that the regeneration of Pt–Ba/Al2O3 monolith NO x trap with hydrogen involves a localized reaction front of the reductant that travels through the catalyst bed with complete regeneration of the trapping sites. The process was limited by the supply of the hydrogen atoms, irrespective of the source of hydrogen (H2 or NH3), and occurred through a complex pathway where at first nitrogen and ammonia were formed according to a parallel scheme. NH3 further reacts with the stored NO x leading to the selective formation of N2.
In order to elucidate the mechanism governing the reduction of the NO x species stored onto a model Pt–Ba/Al2O3 LNT catalyst in the form of nitrates, TPD (temperature programmed desorption), H2-TPSR (temperature programmed surface reaction), NH3-TPSR, H2-ICSC (isothermal concentration step change) and NH3-ICSC experiments have been performed by Lietti et al. [33, 53, 54] under nearly isothermal conditions by using a small concentration of the reductant. The data in Fig. 8 show that the reduction of stored NO x is active at temperatures well below that of NO x thermal decomposition (near 120 °C in panel B vs. near 330 °C in panel A) and accordingly the NO x reduction is a Pt-catalyzed process that does not require the thermal release of stored NO x as the preliminary step. Besides, Fig. 8 shows that the reduction of nitrates by H2 occurs from much lower temperature in the presence than in the absence of H2O (near 50 °C in panel C vs. near 120 °C in panel B). The promoting effect of water might be explained considering that water increases the H2 spillover rate over oxide surfaces [55], thus enhancing the reduction of nitrates stored far away from Pt sites. However specific effects of water on the mobility and reactivity of adsorbed nitrates can be suggested as well considering that water affects the features of nitrate ad-species, as reported by Szanyi and coworkers [56]. This is in line with mechanism involving the NO x spillover from Ba to Pt sites. From Fig. 8 it is also clear that the reduction of nitrates by H2 results in the formation of ammonia as the main product when accomplished at low temperatures (panel C) as opposed to when it’s carried out at high temperatures (e.g. 350 °C). Indeed, in this case N2 represents the most abundant reaction product and NH3 is detected in small amounts and only after N2 evolution (not shown in Fig. 8). Finally, Fig. 8 shows that the reduction of nitrates by ammonia is slower since the onset of this reaction is observed at higher temperature than the one with hydrogen (near 125 °C in panel D vs. near 50 °C in panels C), and is fully selective to nitrogen (panel D).

a TPD in He from r.t. to 600 °C (10 °C/min); b H2-TPSR (2,000 ppm H2) from r.t. to 500 °C (10 °C/min); c H2 + H2O-TPSR (2,000 ppm H2 + 1 % v/v H2O) from r.t. to 500 °C (10 °C/min); d NH3 + H2O-TPSR (1,000 ppm NH3 + 1 % v/v H2O) from r.t. to 500 °C (10 °C/min). Storage conditions: 1,000 ppm NO in 3 % v/v O2 + He at 350 °C over Pt–Ba/Al2O3 catalyst
These data were described by the following molecular reaction scheme:
where reaction (8) is faster than reaction (9).
This scheme together with the integral behavior of the laboratory micro-reactor accounts for the temporal evolution of the products observed during the reduction of the stored nitrates with H2 and with NH3 in the investigated T range 150–350 °C. It also implies the full consumption of H2 and the formation of a hydrogen front travelling along the reactor axis, with ammonia formation that occurs at the H2 front. Although the data could be explained at the quantitative level by the consecutive pathway provided by reactions (8) and (9) [57] a parallel + consecutive reaction scheme, where N2 is also formed directly from nitrates, cannot be ruled out.
A schematic of the regeneration pathway proposed by Ribeiro et al. [51, 52] (and by Phil et al. [58]) is depicted in Fig. 9. The Figure illustrates the propagation of the reductant front along the catalyst bed with complete regeneration of the trap. Once NO x is released it reacts with H2 over Pt to form NH3, N2 and N2O, the selectivity to the individual species depending on the local NO x /H2 concentration ratios. In the beginning of the front where the H2 level is high compared to the NO x NH3 will be mostly formed. The NH3 formed will further react with more NO x to give either N2 or N2O if the temperature is sufficiently high. If the N2O is formed behind the front, it will be reduced to N2 by H2 or NH3. As the front approaches the end of the catalyst the supply of NO x starts to deplete and is insufficient to react with the NH3 formed upstream, leading to the NH3 breakthrough. Before the reaction front reaches the end of the bed, most of the non-selective products will react further to form N2 and maintain the high selectivity of the overall NSR catalyst.

Schematic of the reduction mechanism for a Pt/BaO/Al2O3 monolith regenerated with H2. Bottom panel illustrates a zoomed-in picture of the reaction front (reprinted with permission from [52])
3.3 Pathways of NO x Reduction Using CO and Hydrocarbons
The regeneration of LNT catalysts has also been studied using CO, propane, propene and other hydrocarbons as reductants [56, 59–61]. It has been observed that H2 and CO are highly efficient reductants compared to C3H6, which is somewhat less efficient. C3H8 did not show any NO x reduction ability for NO x stored on Pt/Ba/Al2O3 except at 350 °C.
Szailer et al. [56] reported that during the reduction by CO of NO x stored on Pt/Al2O3 and Pt–Ba/Al2O3 at low temperature and under dry conditions stable isocyanates form at the Pt particles and spill over the basic oxide component of the catalyst, resulting in consumption of a large fraction of gas-phase CO and formation of a significant amount of CO2 but not of N2. At temperatures corresponding to the onset of stored nitrate decomposition these NCO species can readily react with the NO x released by the catalyst to give N2 and CO2 according to the reaction:
N2 formation was significantly enhanced by adding water to the NCO covered catalysts because NCO was hydrolyzed to NH3 and CO2 so that the formed NH3 could react with NO x and form the intermediate that eventually generated N2 and H2O.
Forzatti et al. [62] showed that the reduction of nitrates stored onto Pt–Ba/Al2O3 by CO under dry conditions does not involve the thermal release of stored NO x as the preliminary step, is seen from a temperature where Pt-carbonyls disappear and CO is consumed while N2 is observed from a slightly higher temperature. It was found that the reduction of nitrates by CO occurs via a consecutive reaction scheme with formation of NCO species followed by reaction of these species with nitrates/nitrites and that the reaction of NCO species to give nitrogen is slower than the reduction of nitrates with CO to give NCO species. Large amounts of stable NCO species were formed and retained onto the catalyst surface at the end of the rich phase and were oxidized to give N2 during the subsequent lean phase upon admission of O2 and NO/O2. Lower amounts of isocyanate species were left at the end of the reduction over Pt-K/Al2O3 than over Pt–Ba/Al2O3 likely due their higher mobility [63, 64]. It is worth noting that the C and N balances during the rich and the lean phases of the NSR cycle are consistent with the formation of stable NCO species and their consumption to give N2 respectively. Amiridis and coworkers [61] confirmed that the reaction of surface isocyanates with O2 and NO accounts for a significant fraction of the overall N2 production. The oxidation of isocyanates/cyanates with O2 to give N2 and CO2 was observed previously over Pt–Rh/Ba/Al2O3 and Ag/Al2O3 catalysts by Daturi and coworkers [65, 66] using FT-IR spectroscopy and by Schoulten and coworkers [67] over Pt–Ba/Al2O3 performing lean–rich cycling experiments. Also Toops and coworkers [68] found that Ba–NCO is directly derived from the reduction of Ba nitrate by CO, i.e., Ba–NCO species represent a critical intermediate in the reduction of stored NOx with CO. Under dry cycling conditions with CO as the reductant, N2 is mainly generated via NCO reaction with NO + O2 after the switch to lean phase conditions, rather than being formed during the rich phase. However, the evolution of DRIFT spectra under both temperature-programmed and isothermal reaction modes revealed that H2O is the most reactive species with respect to isocyanate of those tested (H2O, O2, NO, NO/O2).
The role of surface isocyanate species as intermediate in the formation of N2 is also supported by previous literature reports where isocyanates formed on several HC-SCR catalysts were found to be highly reactive towards NO2 and NO + O2 to yield N2 [69–72]. It is worth noting that NCO species are stabilized in the regeneration of NSR catalyst by CO due to the interaction with the alkali or alkaline-earth component of the catalyst.
However, different routes may be active in the reduction of stored NO x species by CO under wet conditions. Indeed the onset temperature for the CO consumption is observed at temperatures slightly lower under wet than under dry conditions and is very close to the onset temperature of the WGS reaction, the NO x removal efficiency is higher, and NH3 and H2 are seen among the products [73]. FTIR showed that bidentate nitrates are transformed with temperature into ionic nitrates while NCO species are observed only in small amounts. The hydrolysis of the surface NCO species to give ammonia and carbon dioxide has also been reported by Daturi and coworkers [65] and the formation of small amounts of NCO species during cycling experiments in the presence of water was observed by Amiridis and coworkers [61]. Therefore, it is concluded that hydrogen formed through the water gas shift (WGS) and/or ammonia formed by hydrolysis of NCO species could be involved in the reduction of stored NO x by CO in the presence of water. Recently Harold and co-workers [74] provided evidence that the surface NCO species are important reaction intermediates at moderate to higher temperatures (≥250 °C) during the cyclic reduction of NO x by CO under wet conditions. Current on-going spatiotemporal studies have been anticipated to identify the best routes to N2.
Propylene and n-heptane were also used in the regeneration of LNT catalysts under dry conditions [35, 75]. Both propylene and n-heptane were found to react well below the onset temperature of nitrate thermal decomposition. Hence, also in this case, as in the case of H2 and CO, the reduction of the stored NO x does not imply the preliminary thermal decomposition of the NO x adsorbed species. The reduction of the stored nitrates is accompanied by the evolution of N2 that represents the main product, and then of minor amounts of ammonia.
However, in reality the NSR is performed in the presence of water and it is commonly accepted that the reduction of stored NO x occurs thanks to the production of H2 via steam-reforming of the hydrocarbon employed as reductant. Then n-C7-TPSR run in the presence of water has been carried out and the onset of the nitrate reduction was observed at temperatures very close to that measured under dry conditions (200–250 °C), i.e. the reaction was not significantly affected by the presence of water. Also the distribution of reduction products was very similar.
Dedicated n-heptane steam-reforming (SR) experiments have been performed, which showed the formation of H2 and CO2 above 250 °C although with poor reactivity. In the SR experiments, the production of H2 is observed from 250 °C. However, the amount of H2 produced via the SR reaction was not sufficient to justify the large amount of N2 produced. Besides no N-containing surface species, in particular isocyanates, could be detected by in situ FT-IR. Accordingly, a different pathway should likely be considered. The pathway may imply cleaning of the Pt sites by the reductant followed by migration-decomposition of the stored NO x at the reduced Pt sites.
3.4 Mechanism of the Reduction of Stored NO x by NH3
As already mentioned, ammonia is seen among the products during reduction of stored NO x in particular at low T when hydrogen is used as reductant and it has been suggested in the literature as intermediate in the production of nitrogen. However, the mechanism of the reduction of stored NO x by NH3 has not been completely clarified so far. A study has been carried out recently in our labs to identify the possible mechanism for N2 formation when ammonia represents the reducing agent of the stored NO x .
Considering that the NSR catalysts have been derived from classical three-way catalysts by adding an alkaline component to the active wash-coat layer, and that during the regeneration of NSR systems NO x are released in the gas phase as NO, one can suppose that a TWC-like reduction chemistry occurs. Accordingly, the following set of elementary reactions can be considered for the reaction between NH3 and NO over Pt-based catalysts, as suggested in the literature [76]:
In this scheme gaseous NO dissociates at Pt sites cleaned by the reductant to form Pt-N and Pt-O species (reactions 11 and 12). Activation of ammonia leads to the formation of Pt-H species (reactions 14) that scavenge the O-ad-atoms leading to the formation of reduced Pt sites and water (reaction 16). The recombination of adsorbed nitrogen atoms follows to form N2 (reaction 15) while reaction of adsorbed nitrogen and hydrogen atoms produces NH x and ultimately NH3 (reverse reactions 14 and 13).
The mechanism of the reduction by ammonia of nitrates stored onto a Pt–Ba/Al2O3 catalyst has been investigated recently using labelled nitrates and unlabelled ammonia [77, 78]. Since in real automobile operation NO will also be present, it is of interest to study how its presence modifies the reaction network for stored NO x reduction to N2, by feeding NO together with the reductant in the regeneration period. To do this, NH3-TPSR and NH3-ICSC of stored nitrates in the presence and absence of NO in the gas phase have been accomplished. The temperature programmed reaction (TPR) between gaseous labelled 15NO and unlabelled 14NH3 over Pt–Ba/Al2O3 was also studied for comparison purposes.
The results of the TPR run between 15NO and 14NH3 are shown in Fig. 10, panel A. The reaction is observed starting near 100 °C, with formation of single- and double-labeled N2O, and of single labeled N2. Above 160–180 °C the conversion of NO is complete, a drop in the concentration of N2O is observed and the reaction becomes fully selective to N2. All N2 isotopes are seen above this temperature, with abundances that do not change significantly with temperature. Notably, the relative abundance of the di-nitrogen isotopes observed at T > 160–180 °C, where 15NO is no longer observed among the products, compares well with that calculated in the case of statistical coupling of N-adspecies formed via dissociation of gaseous 15NO and gaseous 14NH3 according to the stoichiometry:

a TPR 14NH3 (1,000 ppm) + 15NO (1,000 ppm); b TPSR run with 14NH3 (1,000 ppm) + 14NO (1,000 ppm) after 15NO x adsorption at 350 °C (1,000 ppm 15NO + O2 3 % v/v in He); c TPSR run with 14NH3 (1,000 ppm) after 15NO x adsorption at 350 °C (1,000 ppm 15NO + O2 3 % v/v in He)
as shown in Table 2.
The reduction of labeled nitrates with unlabeled ammonia has also been investigated, and results are shown in Fig. 10, panel C. The reaction is detected from slightly above 150 °C, is fully selective to N2 with an isotopic distribution that does not change with temperature. Note that NO has never been detected among the products. The Pt-14N species originated from 14NH3 and the Pt-15N species produced from labelled nitrates participate in the reaction in a ratio 10/6 in line with the stoichiometry constraint provided by the reaction:
leading to the following abundance of N2 isotopes, calculated assuming the statistical coupling of Pt-14N and Pt-15N species: 14N2 = 39 %, 14N15N = 47 % and 15N2 = 14 %. These numbers compare reasonably well with those measured during the 14NH3-TPSR shown in Fig. 10, panel C and also with 14NH3-ICSC experiments of stored labelled nitrates (Table 3). It is concluded that in the reduction of stored nitrates by ammonia, N2 is produced primarily by statistical coupling of adsorbed N-atoms coming from stored NO x and gaseous NH3.
The reaction of 14NH3 with 15NO (TPR experiment, Fig. 10 panel A) is faster than the reaction of 14NH3 with the stored labelled nitrates (TPSR experiment, Fig. 10 panel C). In fact, the onset of the reaction was observed at lower temperatures in the former case (near 100 vs. near 150 °C) and the reaction proceeds till the full consumption of the limiting reactant gaseous 14NO [77]. Besides when both gaseous NO and stored nitrates are present (Fig. 10 panel B) the reaction involving gaseous species prevails [77]. Indeed, when gaseous 14NO is present unlabelled N2O and unlabelled N2 are formed, and when gaseous 14NO has been depleted, 14NH3 reacts with stored labelled nitrates to give 14N15N and 15N2 although unlabelled N2 is by far the most abundant isotope. The reactivity of NH3 with gaseous 14NO seems to be slightly inhibited in the presence of stored nitrates. In fact, the onset of the NH3 + NO reaction is seen at higher temperature in the presence of stored nitrates (near 120 vs. near 100 °C). Based on these results it is concluded that gaseous NO is more reactive towards NH3 than stored nitrates and that the activation of the stored species represents the rate determining step in the reduction of stored nitrates. Besides, when NO is not present in the gas phase, the Pt sites are cleaned by the reducing agent and accordingly reducing conditions prevail. The primary route in the formation of di-nitrogen is the statistical coupling between N-adspecies formed upon release of 15NO from stored nitrates and upon decomposition of 14NH3.
The reduction of labelled nitrites stored from 15NO/O2 at 150 °C on Pt–Ba/Al2O3 with unlabelled ammonia was also investigated [79]. Under temperature programming, the reaction is seen near 120 °C. At low T (T < 160 °C) single labelled 15N14N represents the main product (with minor amounts of 15N14NO). Then, also the other di-nitrogen isotopes are detected. The reaction was also accomplished under isothermal conditions at 150 °C by applying a step change in the concentration of 15NO in the feed. Again single labelled 15N14N represents the main product initially (with minor amounts of 15N14NO). Then also the other N2 isotopes are detected. In both cases NO was never detected among the products.
The abundance of single labelled nitrogen 15N14N obtained during TPR of 15NO + 14NH3, TPSR of stored labelled nitrite with unlabelled NH3 and isothermal concentration step change experiments for the reduction of stored labelled nitrites by unlabelled NH3 at low T (150 °C) is shown in Fig. 11a–c, respectively. It appears that the abundance of single labelled nitrogen 15N14N is significantly higher than that expected in the case of statistical coupling of N-ad-atoms formed from decomposition of gaseous 14NH3 and decomposition of gaseous 15NO or from release of stored nitrite and its further reduction (around 50 % to the best) either at low temperature (TPR and TPSR experiments) and at short time on stream (ICSC experiment at 150 °C).

15N14N relative abundance during a TPR 14NH3 (660 ppm) + 15NO (1,000 ppm); b TPSR run with 14NH3 (1,000 ppm) after 15NO x adsorption at 150 °C (1,000 ppm 15NO + O2 3 % v/v in He); c ICSC run with 14NH3 (1,000 ppm) after 15NO x adsorption at 150 °C (1,000 ppm 15NO + O2 3 % v/v in He)
Accordingly, the statistical coupling of N ad-species cannot be considered here as the unique route responsible for the formation of N2. Another pathway involving the preferential coupling of NO- and NH3-related species must be invoked to explain concentrations of single labeled 15N14N species significantly higher than 50 % (Fig. 12; [79]). It is worth noting that this pathway is also accompanied by the formation of N2O, i.e. an oxidized N-containing species whose presence suggests that the coupling between NO- and NH3-related species operates under slightly oxidizing conditions. Indeed, the presence of PtO sites prevents the NO dissociation.

a TPSR run with 14NH3 (1,000 ppm) after 15NO x adsorption at 150 °C (1,000 ppm 15NO + O2 3 % v/v in He), b TPR run with 14NH3 (660 ppm) and 15NO (1,000 ppm) in He, c TPR run with 14NH3 (660 ppm), 15NO (1,000 ppm), and O2 (3 % v/v) in He over Pt–Ba/Al2O3 catalyst. (reprinted with permission from [79])
Isotopic labelled experiments also provide information on the pathways involved in the formation of N2O [77–79]. As shown in Fig. 12, this species is formed in limited amounts during the reduction of stored nitrites by NH3 (panel A) while amounts higher by one order of magnitude have been observed during the reaction of 15NO (1,000 ppm) present in the feed gas with 14NH3 (panel B). Since unlabelled N2O was not observed, 15NO must be directly involved in the formation of N2O. Accordingly and in line with literature proposals [80, 81] it was suggested that N2O formation involves either the coupling of two adsorbed NO molecule [reaction (19)] or the recombination of an adsorbed NO molecule with an adsorbed NH x fragment originated from NH3 adsorption [reaction (20)]:
The formation of N2O is strongly affected by temperature. Indeed above 180 °C the N2O concentration is always negligible in the absence of oxygen. It was suggested that at high temperature NO dissociation was favored because Pt is kept in a reduced state by ammonia, so that N2O formation is prevented due to the lack of adsorbed NO species. Besides N2O could decompose onto reduced Pt sites to give N2 and PtO [82].
In the presence of oxygen (3 % v/v) in the feed stream, the nitrous oxide concentration is greater (Fig. 12, panel C) because NO dissociation is prevented over the oxygen covered Pt surface and this increases the concentration of NO ad-species. The presence of oxygen in the feed changes the nitrous oxide distribution during the TPR of 14NH3 + 15NO: in the absence of oxygen 15N2O prevails over 14N15NO while in the presence of oxygen the concentration of the double-labelled nitrous oxide is negligible while that of single-labelled nitrous oxide is significantly greater. Indeed adsorbed oxygen drives reaction (19) from right to left, while it was reported to favour ammonia activation yielding reactive NH x adsorbed intermediates [83]; this eventually explains the formation in greater amounts of 14N15NO.
As previously discussed, the presence of NO in the gas phase during the regeneration period could modify the reaction network for NO x reduction to N2. To study the behaviour of a Pt–BaO/Al2O3 catalyst during NO x reduction when NO is also fed together with H2 in the regeneration period, Pereda-Ayo et al. [82] employed isotopically labelled techniques, storing 14N unlabelled nitrates that were reduced during the subsequent rich period, feeding labelled 15NO together with H2. All N2 isotopes were detected, namely 15N2, 15N14N and 14N2. Three different routes for N2 formation were deduced (as illustrated in Fig. 13): (i) in route 1, the incoming hydrogen reacts with stored nitrates to form 14NH3 which further reacts with nitrates stored downstream to form 14N2; (ii) in route 2 the incoming 15NO reacts with hydrogen to form 15NH3 upstream of the hydrogen front, it travels through the catalyst bed and reaches the regeneration front where it gets involved in the reduction of stored nitrates to form 15N14N; (iii) in route 3 incoming 15NO reacts with hydrogen to form 15N2.

Schematic of the proposed regeneration mechanism of the LNT with hydrogen. Storage was achieved with 14NO and regeneration was accomplished with H2 in the presence of 15NO (reprinted with permission from [82])
4 Combined Systems: LNT + SCR and DPNR
4.1 LNT + SCR
It has been shown that NH3 is formed during regeneration of LNT catalysts, while it is well known that SCR catalysts store large quantities of NH3 under reaction conditions [84]. Hence, combining the LNT catalyst with a downstream SCR catalyst offers a potential of capturing NH3 generated over the LNT and using it to convert NO x that slips through the LNT [85]. These LNT + SCR coupled systems result in higher NO x removal and lower NH3 slip compared to the LNT-only systems. The LNT + SCR concept has been commercially demonstrated on the Mercedes E320 Bluetec vehicle [13] which utilized relatively high-loaded PGM LNT in combination with an Fe-based zeolite SCR catalyst. This approach is variously referred to as an in situ, hybrid, booster, or passive LNT + SCR system.
Most of the fundamental studies have involved model catalysts tested in simple laboratory gas mixtures and with hydrogen as the reducing agent. For example, Corbos et al. [86, 87] showed that the NO x removal efficiency can be greatly improved under lean–rich atmosphere if a NSR catalyst is physically mixed with Cu-ZSM-5. Indeed, Fig. 14 shows the NO x removal efficiency during the lean–rich cycles for the different catalyst mixtures (Pt–RhBa and SCR catalysts). The NO x removal activity of Pt–RhBa catalyst shows a volcano type curve and reaches a maximum of 50 % at 300 °C. Figure 14 points out that mixing the Pt–RhBa catalyst with AgAl catalyst has no significant influence on the NO x removal activity as compared to mixing with bare α-Al2O3, regardless of temperature. On the other hand, the NO x removal activity was improved in 200–300 °C temperature range by mixing Pt–RhBa catalyst with CoAl or CuZSM-5. A remarkable difference is observed at 250 and 300 °C. At these temperatures, the activity of Pt–RhBa + CuZSM-5 mixed catalyst is much higher than that obtained with Pt–RhBa + CoAl [85, 86, 88].

NO x removal efficiency (%) during the lean–rich cycles for physically mixed catalysts (diamond) Pt–RhBa + α-Al2O3, (cross symbol) Pt–RhBa + AgAl, (square) Pt–RhBa + CuZSM-5 and (triangle) Pt–RhBa + CoAl. Reaction conditions: lean 100 s, 1 % CO2, 500 ppm NO, 10 % O2, 0.13 % CO/H2 (75/25), 167 ppm C3H6 and rich 10 s with 8.53 % CO/H2 (75/25), 100 ppm NO, 1 % CO2 (reprinted with permission from [86])
In most of these studies, NSR experiments have been performed by alternating long lean phases with short rich periods characterized by high concentration of the reductant. Accordingly, significant temperature effects may have occurred. In view of this, only a qualitative analysis of the data has been generally attempted.
A systematic investigation of the hybrid arrangements LNT + SCR has also been accomplished in our labs [89, 90], which combines reactivity measurements performed under nearly isothermal conditions (i.e. with low concentration of NO and of the reductant) and in situ FT-IR spectroscopy to analyse the species formed in the gas phase and at the catalyst surface during the lean–rich cycles, covering the temperature range 150–350 °C and also considering the presence of H2O and CO2 in the exhausts. Pt–Ba/Al2O3 LNT catalyst and Fe-ZSM-5 SCR catalyst have been tested alone in a typical NSR adsorption-reduction cycle. Then both LNT + SCR dual bed and LNT/SCR physical mixture have been tested in the same conditions. NO x removal efficiency, N2 production during the lean and rich periods, NH3 oxidation upon O2 admission during the lean phase and NH3 slip were determined and explained at the quantitative level for all the mentioned arrangements and the results obtained during lean–rich experiments performed at 250 °C are reported in Table 4. From this quantitative analysis, and the FT-IR evidences, it has been possible to provide a picture of the working mechanism of these combined catalytic systems, depicted in Fig. 15. During the lean phase, the storage of NO x (NO x stored in Table 4) occurs primarily on Pt–Ba/Al2O3 and results in the formation of nitrate species, well recognized by FTIR at the end of the lean phase (bands at 1,416, 1,328 and 1,035 cm−1). Moreover, the FT-IR study showed that the Fe-ZSM-5 SCR catalyst is able to adsorb large amounts of ammonia, the trapping of NH3 being most favoured at low temperatures. Accordingly, during the rich phase of a NSR cycle the ammonia produced in the stored NO x reduction can be adsorbed by Fe-ZSM-5 SCR catalyst of the combined LNT + SCR catalytic system (both physical mixture or dual bed).

Sketch of the behavior of hybrid LNT-SCR catalytic systems, physical mixture and dual bed
As it clearly appears from the sketch of Fig. 15, and the data of Table 4, the NO x removal efficiency (NO x removed in Table 4) is always higher for both hybrid LNT + SCR systems due to the contribution of N2 produced by the SCR reactions over the Fe-ZSM-5 catalyst during the lean phase (N2 lean) in addition to NO x stored onto the LNT catalyst during the lean phase (NO x stored in Table 4). For all investigated systems (LNT, LNT + SCR physical mixture and LNT + SCR dual bed) the amounts of NO x removed correspond to amounts of NO x stored plus those of N2 lean.
The ammonia species adsorbed during the rich phase are completely consumed by the SCR reactions upon subsequent admission of NO/O2 to produce N2 (N2 lean in Table 4). Until the ammonia is present on the surface of the SCR catalyst, the only observable NO x surface species are Fe3+(NO) indicating that NO is directly bonded at Fe sites (band at 1,878 cm−1). After adsorbed ammonia is completely consumed, the increase of NO+ (peak at 2,132 cm−1) and nitrate (main peaks at 1,628 cm−1 and at 1,573 cm−1) species is observed together with the erosion of the band related to Fe3+(NO) species. This indicates that at first SCR reaction takes place and then small amounts of NO x are stored at the catalyst surface as nitrates [91].
When the hybrid LNT + SCR systems are considered, in the reduction of nitrates stored over Pt–Ba/Al2O3, N2 is formed during the rich phase (N2 rich in Table 4) together with NH3, that is trapped onto the Fe-ZSM-5 catalyst in the form of coordinated NH3 (absorptions in the region 3,500–3,100 cm−1 and at 1,616 cm−1) and of protonated NH4 + (absorptions in the region 3,000–2,250 cm−1 and at 1,447 cm−1).
In the NSR cycle, NH3 trapped onto Fe-ZSM-5 during the rich phase is oxidized during the subsequent lean phase immediately after O2 admission to give N2, N2O, and NO x (N2 + N2O + NO x at O2 admission in Table 4) and upon subsequent NO admission to give N2 according to standard and fast SCR reactions (N2 lean). Therefore, the amount of NH3 stored during the rich phase over the Fe-ZSM-5 particles is obtained by the sum of amounts of N2 + N2O + NO x at O2 admission and of amounts of N2 lean; this sum is always higher for the physical mixture than for the dual bed. It turns out that the trapping of NH3 is most favoured when the particles of Pt–Ba/Al2O3 and of Fe-ZSM-5 are evenly distributed in the catalyst bed, as in the case of the LNT + SCR physical mixture. Trapping the NH3 onto the Fe-ZSM-5 particles prevents the reaction of NH3 with NO x stored downstream onto Pt–Ba/Al2O3 particles to give N2 during the rich phase (N2 rich in Table 4), that in fact is lower for the physical mixture than for the dual bed.
It is worth noting that the oxidation of NH3 trapped onto the Fe-ZSM-5 particles after admission of O2 was observed only in the case of the physical mixture and was ascribed to desorption of ammonia from the Fe-ZSM-5 particles followed by reaction over the Pt–Ba/Al2O3 in close proximity. The selectivity to nitrogen for this reaction increases significantly with temperature and is almost complete at 350 °C. The adsorption of NH3 over the zeolite catalyst during the rich phase and its consumption during the subsequent lean phase is documented by the FT-IR study. No other intermediate species have been detected.
In conclusion, due to the fact that ammonia, formed in the reduction of nitrates by hydrogen over the LNT catalyst, can be effectively stored onto the SCR catalyst, the NO x removal efficiency is increased and the ammonia slip is reduced over the coupled LNT + SCR systems. The ammonia slip is reduced to the best over the dual bed arrangement (LNT ahead of SCR). The synergy becomes more important with the aged NSR catalyst where generally higher NH3 yields are observed in comparison with the fresh NSR catalyst [91].
The class of combined LNT + SCR catalysts also comprises the system developed by Sato and coworkers [92] consisting of a solid acid SCR catalyst and Pt/OSC (oxygen storage component) such as CeO2/ZrO2 as NSR catalyst.
4.2 DPNR
The DPNR (Diesel Particulate-NO x Reduction) technology aimed at the simultaneous reduction of PM and NO x was developed by Toyota [16, 93]. The catalytic converter for DPNR is a porous ceramic wall-flow filter coated with a NSR catalyst. Like the NSR technology, this system accomplishes the removal NO x under cyclic lean/rich conditions; in addition the simultaneous soot abatement occurs, mainly under lean conditions due to the presence of NO x and the excess oxygen in the exhaust gas. Of note, Toyota researchers also proposed that active oxygen species, which are formed during NO x adsorption, are effective in soot oxidation under rich conditions as well.
Castoldi et al. [94] showed that under cycling conditions, i.e. alternating lean/rich phases, the Pt–Ba/Al2O3 NSR system is able to simultaneously remove NO x and soot.
Soot combustion was primarily ascribed to the NO2 formed upon NO oxidation over Pt, and to the occurrence of the NO recycling to NO2 [95]. Moreover, based on a comparative study of Pt–Ba/Al2O3 and Pt/Al2O3, a role of the stored NO x in the oxidation of soot has been suggested [96], which is in agreement with the results of Suzuki and Matsumoto [16] who noted a higher combustion activity of soot when a NO x trap material is incorporated in the catalyst. Actually, there is a general consensus on the fact that the NO x storage function of the LNT catalysts positively affects the combustion of soot by providing an additional path for oxidation of the carbonaceous material in addition to the well-established gas-phase reactions involving O2 and NO2. Kustov et al. [97] reported that the stored nitrates may decrease the onset temperature of soot oxidation when nitrate decomposition occurs in a proper temperature range, due to the release of NO2 in the gas phase. Along similar lines, Sullivan et al. [98] reported that the presence of a NO x trapping component like BaO in Pt/SiO2 systems does not promote per se the particulate combustion, but favors soot combustion due to the increase in the NO2 gas phase concentration, observed upon nitrates decomposition during LNT catalyst regeneration.
However more recently, some of us investigated the reactivity of stored nitrates during TPD (temperature programmed desorption)/TPO (temperature programmed oxidation) experiments carried out over Ba- and K-based LNT systems and suggested that stored nitrates take a direct part in soot oxidation, which indeed is observed below the temperature threshold for nitrates thermal decomposition [96, 99, 100]. In particular a redox mechanism occurring between nitrates, acting as oxidant agents toward soot, and soot particles, acting as reducing centres, has been suggested. This hypothesis is in agreement with the results obtained by Ito et al. [101] over Cs-MnO x –CeO2 catalysts and by Sanchez et al. [102, 103] over lanthanum supported catalysts containing noble metals.
Several authors reported that the activity of LNT catalysts for soot combustion is higher in the presence of K rather than of Ba [104–106]. A direct role of the alkali or alkaline-earth component in the oxidation of soot has been pointed out, which seems to be superimposed to the gas phase soot combustion activity by NO2. It has been suggested [107, 108] that potassium may favor the soot oxidation through the formation of low melting point compounds, thus improving the mobility of the active surface species and favoring the contact between the soot and the catalyst which has been claimed to be a key factor in the oxidation of soot [109]. More recently, Castoldi et al. [110] investigated the intrinsic reactivity of alkaline (Na, K, Cs)—and alkaline/earth (Ca, Ba, Mg)—based catalysts in the oxidation of soot, and prepared samples in which the active elements were deposited directly onto the soot particles (‘‘full contact’’ conditions). The reactivity in the soot combustion of the selected elements has been correlated with their electronegativity, with the overall activity ranking (Cs ≥ K > Ba > Na > Ca ≫ Mg) which follows the electropositivity order of the investigated elements. Notably, under these conditions, the performance of the poorly reactive Ba-containing sample became more comparable to that of the very active K-based system.
It is worth noting that, when considering the loose contact systems the correlation between electropositivity and activity in the soot combustion is not more apparent. This indicates that under these conditions other parameters play a major role in the activity, thus confirming that the high activity of K-based systems, even under loose conditions, is likely due to the high mobility of the K surface species. It has been noted that NSR catalysts can suffer for the loss of the alkali or alkaline-earth metal oxide in view of its surface mobility. Indeed Krishna and Makkee [104] found Pt-K/Al2O3 catalyst to be more active but less stable than Pt–Ba/Al2O3.
Finally, several studies have pointed out the negative effect of soot on the NO x storage behavior of the LNT catalysts [99, 100, 105, 106]. Sullivan et al. [111] showed that the presence of soot decreases the NO x storage capacity of a Pt–Ba/SiO2 system at 400 °C; in particular they hypothesized that soot may compete with Ba sites for reaction with NO2 by offering another reaction path for the utilization of NO2 rather than the desired formation of surface nitrates. Along similar lines, Artioli et al. [99] observed that soot decreases the NO x storage capacity of Pt–Ba/Al2O3 system and favors the decomposition and reduction of the stored nitrates, while soot is being oxidized. A direct reaction between the stored nitrates and soot was suggested, that has been explained with the surface mobility of the adsorbed nitrates. More recently Klein et al. [112, 113] attributed the decrease in the NO x storage capacity of Pt–Ba/Al2O3 catalysts to the destabilization of stored nitrates via a direct surface reaction with carbon particles.
In conclusion, very few investigations deal with the effect of soot on the regeneration of LNT catalysts. Nakatani et al. [93] observed that the NOx conversion efficiency of DPNR catalysts is almost similar to that of the soot-free NSR catalysts. This is confirmed by investigations performed within our group [96, 99, 100].
5 Perspectives
The development of successful catalytic technologies for the control of NO x emissions from vehicles under lean conditions over the last 20 years has been a remarkable technical achievement. Catalytic lean NO x removal converters are of commercial importance and extensive research has been done on several aspects of their formulation, synthesis and performance. Presently SCR and NSR represent the top contenders for lean NO x removal. SCR is a more mature technology that requires an on board urea tank so that it is used mostly for HD and high segment vehicles but it is considered as well for LD vehicles and passenger cars. NSR does not require big layout modification because it uses the exhaust as the reducing agent and is the favored approach for low segment vehicles.
In spite of the fact that the purpose of this paper was not to compare one technology with the other, but to address the reaction process fundamentals of the NSR technology, still a comparison between NSR and SCR technologies is worthwhile in order to enlighten analogies and differences in the associated chemistries and catalyst functionalities.
History teaches us that the advances in technology will come mostly from the application and that fundamental insight of such advances usually comes later. Experimental and theoretical tools are now available that allow us to better understand the details of the catalytic chemistry of NSR. Among these special importance has been (and it is expected that more will be in the future) the use of spatially resolved analytical tools such as capillary inlet mass spectrometry (SpaciMS) to provide a spatiotemporal resolution during transient storage and regeneration processes occurring over NSR catalysts, of isotopic experiments to address the mechanism of NO x release and of N2 formation and of spectro- kinetics to combine the analysis of gas phase and surface concentrations.
It is noted that coupled LNT + SCR systems, that use the LNT to generate ammonia during the rich period of the cycle and then store and use ammonia in a downstream SCR catalyst, are of special interest nowadays. They are being optimized to ensure high NO x removal efficiencies while reducing the PGM usage and are moving onto much more vehicle testing. Ammonia generation from the NSR and finding optimum system configurations are at the forefront [12].
Recently a very interesting phenomenon that achieves high NO x conversion was observed over NSR catalysts by Toyota researchers [114]. This phenomenon occurs when continuous short cycle injections of hydrocarbons are supplied at a predetermined concentration in lean conditions. The system that uses this concept was named Di-Air (diesel NO x after-treatment by adsorbed intermediate reductant) because it was concluded that such a phenomenon is likely due to the generation of reactive intermediates formed by adsorbed NO x over the base catalysts and HCs partially oxidized. It is recommended that the Di-Air concept should be studied more deeply by the scientific community.
Besides, the nitrite route, which was found to dominate at low temperatures during the storage of NO/O2 over LNT catalysts, suggests we should revisit the chemistry of NO oxidation to NO2 since in this case the reaction proceeds through a stepwise oxidation to form nitrites that are stabilized against further reaction to form NO2 by the interaction with nearby Ba sites.
Finally, it has been shown that nitrites and nitrates stored species exhibit different stability and reactivity during the reduction process. These aspects could be investigated in details and the possible implications and/or challenges explored.
References
http://delphi.com/manufacturers/auto/powertrain/emissions_standards/. Accessed 18 Aug 2014
Johnson T (2008) Platin Met Rev 52(1):23
Twigg MV (2013) Platin Met Rev 57(3):192
Forzatti P (2001) Appl Catal A 222:221
Kato A, Matsuda S, Kamo T, Nakajima F, Kuroda H, Narita T (1981) J Phys Chem 85:4099
Koebel M, Elsener M, Madia G (2001) Ind Eng Chem Res 40:52
Takahashi N, Shinjoh H, Iijima T, Suzuki T, Yamazaki K, Yokota K, Suzuki H, Miyoshi M, Matsumoto S, Tanizawa T, Tanaka T, Tateishi S, Kasahara K (1996) Catal Today 27:63
Matsumoto S (2000) CATTECH 4:102
Epling WS, Campbell IE, Yezerets A, Currier NW, Park JE (2004) Catal Rev Sci Eng 46:163
Engstrom P, Ambernstsson A, Skoglundh M, Fridell F, Smedler G (1999) Appl Catal B 22:241
Sedlmair C, Seshan K, Jentys A, Lercher JA (2002) Catal Today 75:413
Johnson T (2010) Platin Met Rev 54:216
Weibel M, Waldbüβer N, Wunsch R, Chatterjee D, Bandi-Konrad B, Krutzsch B (2009) Top Catal 52:1702
Jelles SJ, Makkee M, Moulijn JA (2001) Top Catal 16:269
Setiabudi A, Makkee M, Moulijn JA (2004) Appl Catal B 50:185
Suzuki J, Matsumoto S (2004) Top Catal 28:171
Fridell E, Persson H, Westerberg B, Olsson L, Skoglundh M (2000) Catal Lett 66:71
Mahzoul H, Brilhac JF, Gilot P (1999) Appl Catal B 20:47
Prinetto F, Ghiotti G, Nova I, Lietti L, Tronconi E, Forzatti P (2001) J Phys Chem B 105:12732
Lietti L, Daturi M, Blasin-Aubé V, Ghiotti G, Prinetto F, Forzatti P (2012) Chem Cat Chem 4:55
Westerberg B, Fridell E (2001) J Mol Catal A 165:249
Schmitz P, Baird R (2002) J Phys Chem B 106:4172
Prinetto F, Ghiotti G, Nova I, Castoldi L, Lietti L, Forzatti P (2003) Phys Chem Chem Phys 105:4428
Nova I, Castoldi L, Prinetto F, Ghiotti G, Lietti L, Tronconi E, Forzatti P (2004) J Catal 222:377
Visconti CG, Lietti L, Manenti F, Daturi M, Corbetta M, Pierucci S, Forzatti P (2013) Top Catal 56:311–316
Morandi S, Prinetto F, Ghiotti G, Castoldi L, Lietti L, Forzatti P, Daturi M, Blasin-Aubé V (2014) Catal Today 231:116–124
Weiss BM, Caldwell KB, Iglesia E (2011) J Phys Chem C 115:6561
Epling WS, Peden CHF, Szanyi J (2008) J Phys Chem C 112:10952
Chaugule SS, Kispersky VF, Ratts JL, Yezerets A, Currier NW, Ribeiro FH, Delgass WN (2011) Appl Catal B 107:26
Lietti L, Forzatti P, Nova I, Tronconi E (2001) J Catal 204:175
Lindholm A, Currier NW, Friedell E, Yezerets A, Olsson L (2007) Appl Catal B 75:78
Cant NW, Liu IOY, Patterson MJ (2006) J Catal 243:309
Nova I, Lietti L, Castoldi L, Tronconi E, Forzatti P (2006) J Catal 239:244
Castoldi L, Nova I, Lietti L, Tronconi E, Forzatti P (2004) Catal Today 96:43
Clayton RD, Harold MP, Balakotaiah V, Wan CZ (2009) Appl Catal B 90:662
Tuttlies U, Schmeisser V, Eigenberger G (2004) Chem Eng Sci 59:4731
Kabin KS, Muncrief RL, Harold MP (2004) Catal Today 96:79
Muncrief RL, Kabin KS, Harold MP (2004) AIChE J 50:2526
Amberntsson A, Persson H, Engström P, Kasemo B (2001) Appl Catal B 31:27
Liu Z, Anderson JA (2004) J Catal 224:18
Cant NW, Patterson MJ (2003) Catal Lett 85:153
Poulston S, Rajaram R (2003) Catal Today 81:603
Zho GU, Luo T, Gorte RJ (2006) Appl Catal B 64:88
Epling WS, Parks JE, Campbell GC, Yezerets A, Currier NW, Campbell LE (2004) Catal Today 96:21
Otto K, Shelef M, Kummer JT (1970) J Phys Chem 74:2690
Bathia D, Harold MP, Balakotaiah V (2010) Catal Today 151:314
Coronado J, Anderson J (1999) J Mol Catal A 138:83
Righini L, (2014) NOx Storage‐Reduction (NSR) catalytic process for NOx removal from mobile sources, PhD Thesis, Politecnico di Milano
Clayton RD, Harold MP, Balakotaiah V (2008) Appl Catal B 84:616
Partridge WP, Choi JS (2009) Appl Catal B 91:144
Mulla SS, Chaugule SS, Yezerets A, Currier NW, Delgass WN, Ribeiro FH (2008) Catal Today 136:136
Cumaranatunge L, Mulla SS, Yezerets A, Currier NW, Delgass WN, Ribeiro FH (2007) J. Catal. 246:29
Nova I, Lietti L, Forzatti P (2008) Catal Today 136:128
Lietti L, Nova I, Forzatti P (2008) J Catal 257:270
Stoica M, Caldararu M, Ionescu NI, Auroux A (2000) Appl Surf Sci 153:218
Szailer T, Kwak JH, Kim DH, Hanson JC, Peden CHF, Szanyi J (2006) J Catal 239:51
Forzatti P, Lietti L, Gabrielli N (2010) Appl Catal B 99:145
Pihl JA, Parks JE, Daw CS, Root TW (2006) SAE Technical Paper 01-3441
Abdulhamid H, Fridell E, Skoglundh M (2006) Appl Catal B 62:319
Abdulhamid H, Fridell E, Skoglundh M (2004) Top Catal 30(31):161
Di Giulio C, Komvokis VG, Amiridis MD (2011) Catal Today 184:8
Forzatti P, Lietti L, Nova I, Morandi S, Prinetto F, Ghiotti G (2010) J Catal 274:163
Castoldi L, Lietti L, Bonzi R, Artioli N, Forzatti P, Morandi S, Ghiotti G (2011) J Phys Chem C 115:1277
Morandi S, Ghiotti G, Castoldi L, Lietti L, Nova I, Forzatti P (2011) Catal Today 176:399
Lasage T, Verrier C, Bazin P, Saussey J, Daturi M (2003) Phys Chem Chem Phys 5:4435
Bion N, Saussey J, Haneda M, Daturi M (2003) J Catal 217:47
Scholz CMI, Maes BHW, de Croon MHJM, Schoulten JC (2007) Appl Catal A 332:1
Ji Y, Toops TJ, Crocker M (2013) Appl Catal B 140–141:265
Okuhara T, Hasada Y, Misono M (1997) Catal Today 35:83
Kameoka S, Chafik T, Ukisu Y, Miyadera T (1998) Catal Lett 55:211
Ukisu Y, Miyadera T, Abe A, Yoshida K (1996) Catal Lett 39:265
Sumiya S, He H, Abe A, Takezawa N, Yoshida K (1998) J Chem Soc Faraday Trans 94:2217
Nova I, Lietti L, Forzatti P, Prinetto F, Ghiotti G (2010) Catal Today 151:330
Dasari P, Muncrief R, Harold M (2013) Top Catal 56:1922
Castoldi L, Lietti L, Righini L, Forzatti P, Morandi S, Ghiotti G (2013) Top Catal 56:193
van Tol MFH, Siera J, Cobden PD, Nieuwenhuys BE (1992) Surf Sci. 274(1):63
Lietti L, Righini L, Castoldi L, Artioli N, Forzatti P (2013) Top Catal 56:7
Righini L, Castoldi L, Lietti L, Da Costa P, Sauce, Forzatti P (2013) Top Catal 56:1906
Lietti L, Artioli N, Righini L, Castoldi L, Forzatti P (2012) Ind Eng Chem Res 51:7597
Burch R, Shestov AA, Sullivan JA (1999) J Catal 188:69
Burch R, Daniells ST, Hu P (2002) J Chem Phys 117:2092
Pereda-Ayo B, González-Velasco JR, Burch R, Hardacre C, Chansai S (2012) J Catal 285:177
Perez-Ramirez J, Kondratenko EV, Kondratenko VA, Baerbs M (2005) J Catal 229:303
Tronconi E, Lietti L, Forzatti P, Malloggi S (1966) Chem Eng Sci 51:2965
Can F, Curtois X, Royer S, Blanchard G, Rousseau S, Duprez D (2012) Catal Today 197:144
Corbos EC, Haneda M, Courtois X, Marecot P, Duprez D, Hamada H (2008) Catal Commun 10:137
Corbos EC, Haneda M, Courtois X, Marecot P, Duprez D, Hamada H (2009) Appl Catal A 365:187
Wang J, Ji Y, He Z, Crocker M, Dearth M, McCabe RW (2012) Appl Catal B 111(112):562
Forzatti P, Lietti L (2010) Catal Today 155:131
Castoldi L, Bonzi R, Lietti L, Forzatti P, Morandi S, Ghiotti G, Dzwigaj S (2011) J Catal 282:128
Chatterjee D, Koci P, Schmeiβer V, Marek M (2010) Krutzch B 151:395
Nakatsuji T, Matsubara M, Rouistenmaki J, Sato N, Ohno H (2007) Appl Catal B 77:190
Nakatani K, Hirota S, Takeshima S, Itoh K, Tanaka T (2002) SAE Technical Paper SP- 1674 01-0957
Castoldi L, Matarrese R, Lietti L, Forzatti P (2006) Appl Catal B 64:25
Setiabudi A, van Setten BAAL, Makkee M, Moulijn JA (2002) Appl Catal B 35:159
Matarrese R, Castoldi L, Lietti L (2012) Catal Today 197:228
Kustov AL, Makkee M (2009) Appl Catal B 88:263
Sullivan JA, Keane O, Cassidy A (2007) Appl Catal B 75:102
Artioli N, Matarrese R, Castoldi L, Lietti L, Forzatti P (2011) Catal Today 169:36
Matarrese R, Artioli N, Castoldi L, Lietti L, Forzatti P (2012) Catal Today 184:271
Ito K, Kishikawa K, Watajima A, Ikeue K, Machida M (2007) Catal Commun 8:2176
Sánchez BS, Querini CA, Miró EE (2009) Appl Catal A 366:166
Sánchez BS, Querini CA, Miró EE (2011) Appl Catal A 392:158
Krishna K, Makkee M (2006) Catal Today 114:48
Pieta IS, García-Diéguez M, Herrera C, Larrubia MA, Alemany LJ (2010) J Catal 270:256
Matarrese R, Castoldi L, Lietti L, Forzatti P (2007) Top Catal 42(43):293
Querini CA, Cornaglia LM, Ulla MA, Miró EE (1999) Appl Catal B 20:165
An H, McGinn PJ (2006) Appl Catal B 62:45
Neeft JPA, Makkee M, Moulijn JA (1996) Appl Catal B 8:57
Castoldi L, Matarrese R, Lietti L, Forzatti P (2009) Appl Catal B 90:278
Sullivan JA, Dulgheru P (2010) Appl Catal B 99:235
Klein J, Fechete I, Bresset V, Garin F, Tschamber V (2012) Catal Today 189:60
Klein J, Wu D, Tschamber V, Fechete I, Garin F (2013) Appl Catal B 132(133):527
Bisaiji Y, Yoshida K, Inoue M, Umemoto K, Fukuma T (2012) SAE Int J Fuels Lubr. 5(1):380
Acknowledgments
The authors thank Dr. William Epling for useful discussion and suggestions.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Forzatti, P., Lietti, L. & Castoldi, L. Storage and Reduction of NO x Over LNT Catalysts. Catal Lett 145, 483–504 (2015). https://doi.org/10.1007/s10562-014-1343-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-014-1343-0