
Abstract
Methanol, like ammonia, is one of the key industrial chemicals produced by heterogeneous catalysis. As with the original ammonia catalyst (Fe/K/Al2O3), so with methanol, the original methanol synthesis catalyst, ZnO, was discovered by Alwin Mittasch. This was translated into an industrial process in which methanol was produced from CO/H2 at 400 °C and 200 atm. Again, as with the ammonia catalyst where the final catalyst which is currently used was achieved only after exhaustive screening of putative “promoters”, so with methanol, exhaustive screening of additives was undertaken to promote the activity of the ZnO. Early successful promoters were Al2O3 and Cr2O3 which enhanced the stability of the ZnO but not its activity. The addition of CuO was found to increase the activity of the ZnO but the catalyst so produced was short lived. Current methanol synthesis catalysts are fundamentally Cu/ZnO/Al2O3, having high CuO contents of ~60 % with ZnO ~ 30 % and Al2O3 ~ 10 %. Far from promoting the activity of the ZnO by incorporation of CuO, the active component of these Cu/ZnO/Al2O3 catalysts is Cu metal with the ZnO simply being involved as the preferred support. Other supports for the Cu metal, e.g. Al2O3, MgO, MnO, Cr2O3, ZrO2 and even SiO2 can also be used. In all of these catalysts the activity scales with the Cu metal area. The original feed has now changed from CO/H2 to CO/CO2/H2 (10:10:80), radiolabelling studies having provided the unlikely discovery that it is the CO2 molecule which is hydrogenated to methanol; the CO molecule acts as a reducing agent. The CO2 is transformed to methanol on the Cu through the intermediacy of an adsorbed formate species. These Cu/ZnO/Al2O3 catalysts now operate at ~230° and between 50 and 100 atm. This important step change in the activity of methanol synthesis has resulted in a significant reduction in the energy required to produce methanol. The “step change” however has been incremental. It has been obtained on the basis of fundamental knowledge provided by a combination of surface science techniques, e.g. LEED, scanning tunnelling microscope, TPD, temperature programmed reaction spectroscopy, combined with catalytic mechanistic studies, including radiolabelling studies and chemisorption studies including reactive chemisorption studies, e.g. N2O reactive frontal chromatography.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The industrial synthesis of methanol is a process whose mode of operation is completely counter intuitive. Originally methanol was produced by felling trees and distilling off the small amount of methanol in the wood. Given that currently 40 million tons of methanol are produced annually, it is obvious that that means of production was unsustainable. It was in 1923 that Alwin Mittasch who, working in Badische Anilin und Soda Fabrik [1, 2], following his discovery of the Fe/K/Al2O3 catalyst for ammonia synthesis, found that methanol could be produced from CO/H2 mixtures over a ZnO catalyst. The process developed from this discovery, however, was highly inefficient, requiring high temperatures (400 °C) and high pressures (200 atm). While this was a massive advance on the dry distillation of wood, the search was then on for an improved catalyst.
Although the addition of Al2O3 or Cr2O3 or both was found to improve the stability of the catalyst and its activity somewhat, their addition made little or no impact on the operating temperatures and pressures of the original ZnO catalyst. The addition of Cu was found to make a significant improvement to the activity of both the ZnO/Al2O3 or ZnO/Cr2O3 catalysts [3], but the activity of Cu/ZnO/Al2O3 catalysts was found to be short lived due to their being prone either to poisoning or sintering. In 1955 therefore, Giulio Natta [4],the winner of the 1963 Nobel Prize in chemistry for catalysis stated that Cu/ZnO/Al2O3 catalysts were “of only slight interest” and would not be of any industrial import. This statement held for 11 years. In 1966, however, Imperial Chemical Industries (ICI) patented a co-precipitation method for the preparation of a stable, highly active Cu/ZnO, Al2O3 catalyst for the synthesis of methanol. This was a significant advance since this new catalyst lowered the operating temperature from 400 to 230 °C and the operating pressure from 200 atm to between 50 and 100 atm [5]. Originally the reactant mixture was CO/H2. So convinced were operators at that time that the process was the hydrogenation of CO, that CO2 was scrubbed from the reactant mixture. It was only when, after the CO2 scrubber failed and CO2 was accidentally admitted to the CO/H2 mixture, that a large increase in the methanol make was observed, that CO2 was incorporated into the reactant mixture [6].
It is at this point that the counter intuitive aspects of the process become apparent. A priori, it seems to be self-evident that in a CO/CO2/H2 feed it would be the CO molecule which produced the methanol since making methanol from CO2 would require the breaking of a CO bond, a process which would require >400 kJ mol−1 [7]. In addition it was well known that ZnO, while being the original methanol synthesis catalyst, was not particularly active. Cu metal was found to have little or no activity [8–12]. It seemed logical therefore to conclude that there was something unique about the Cu/ZnO combination. This uniqueness was termed “synergy” [8–12].This synergy was considered to derive from the nature of the active centre in Cu/ZnO catalysts which was considered to be a Cu+ ion substituted for a Zn2+ ion in the ZnO lattice, this substitution being regarded as entirely reasonable since Cu+ and Zn2+ were isoelectronic.
There was some experimental evidence for this postulate. In a series of highly influential papers [8–12], Klier and co-workers [8], using quantified X-ray diffraction, reported that a small amount of copper (2 % in a Cu/ZnO [15 % Cu] catalyst) was dissolved in the ZnO component of the catalyst. After reduction, the amount of Cu dissolved in the ZnO was found to have increased to 12 % in the Cu/ZnO (15 % Cu) [10] catalyst and to 16 % in a Cu/ZnO/Al2O3 catalyst [10]. The Cu+ ion was deemed to be the active centre for the adsorption and hydrogenation of CO, the dissociative adsorption of hydrogen taking place on the vicinal Zn2+–O2− ion pair [8]. A schematic of this reaction mechanism is shown in Fig. 1. The role of the CO2 in the CO/CO2/H2 feed was considered to be that of an oxidant, maintaining the active centre in the Cu+ state [8].

Proposed mechanism of methanol synthesis from CO/H2 exemplifying the concept of synergy between Cu+ and ZnO
The experimental discoveries which totally overturned the synergistic argument were:
-
(i)
It was the CO2 molecule of the CO/CO2/H2 feed which was hydrogenated to methanol [13],
-
(ii)
Cu metal was the active centre and
-
(iii)
The turnover number (TON) (frequency) of Cu metal in methanol synthesis from CO2, which is defined as, molecule CH3OH, per Cu s−1 site−1, was the same for a range of supports which included, MnO, MgO, SiO2, Al2O3 as well as ZnO and ZnO/Al2O3 and so no synergy accrued to the Cu/ZnO pairing [14]. The term turnover frequency (TOF) is used synonymously and interchangeably throughout.
2 Radiolabelling Studies
The discovery that it was the CO2 molecule which was the immediate precursor to methanol was made by Rozovsky and co-workers [13]. Believing, as did everyone else at the time, and to some extent do some still, that it was the CO molecule which was hydrogenated to methanol, they labelled the CO molecule of a CO/CO2/H2 feed with 14C, expecting to find the same specific radioactivity in the methanol produced. Instead, and completely against expectation, they found that the specific activity of the methanol produce coincided with that in the exit CO2. They posited that the radioactive 14CO2 was produced by the shift reaction
This discovery went largely unnoticed, principally because it contradicted conventional wisdom. In addition, the nature of the experiment was confusing, since the “wrong” molecule—the CO molecule—was labelled and it was difficult to see how the forward shift reaction could produce labelled CO2 to any extent in a CO/CO2/H2 stream.
A considerable body of unreported data existed in internal documents in ICI at the time (~1982). This included a comprehensive study of the interaction of the components of the gaseous reactants CO, CO2 and H2 separately, then in pairs and finally in total combination with the components of the catalyst Cu, ZnO and Al2O3 again separately, in pairs and then the total Cu/ZnO/Al2O3 catalyst. It was found that a CO2/H2 (10 % CO2, 101 kPa) stream produced methanol >102 faster than a CO/H2 stream of the same composition. The obvious conclusion was that in a CO/CO2/H2 stream over a Cu/ZnO/Al2O3 catalyst, it was the CO2 molecule of the feed which predominantly produced the methanol. This conclusion was confirmed by labelling experiments in which the CO2 molecule was labelled with 14C. The work was carried out at industrial conditions, i.e. at 50 atm and 250 °C over a standard Cu/ZnO/Al2O3 (60:30:10) catalyst as a function of the flow rate of the reactant mixture (CO, CO2, H2 [10:10:80]) [15]. The results are shown in Fig. 2 in which the specific radio activity of the methanol produced is plotted as a function of the gas hourly space velocity. At high space velocities, the specific radioactivity of the exit CH3OH was the same as that of the inlet CO2, while at intermediate space velocities the specific radioactivity of the exit CH3OH was the average of the inlet and exit CO2 radioactivity. All of the CH3OH was produced from the CO2. In addition, by using the labelled 14CO2 in different CO:CO2 ratios, it was possible to determine the fraction of CH3OH made from the 14CO2. For a 1 % CO2 in the CO roughly 70 % of the CH3OH was made from the CO2 in agreement with the separate rate measurements referred to above (Fig. 3).

Radiolabelling studies of methanol synthesis from CO/CO2/H2 (10:10:80) at 50 atm and 250 °C over a Cu/ZnO/Al2O3 (60:30:10) catalyst using 14CO2

Fraction of the CH3OH produced from the CO2 molecule in CO/CO2/H2 mixture of different CO:CO2 ratios determined using 14CO2
3 Measurement of the Copper Metal Area
A key measurement which had not been carried out at the time, probably because of the certainty that it was the Cu+ ion which was the active centre for the reaction, was that of the Cu metal area. Nevertheless this was both surprising and a serious omission since the composition of the optimal industrial Cu/ZnO/Al2O3 catalyst was 60:30:10, while the maximum incorporation of the Cu+ into the ZnO lattice was considered to be between 12 and 16 %. The Cu loading therefore was considerably in excess of that required for 16 % loading in the ZnO lattice. It was inevitable that this apparent excess Cu would produce Cu metal on reduction of the catalyst.
The method developed for the in situ measurement of the Cu metal area derived from the well known fact that N2O decomposed on Cu. Indeed, Ertl had used the decomposition of N2O on Cu to determine the structure of the oxygen overlayer on Cu(110). The structure was a (2 × 1)-O overlayer and so saturation coverage was ½ monolayer (ML) [16]. (This was also found to be the case for Cu(111) and Cu(110).) Vandervell and co-workers [17] used this information as the basis for the in situ measurement of the Cu metal area of supported Cu catalysts. They used a variant of the frontal uptake method they had used for the measurement of the total area of a catalyst [18]. The N2O was dosed on to reduced unsupported polycrystalline Cu whose total area had been measured in situ by N2(10 % N2 in He) frontal uptake at 77 K. The experiment was repeated as a function of N2O partial pressure and temperature of dosing. The optimum conditions were 60 °C (333 K) for the temperature of dosing and 5 % N2O in He as the optimum pressure. These conditions produced 100 % decomposition of the frontally dosed N2O, the reaction being found to cut off abruptly at completion of the half ML. The method was termed “reactive” frontal chromatography (RFC) [17]. A reactive frontal chromatogram is shown in Fig. 4.

The reaction of N2O (5 % in He, 101 kPa, 25 cm3 min−1) with unsupported polycrystalline Cu at 60 °C—RFC
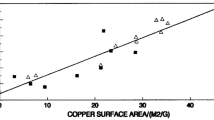
It was then suggested that the method should be applied to determine the origin of the loss of activity of the Cu/ZnO/Al2O3 methanol synthesis catalysts with poisoning being the expected cause [19]. A series of differently decayed Cu/ZnO/Al2O3 catalysts was therefore examined. This revealed that their activity was a linear function of the Cu metal area, excluding poisoning but implicating sintering as the cause of the loss of activity [14] (Fig. 5). The research was then broadened to determine if Cu metal area was the sole determinant of activity of supported Cu catalysts by measuring the methanol synthesis activity of differently supported (MgO, Al2O3, ZnO, MnO, SiO2) Cu catalysts and this was found to be the case [14]. Indeed, a SiO2 supported Cu was found to be the most active catalyst but one which was totally impractical because of its sintering rapidly [14]. While not included in Fig. 5 when it was originally published due to its proximity to the origin, the activity of unsupported Cu was found to lie on the line.

The relationship between methanol synthesis activity and Cu metal area
The TON for methanol synthesis at 240 °C (513 K) and 50 atm from CO/CO2/H2/He mixtures in which the CO partial pressure ranged from 10 to 14 %, the CO2 partial pressure ranged from 11 to 14 % and the H2 partial pressure ranged from 46 to 60 % calculated from the gradient of Fig. 5, was 1.6 × 10−2 molecule CH3OH s−1 site−1, assuming a value of 1019 sites m−2.
Other groups also found a linear relationship between methanol synthesis activity and Cu metal area. Most notably, Roberts and co-workers [20] determined a TON for methanol synthesis of 0.03 g CH3OH/m2 Cu/h at 523 K and 50 atm pressure from a CO/CO2/H2 (70/22/6) stream. These conditions of temperature, pressure and composition are close to those chosen by Chinchen et al. [17]. Re-calculating the data of Roberts and Griffin gives a TON of 1.6 × 10−2 molecule CH3OH s−1 site−1—identical to that of Chinchen et al.
Topsoe and co-workers [21, 22] argued that, because of potential complications associated with the reaction of N2O with Cu, the temperature programmed desorption of H2 should be used to determine the Cu metal area. The complications they suggested could occur principally were: (i) difficulty in achieving half ML coverage of supported polycrystalline Cu without also incurring bulk oxidation of the Cu and (ii) sintering of the Cu as a result of the heat of reaction of the N2O with the Cu. In their original work on N2O decomposition on Cu, Dell et al. [23] found that bulk oxidation of the only occurred at temperatures >110 °C (383 K), and in the original experiments aimed at optimising the conditions for N2O RFC, Chinchen and co-workers found that the temperature of 60 °C gave rapid and quantitative decomposition of the N2O without causing bulk oxidation of the Cu. Re-examination of the original data where the reported Cu metal area decreased from 4.11 to 3.51 m2 g−1 on increasing the N2O partial pressure to 22 % might suggest that the heat of reaction may have caused sintering [17]. However, using the recommended low partial pressure of N2O of 5 % resulted in no change in Cu metal area nor even in the morphology of the Cu as can be seen from the CO desorption spectra from a reduced fresh Cu/Al2O3 catalyst and from the same catalyst which had been oxidised by N2O (2.5 % in He) and re-reduced. They are identical (Fig. 6). The heat of adsorption of CO on Cu is a function of the Cu surface crystal structure and so the CO desorption spectrum in Fig. 6 is a fingerprint of the surface morphology of the Cu.

The CO desorption spectra from freshly reduced Cu/Al2O3 and from the same catalyst which had undergone oxidation by N2O at 333 K followed by reduction in H2
4 The Reaction Mechanism
Knowing that it was the CO2 molecule which was converted to CH3OH, the problem was determining how this was accomplished. At that time CO2 was regarded as a highly unreactive molecule. The first indication that this might not be the case was the finding that CO2 decomposed on unsupported polycrystalline Cu and in doing so left an oxygen atom on the surface of the Cu [24]. This discovery was made by accident. The intention was to determine the adsorption isotherms of CO2 on unsupported polycrystalline Cu by gas adsorption chromatography. At the end of each experiment, however, the Cu was always found to be oxidised. Extensive efforts were made to remove the trace quantities of O2 from the CO2/He stream, trace quantities of O2 in the CO2/He being thought to be the only reason for the Cu being oxidised. It was only after it was certain that no O2 impurity existed in the feed that the possibility that the CO2 was decomposing on the Cu was contemplated. It was found that the maximum coverage of the Cu by O atoms produced by CO2 decomposition was limited to 10 % of a ML and so the reaction was concluded to be structure sensitive [24]. Additionally, the extent of CO2 decomposition was found to increase at lower temperatures [24]. The reaction was therefore precursor state mediated. Follow-up studies on CO2 decomposition on Cu using RFC showed that the reaction had an apparent negative activation energy of 4 kJ mol−1, which confirmed that the reaction was precursor state mediated [25]. Figure 7 shows the CO2 reactive frontal chromatograms.

The reactive frontal chromatograms of CO2 with a reduced Cu/ZnO/Al2O3 (60:30:10) catalyst at 333 K (a), 299 K (b), 213 K (c) and 173 K (d)
The conclusion that CO2 decomposed on Cu proved to be highly controversial. Campbell and co-workers [26] wrote a paper entitled, “Does CO2 Decompose on Copper?”. He concluded that it did, but only on Cu(110). Schneider and Hirschwald [27] found that CO2 decomposition on Cu(110) was facile, occurring at 85 K, while Fu and Somorjai [28, 29] stated that CO2 did not decompose on Cu(110) but did so on Cu(311). It was certain, therefore, that CO2 did decompose on Cu and that, while there was no consensus on the exact crystal face on which CO2 decomposed, the reaction was structure sensitive.
The stable intermediate on the reaction coordinate from CO2 to CH3OH was found to be a formate [30]. This was discovered by the use of temperature programmed reaction spectroscopy (TPRS) [30]. The temperature programmed reaction spectrum obtained after having dosed HCHO on to a partially oxidised Cu/ZnO/Al2O3 at 300 K is shown in Fig. 8. It shows peaks at 380 and 440 K. The 380 K peak is characterised by the coincident desorption of CO, H2 and CH3OH. This results from the desorption/decomposition of a adsorbed methoxy (CH3O(a)) species. The methoxy species was first characterised by TPRS by Wachs and Madix [31] following adsorption of CH3OH on to Cu(110). The 440 K peak is characterised by the coincident desorption of CO2, H2 and CH3OH, and is the result of the desorption/decomposition of an adsorbed formate species. This was identified by Ying and Madix [32]. They dosed formic acid on to partially oxidised Cu(110) to produce an adsorbed formate species and found CO2 and H2 desorbing coincidently at 440 K on temperature programming [32].

The temperature programmed reaction spectrum of formaldehyde adsorbed on a partially oxidised Cu/ZnO/Al2O3 (60:30:10) catalyst
Figure 9 is the temperature programmed reaction spectrum obtained on temperature programming following the adsorption of CH3OH on a partially oxidised Cu/ZnO/Al2O3 catalyst. The CO, H2, CH3OH peak at 380 K is the adsorbed methoxy species seen in Fig. 8. The CO2/H2/CH3OH peak at 440 K is the formate species also seen in Fig. 8. The CO, CO2, H2 peak desorbing at 580 K derives from the desorption/decomposition of a formate species adsorbed on the ZnO component of the catalyst [33].

The temperature programmed reaction spectrum of methanol adsorbed on a partially oxidised Cu/ZnO/Al2O3 (60:30:10) catalyst
Two important conclusions were drawn from Figs. 8, 9. Since the adsorbed formate species is the highest temperature desorbing species on Cu and is formed both on CH3OH formation and decomposition it is: (i) a true intermediate in methanol synthesis and (ii) it is the most stable and long-lived intermediate in CH3OH synthesis on Cu. Since CH3OH is desorbed at the same temperature as the adsorbed formate decomposes to CO2 and H2, the rate determining step in CH3OH synthesis on Cu is the hydrogenation of an adsorbed formate species. The decomposition of the most stable intermediate on the reaction coordinate in CH3OH synthesis to CO2 and H2 corroborated earlier conclusions that it is the CO2 molecule which is the precursor to CH3OH. Indeed, co-adsorption studies in which: (i) CO2/H2/He (8 % CO2, 8 % H2, 84 % He, 101 kPa, 305 K, 10 min) and (ii) CO/H2/He (8 % CO, 8 % H2, 84 % He, 101 kPa, 305 K, 10 min) mixtures were dosed on to polycrystalline Cu showed that the formate was formed from CO2/H2 co-dosing and not from CO/H2 co-dosing [30].
Time/temperature dependent infrared studies elucidated the mechanism of the reaction of H2/CO2 on the surface of the Cu to form the formate [34]. Co-dosing a CO2/H2 (50 % CO2, 50 % H2, 101 kPa) on to a reduced Cu/SiO2 catalyst at 295 K over 105 min showed that a carbonate species, evidenced by a band at 1,410 cm−1, gradually developed on the surface (Fig. 10). Heating the CO2/H2 to 388 K resulted in the loss of the carbonate band and the gain in the formate band at 1,350 cm−1 (Fig. 11). There was a linear relationship between the loss of the CO3(a) (1,410 cm−1) band and the gain in the HCO2(a) (1,350 cm−1) band. The reaction sequence is: (i) the dissociation of CO2 on Cu to form gaseous CO and surface O(a) atoms, (ii) the adsorption of CO2 on to the O(a) atoms to form adsorbed carbonate (CO3(a)) and (iii) the reaction of the adsorbed CO3(a) with adsorbed H atoms to form the adsorbed formate species

The development with time of an adsorbed carbonate species produced by exposing a Cu/SiO2 catalyst to CO2/H2 (1:1, 101 kPa) at 295 K shown by the increase in the 1,410 cm−1 band

The development with time of the formate species and the concomitant loss of the carbonate species produced by heating the Cu/SiO2 which had been exposed to CO2/H2 (1:1, 101 kPa) at 295 K (Fig. 10) to 388 K. The gain in the formate species is seen by the increase in the 1,350 cm−1 band
It is not clear how the adsorbed H atoms react with the adsorbed CO3(a) to form the formate. It could be that the initial interaction is to produce an adsorbed bicarbonate HCO3(a) species. Even if the bicarbonate were formed, the exact mechanism of the decomposition of an adsorbed HCO3(a) to HCO2(a) and O(a) is also not clear. The formation of an adsorbed carbonate species is an important elementary reaction in the overall synthesis of methanol. Its importance lies in the fact that it provides a large steady state coverage of adsorbed CO2 under reaction conditions. This would not be the case if molecularly adsorbed CO2, whose heat of adsorption is 18 kJ mol−1 [25], were the only source of CO2 for the reaction.
Additional information about the identity and population of the adsorbed species existing during the reaction was provided by an in situ DRIFTS study in which the infrared spectra of the adsorbates existing on the surface of a Cu/ZnO/Al2O3 catalyst at 373, 413, 443 and 473 K under a H2/CO2 (2.4:1) mixture at 30 bar was measured [35]. At 373 K the spectrum showed mostly adsorbed carbonates with a small formate signal. The population of the formate species maximised at 413 K. Under reaction conditions at 473 K, when significant quantities of gas phase methanol were detected by DRIFTS, the population of adsorbed carbonate and formate species were roughly equal [35].
The Topsoe group posited that it was the dioxy methylene species and not the formate which was the intermediate to methanol. They also claimed that hydrogenation of the formate produced either formaldehyde and/or formic acid with only small amounts of methanol [36]. This proposition was addressed by Sakakini et al. [37]. They pre-dosed a Cu/ZnO/Al2O3 (40:40:20) catalyst with adsorbed formate species by temperature programmed reaction of a CO2/H2 (10 % CO2, 101 kPa) stream raising the temperature from ambient to 493 K at 2.5 K min−1 (Fig. 12). This had shown CH3OH to have been formed at an onset temperature of 150 °C (423 K), its rate maximising at 190 °C (463 K). Post reaction TPD showed the existence of two formate species: (i) the bidentate formate species on the Cu, identified by the coincident desorption of CO2 and H2 at 440 K and (ii) a formate species on ZnO identified by the coincident desorption of CO2, CO and H2 at 530 K [37] (Fig. 13). Temperature programmed reaction of these pre-adsorbed formate species with H2 produced CH3OH at peak maximum temperatures of 390 and 530 K (Fig. 14). There was a carbon balance between the amount of formate species desorbing at 440 K and the amount of CH3OH produced at 390 K. Methanol was formed quantitatively by hydrogenation of the formate species adsorbed on Cu [36].

Temperature programmed reaction (TPR) of a CO2/H2 (10 % CO2, 101 kPa) over a Cu/ZnO/Al2O3 (40:40:20) catalyst

Temperature programmed desorption spectrum from a Cu/ZnO/Al2O3 (40:40:20) catalyst after the CO2/H2 TPR shown in Fig. 12

Temperature programmed hydrogenation using a H2 stream (101 kPa) of the different types of formate species shown in Fig. 13
5 Role of CO in Determining the Coverage of the Cu with O atoms and the Induction Period
The role of the CO molecule in a CO/CO2/H2 mixture in methanol synthesis was elucidated by the unlikely method of N2O RFC. Methanol was synthesised at 1, 50 and 100 atm pressure and between 240 and 300 °C using CO/CO2/H2/N2 and CO/CO2/H2/He mixtures of different composition in which the H2 partial pressure varied between 46 to 67 %, the CO partial pressure varied between 10 to 14 % and the CO2 partial pressure varied between 3 to 14 % [14, 38] for between 6 and 140 h on line. The Cu metal area was measured before reaction by N2O RFC. After reaction, the system was depressurised, swept out with He and cooled to 60 °C and the Cu metal area was measured again. The difference between the Cu metal area of the fresh catalyst and that post reaction was the coverage of the Cu by adsorbed O atoms. Confirmation that this lowering of the Cu metal area post reaction was not caused by sintering of the Cu as a result of reaction was provided by re-reducing the catalysts and re-determining the Cu metal areas by N2O RFC. The areas of these regenerated catalysts were found to be the same as those of the fresh catalysts. Figure 15 shows that the steady state coverage of Cu by O as a result of methanol synthesis from CO/CO2/H2 mixtures is linearly dependent on the CO2:CO ratio. Therefore under industrial conditions of 50–100 atm pressure and temperatures between 240 and 300 °C in CO/CO2/H2 mixtures in which the H2 partial pressure varies between 1 to 67 atm, the role of the CO is that of a reducing agent.

The coverage of the Cu component with O(a) as a function of the CO:CO2 ratio following reaction in CO/CO2/H2/He streams of different composition at 513 K and 50 atm pressure
The steady state oxygen coverage of the Cu component of a Cu/ZnO/Al2O3 (60:30:10) catalyst which had been producing CH3OH at a rate of 1.9 × 10−9 mol s−1 g−1 from a H2CO2/He stream (80:110:10 101 kPa, 100 cm3 min−1) at 500 K was determined directly by the following method [30]: (i) the flow was switched from the H2/CO2/He stream to He and the temperature was lowered to 470 K over a period of 30 min (All of the adsorbed intermediates would have desorbed in that time at that temperature; the formate which was the most stable intermediate was calculated to have a desorption half life of 5 s at 470 K [30].) and (ii) after 30 min the flow was switched to CO/He (10:90, 101 kPa, 60 cm3 min−1) and CO2, H2O and H2 were followed continuously on the mass spectrometer. The result is shown in Fig. 16.

Temperature programmed reaction of CO with the surface oxidised Cu of a Cu/ZnO/Al2O3 (60:30:10 catalyst at 470 K following methanol synthesis at a rate of 1.9 × 10−9 mol s−1 g−1 at 500 K from a CO2/H2/He (10:80:10), 101 kPa, 25 cm3 min−1)
CO2 was produced immediately upon contact of the CO with the catalyst. The coverage of the Cu with O atoms calculated from the integral of the CO2 produced was 0.4 ML. Remembering that saturation coverage by O atoms is 0.5 ML, this coverage by O atoms is 80 % of saturation. However, this is the same coverage of O(a) atoms found for a CO/CO2/H2/He (10:13:52:28) stream at 50 bar and so reduction by H2 does appear to be occurring to a small extent.
A surprising result of the reaction of CO with the surface oxygen was the observation of a sudden evolution of H2 after ~40 % of the adsorbed oxygen had been removed. Since all of the adsorbed species, including the adsorbed H atoms, had been removed in the 30 min period of cooling the system in He from 500 to 470 K after the CO2/H2 reaction, it was concluded that this H was subsurface. This subsurface H corresponded to ~1 ML of the Cu atoms. Subsurface H atoms had been observed to desorb from Cu(100) by Chorkendorff and co-workers [39].
The adsorbate composition of the existing on the surface of the Cu as a result of CH3OH formation from the CO2/H2/He reaction at 500 K was determined by post reaction TPD. The system was cooled from 500 K to ambient under the reaction mixture, the flow was switched to He and temperature programming was begun at 5 K min−1, raising the temperature from ambient to 700 K. The desorption spectrum obtained is shown in Fig. 17. The coincident desorption of CO2 and H2 at 420 K is evidence of the existence of a monodentate formate on the Cu [34]. The broad H2 signal maximising at 430 K derives from H atoms adsorbed on the ZnO [33].

Temperature programmed desorption of the adsorbates extant on the surface of a Cu/ZnO/Al2O3 (60:30:10) catalyst following methanol synthesis at a rate of 1.9 × 10−9 mol s−1 g−1 at 500 K from a CO2/H2/He (10:80:10, 101 kPa, 25 cm3 min−1) at 500 K
After removal of the adsorbates, the temperature was lowered from 700 to 470 K, the flow was switched to CO/He (10 % CO, 101 kPa). This produced an identical reaction spectrum to that shown in Fig. 15. The same quantity of adsorbed O atoms was obtained as was the amount of subsurface H atoms. The coverage of the Cu surface by O atoms at 80 % of saturation appears to lock in the subsurface H atoms. It is only when ~40 % of these O atoms are removed by reaction with CO that the H finds a route for its evolution.
Ertl and co-workers [40] published a model of the (2 × 1)-O overlayer shown in Fig. 18. Scanning tunnelling microscope (STM) studies confirmed that model and added the additional information that the (2 × 1) O–Cu–O rows grow in the (001) direction from the step edges [41]. A video of this reconstruction produced by overlaying successive STM images taken at 90 °C, vividly shows this surface reconstruction. Some form of collective rearrangement of this structure on the removal of 40 % of the O overlayer appears to occur which allows the subsurface H atoms to desorb in this explosive manner.

The structure of the (2 × 1)-O overlayer on Cu(110)
As in all things in methanol synthesis catalysis, the conclusion that the surface adsorbate structure of the operating catalyst was a formate species adsorbed on an oxidised Cu surface was controversial [42–45]. There was total agreement that a formate species was adsorbed on the Cu surface, the Cu surface however was proposed to be in the metallic state with no O(a) adsorbed on it.
Josihara and Campbell [41] studied methanol synthesis kinetics over a poly crystalline Cu foil [41] and over Cu(110) using a CO2/H2 (1/11) feed at 5.1 bar [42]. They found an activation energy of 77 ± 17 kJ mol−1 for Cu(110). The specific rate of methanol production at 510 K in 5.1 bar CO2/H2 was 6 × 10−3 molecule site−1 s−1 which is comparable to that (1.6 × 10−2 molecule site−1 s−1) found for Cu/ZnO/Al2O3 catalysts at 50 bar [14, 20]. Post reaction analysis of the surface by TPD and XPS showed an almost full ML coverage of adsorbed formate species, identified by the coincident desorption of CO2 and H2 at 475 K, but the XPS analysis showed no adsorbed O atoms [42, 43]. The addition of CO up to 0.8 bar, by substituting some of the H2 while keeping the CO2 pressure fixed at 0.41 bar and the total pressure constant at 5.1 bar resulted in a doubling in the TOF at the highest CO pressure [43]. The increase in the TOF was linear with the increase in CO partial pressure. Post reaction analysis of the Cu(110) surface which had been exposed to the highest CO partial pressure in the CO/CO2/H2 (0.8:0.41:3.99) mixture showed two CO2 peaks, one at 400 K and the other at 475 K, in addition to a m/z = 28 peak at 380 K, which the authors state is “clearly not simply due to molecularly adsorbed CO” [43], suggesting that it might be CO stabilised by co-adsorbed formate. The authors finally conclude that, “CO has a weak positive influence on the net rate” [43], with no further expansion on the nature of its role.
The reverse water–gas shift (WGS) was observed to proceed in parallel with methanol synthesis [43]. Campbell and co-workers [26, 44, 45] agree with the general consensus that the shift reaction proceeds through a redox mechanism in which the surface of the Cu is first oxidised by H2O forming H2 in the gas phase and O(a). The role of the CO is to remove that O(a) forming CO2
Under industrial conditions extant during the WGS reaction (50 bar ~493 K, 3 % CO, 10 % CO2, 37 % H2 and 50 % H2O), the role of the CO therefore is to remove O(a) from the surface of the Cu under a large partial pressure of H2. The H2 obviously does not scavenge all of the adsorbed O(a). This is exactly what has been proposed for methanol synthesis. It is therefore inconsistent to suggest that O does not exist on the surface of the Cu under methanol synthesis conditions while it clearly does under shift reactions which are not dissimilar to methanol synthesis conditions.
Muhler and co-workers [46] also found no support for the role of adsorbed O atoms in methanol synthesis. Injecting pulses of CO into a CO2/H2 stream which was producing methanol, they observed no significant reaction of the CO to form CO2 and concluded that the surface was essentially Cu metal. The figure they showed, however, did reveal a sharp drop in the H2O which confirmed that CO was a more powerful reducing agent than H2, but no comment was made on this. Pulsing CO2 into a CO/H2 stream which was producing methanol at a steady state value of 100 ppm resulted in an instantaneous increase in methanol production. No induction period was observed as had been reported by Chinchen and co-workers [47].
Resolution of this apparent impasse was found in the work of Hinrichsen and co-workers [48, 49]. They found that the response of the Cu/ZnO/Al2O3 catalyst to flow changes depended critically on the pre-treatment which the catalyst was subjected to. Pre-treatment in a reducing atmosphere (CO/He [10:90], 473 K, 0.5 h) resulted in an instantaneous production of methanol on switching flows to CO/CO2/H2/He (10:4:72/14). Pre-treatment in CO2/He (4:96) stream resulted in an induction period of >1 h before steady state production of methanol was achieved. The authors concluded that the temperature programmed methods of collecting kinetic date in methanol synthesis “was limited due to a slow change of the state of the working Cu/ZnO/Al2O3 catalyst” [49].
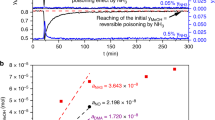
Two points should be made here. The first is that there is no question that methanol synthesis over Cu/ZnO/Al2O3 catalysts from CO2/H2 or CO/CO2/H2 feeds has a temperature dependent induction period. Figure 19 is the time dependence of the TON of methanol synthesis from a CO2/H2 (10:90, 101 kPa, 25 cm3 min−1) feed over a Cu/ZnO/Al2O3 (60:30:10) catalyst at the temperatures listed in the figure. (The reaction was carried out in a pyrex microreactor. The concentration of methanol in the product stream was determined by injecting 2 cm3 of the product stream into a gas chromatographic column with a flame ionization detector using a 6 port sample valve.) It shows an induction period of >200 min at 433 K which diminishes to about 150 min at 473 K. (The excursions in the TON at 453 K at >130 min, which were reproducibly observed, are outside the experimental error for the system and may result from the catalyst having two steady states.) The validity of Hinrichsen’s conclusion that temperature programmed methods for collecting kinetic data in methanol synthesis should not be used due to the slow change in the state of the working Cu/ZnO/Al2O3 catalyst [49] is corroborated by this data. The activation energy for the production of methanol from CO2/H2 calculated from Fig. 19 at 433, 443 and 453 K falls from 108 kJ mol−1 at 30 min, to 58 kJ mol−1 at 50 min, to 33 kJ mol−1at 180 min.

The time dependence of the TON of methanol from a CO2/H2 (10:90, 101 kPa, 25 cm3 min−1) over a Cu/ZnO/Al2O3 catalyst at the temperatures listed
Figure 20 is the time dependence of the TON of methanol synthesis from a CO/CO2/H2 (10:10:80, 101 kPa, 25 cm3 min−1) over the same Cu/ZnO/Al2O3 catalyst (60:30:10) at the temperatures listed in the figure. It too shows an induction period. In this case the induction period is longer, having a value of >200 min at 453 K which reduces to ~150 min at 463/473 K.

The time dependence of the TON of methanol synthesis from a CO/CO2/H2 (10:10:80, 101 kPa, 25 cm3 min−1) over a Cu/ZnO/Al2O3 catalyst at the temperatures tested
In addition, adding CO to the CO2/H2 feed causes the temperature at which equilibrium occurs to be increased from 453 K to between 463 and 473 K. (Evidence that equilibrium had been established is provided by the lowering of the steady state rate of methanol synthesis as the temperature is increased.) This can only occur if the addition of CO to the CO2/H2 feed causes the value of the equilibrium constant to increase. CO, far from having a “weak positive influence” is involved in the reaction.
The heat of reaction 8
is −49 kJ mol−1. If CO acts as a reducing agent (reaction 10)
so that the net reaction is reaction 11,
the heat of reaction increases to −90.2 kJ mol−1. This is additional evidence for CO acting as a reducing agent.
The second point to note is that there is no question that the surface of the Cu of an operating methanol synthesis catalyst has both O atoms and formate species adsorbed on to it. The removal of the O atoms after methanol synthesis by reaction with CO is entirely reproducible. No reason for that measured O atom coverage to exist other than as a result of the methanol synthesis reaction can be conjectured.
6 Surface Morphology of the Copper
An important side effect of the removal of the surface O atoms by reaction with CO at 473 K is the sudden evolution of H2 where ~40 % of the adsorbed O atoms have been removed. This H, which was attributed to evolving from subsurface Cu [30], has been shown to play an important role in determining the surface morphology of the Cu [50].The CO desorption spectrum obtained after dosing CO (10 % in He) at 77 K on to a Cu/ZnO/Al2O3 catalyst which had been reduced by the standard method and so had H atoms subsurface in the Cu, showed peaks at 168 and 200 K, corresponding to desorption from the Cu(110) face. Oxidation of this surface by reaction with CO2 at 213 K followed by removal of the adsorbed O atoms by reaction with CO at 473 K resulted in the loss of these CO desorption states. Loss of the subsurface H atoms had therefore resulted in the loss of the Cu(110) face [50].
An indication of the surface coverage population of the Cu(211) face of a Cu/ZnO/Al2O3 (60:30:10) catalyst after reduction was obtained by CO infrared spectroscopy [51]. The method of dosing (CO/He 10 %, 101 kPa, 25 cm3 min−1, 285 K, 15 min followed by flushing the system in He for 40 min) ensured that only CO bound to Cu with heats of adsorption >90 kJ mol−1 existed on the surface. A band at 2,100 cm−1 which Pritchard et al. [52] in single crystal studies attributed to CO bonded to Cu(211) was observed. This state was lost at ~350 K on temperature programming in He.
Oxidation of this surface by reaction with CO2 followed by reduction with CO and 473 K and then re-dosing the CO at 285 K as described above resulted in a threefold increase in the intensity of the 2,100 cm−1 band, i.e. a threefold increase in the surface population of the Cu(211) face. Figure 21 is a structural model of the Cu(211) surface. It can easily be seen how removal of O atoms from their bridged sites on the (2 × 1)O-Cu surface (Fig. 18) by reaction with CO will result in the development of the Cu(211) face, assuming the added Cu atoms remain in their fourfold hollow sites. Exposure of this catalyst to H2/He under normal reducing conditions (5 % H2, 101 kPa, 513 K, 16 h) resulted in the partial restoration of the original Cu(110) morphology [50].

Structural model of the Cu(211) surface
The over-riding importance of the morphology of the surface of the Cu in determining the reactivity of Cu was elegantly demonstrated by Hadden et al. [53] who showed that the TON of the Cu in the shift reaction could be changed by a factor of 2 as a result of minor changes in pH and temperature in the process of Na2CO3 precipitation of the Cu/Zn/Al nitrate precursor. The catalyst having the higher TON (catalyst A in Fig. 22) was precipitated at pH 6.8 and 70 °C while that with the TON which was half that of the higher one (catalyst B in Fig. 22) was precipitated at pH 6.5 and 65 °C. This could indicate that catalyst A had a higher surface population of the Cu(110) face than catalyst B. Previous studies have shown that a key elementary step in the forward shift reaction, namely the adsorption and decomposition of H2O is structure sensitive [45, 54–57] with the Cu(110) face being the most active, while the reverse WGS reaction in which the adsorption and decomposition of CO2 is the rate limiting step, here also the Cu(110) face has been shown to be the most active [26, 27].

The relationship between the copper metal surface area and WGS activity of two discrete groups of Cu/ZnO/Al2O3 catalysts
Joshihara and Campbell [43], however, have reported that methanol synthesis is only “mildly structure sensitive” with the apparent activation energies for the reaction being 69, 77 and 67 kJ mol−1 for Cu(100), Cu(poly) and Cu(110) respectively. They are unable to explain however why, under comparable conditions, the TON for methanol synthesis in Cu(110) is 30 times greater than that on Cu(100) and 2 times greater on Cu(poly).
It might be expected then that methanol synthesis would be structure sensitive since one of the key initial reactions in the mechanism is the adsorption and decomposition of CO2 to produce an adsorbed O(a) and CO, the adsorbed O(a) being the site for the adsorption of CO2 to form a carbonate species which is subsequently hydrogenated to a formate. This set of reactions explains the induction period for the establishment of the steady state rate of methanol synthesis shown in Figs. 19, 20. The slow establishment of an optimal coverage of adsorbed O(a) in the CO2/H2 reaction will be because the H2 will remove the O(a) in the reverse shift reaction (but not quantitatively). Similarly the even slower establishment of the optimal steady state oxygen atom coverage in a CO/CO2/H2 feed is because the CO as well as the H2 removes the adsorbed O(a). All of the Cu atoms appear to be involved in the reaction. This could be because the key intermediate in the reaction, the formate species, is mobile on the surface or because the O(a) ultimately causes a complete reconstruction of all the low index Cu faces to a (2 × 1)-O structure.
7 Conclusion
The production of methanol from CO/CO2/H2 mixtures over Cu/ZnO/Al2O3 catalysts is a well established and, to some extent, a mature process. Roughly 40 million tons/year are produced by it. While it is well established it is not without its on-going controversies. These include: (i) whether, at steady state, the Cu component of the catalyst is partially covered with O atoms as well as with formate species, (ii) if the reaction has an induction period and, if so, whether the induction period is brought about by a reconstruction of the Cu and (iii) if the reaction is structure sensitive, or if all of the Cu surface is active, deriving from the formate intermediate being mobile on the Cu. These on-going controversies are the grist of the future research mill.
References
Anilin B, Fabik S (1923) DR Patent 415,686; 441,443; 462,837
Anilin B, Fabik S (1923) US Patent 1,558,559; 1,569,775
Schmidt O, Ufers K (1928) D Patent 571,355; 571,356; 580,705
Natta G (1955) In: Emmett PH (ed) Catalysis, vol III. Reinhold Publishing Corp, New York, p 373
Davies P, Snowdon FF, Bridger GW, Hughes DO, Young PW (1966) UK Patent 101087
Andrew SPS, private communication
Luo Y-R (2007) Comprehensive handbook of chemical bond energies. CRC Press, Taylor Francis Group, Boca Raton
Herman RG, Klier K, Simmons GW, Finn BP, Bulko JB, Kobylinski TP (1979) J Catal 56:407
Bulka JB, Herman RG, Klier K, Simmons GW (1979) J Phys Chem 83:3118
Mehta S, Simmons GW, Klier K, Herman RG (1979) J Catal 57:339
Klier K (1982) Adv Catal 31:243
Klier K (1984) Appl Surf Sci 19:267
Kagan YuB, Liberov LG, Slivinsky EV, Lockev SM, Lin GI, Ya Rozovsky A, Bashkirov AN (1975) Dokl Acad Nauk SSSR 222:1093
Chinchen GC, Waugh KC, Whan DA (1986) Appl Catal 25:101
Chinchen GC, Denny PJ, Parker DG, Short GD, Spencer MS, Waugh KC, Whan DA (1984) Prepr Am Chem Soc Div Fuel Chem 29:178
Ertl G (1967) Surf Sci 6:208
Chinchen GC, Hay CM, Vandervell HD, Waugh KC (1987) J Catal 103:79
Waugh KC (1988) Appl Catal 43:315
Whan DA, private communication
Pan WX, Cao R, Roberts DL, Griffin GL (1988) J Catal 114:440
Muhler M, Nielsen LP, Tornqvist E, Clausen BS, Topsoe H (1992) Catal Lett 14:241
Topsoe N, Topsoe H (1999) Top Catal 8:267
Dell RM, Stone FS, Tiley PF (1953) Trans Faraday Soc 49:195
Haddon RA, Vandervell HD, Waugh KC, Webb G (1988) Catal Lett 1:27
Elliott AJ, Hadden RA, Tabatabaei J, Waugh KC, Zemicael FW (1995) J Catal 157:153
Nakamura J, Rodriguez JA, Campbell CT (1989) J Phys Condens Matter 1:SB149
Schneider T, Hirschwald W (1992) Catal Lett 14:192
Fu SS, Somorjai GA (1990) Surf Sci 237:87
Fu SS, Somorjai GA (1992) Surf Sci 262:68
Bowker M, Hadden RA, Houghton H, Hyland JNK, Waugh KC (1988) J Catal 109(2):263
Wachs IE, Madix RJ (1978) J Catal 53:208
Ying DHS, Madix RJ (1980) J Catal 61:48
Bowker M, Houghton H, Waugh KC (1981) J Chem Soc Faraday Trans 1 77:3023
Millar G, Rochester CH, Howe C, Waugh KC (1991) Mol Phys 76:833
Bailey S, Froment GF, Snoeck GW, Waugh KC (1995) Catal Lett 30:99
Taylor PA, Rasmussen PB, Chorkendorff I (1995) J Chem Soc Faraday Trans 91:1267
Sakakini BH, Tabatabaei J, Watson MJ, Waugh KC, Zemicael FW (1966) Faraday Discuss 105:369
Chinchen GC, Waugh KC (1986) J Catal 97:280
Rasmussen PB, Holmblad PM, Christoffersen H, Taylor PA, Chorkendorff I (1993) Surf Sci 287/288:71
Wintterlin J, Ertl G, Behn RJ (1990) Phys Rev Lett 64:1761
Besenbacher F, Springer PT, Raun L, Olsen L, Stensgaard I, Loegsgaard E (1994) Top Catal 1:325
Joshihara J, Parker SC, Schafer A, Campbell CT (1995) Catal Lett 31:313
Joshihara J, Campbell CT (1996) J Catal 161:776
Nakamura J, Campbell JM, Campbell CT (1990) J Chem Soc Faraday Trans 86:2725
Campbell CT, Daube KA (1987) J Catal 104:109
Muhler M, Tornqvist E, Nielsen LP, Clausen BS, Topsoe H (1994) Catal Lett 25:1
Chinchen GC, Spencer MS, Waugh KC, Whan DA (1987) J Chem Soc Faraday Trans 183:2193
Bielawa H, Kurtz M, Genger J, Hinrichsen O (2001) Ind Eng Chem Res 40:2793
Wilmer H, Hinrichsen O (2002) Catal Lett 82:117
Hadden RA, Sakakini BH, Tabatabaei J, Waugh KC (1997) Catal Lett 44:145
Sakakini BH, Tabatabaei J, Watson MJ, Waugh KC (2000) J Mol Catal A 162:297
Pritchard J, Catterick T, Gupta RG (1975) Surf Sci 53:1
Hadden RA, Lambert PJ, Ranson C (1995) Appl Catal A 122:L1
Spitzer A, Luth H (1985) Surf Sci 152/153:543
Au CT, Breza J, Roberts MW (1979) Chem Phys Lett 66:340
Spitzer A, Luth H (1982) Surf Sci 120:376
Heras JM, Viscido L (1988) Catal Rev Sci Eng 30(2):306
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Waugh, K.C. Methanol Synthesis. Catal Lett 142, 1153–1166 (2012). https://doi.org/10.1007/s10562-012-0905-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-012-0905-2