
Abstract
We demonstrate a technique to prepare high surface area ZnO powders that preferentially favor a plate-like morphology, exposing the \((0001)/(000\bar{1})\) facets. A solution-based synthetic route was used to decompose zinc acetate in the presence of amine and citrate ions to block the \((0001)/(000\bar{1})\) facets and favor growth from the pyramid planes. The ZnO platelets remained stable upon heating to 250 °C as evidenced by electron diffraction patterns. The high surface area (75 m2/g) and surface energetics of the (0001) plane make these powders suitable as supports for heterogeneous catalysts.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Zinc oxide (ZnO) is an important component of supported catalysts such as those for methanol synthesis [1] and steam reforming [2]. Karim et al. [3] have suggested that faceted ZnO powders play an important role in the activity of Pd/ZnO steam reforming catalysts. Of these facets, the polar (0001)-Zn and \((000\bar{1})\)-O surfaces are considered [4–7] the most active ZnO surfaces and are reportedly responsible for the entirety of methanol decomposition activity in a prepared catalyst [4]. Vohs and Barteau [6] have argued that the geometry of the (0001)-Zn surface allows for the exposure of active ion pair sites, while the oxygen terminated surface completely obscures those sites. In practical use, such a geometry is difficult to produce as commercially available powders consist of prisms that do not exhibit pronounced (0001) and \((000\bar{1})\) surfaces. These prisms primarily expose non-polar \((10\bar{1}0)\) and \((11\bar{2}0)\) facets with the only polar facets at the end caps as a result of the unbalanced Zn2+ or O2− terminated faces. A typically synthesized ZnO powder possesses an aspect ratio of 6, where the length of the prism along the [0001] direction is compared to the thickness of the prism in a direction perpendicular to the [0001]. This morphology implies that the majority of the exposed surface of a typical ZnO powder is inactive [8]. To improve the activity, ZnO supports with lower aspect ratios exposing more polar facets are desired.
Polar surfaces are typically prepared by polishing single-crystal wafers [6, 7, 9, 10] or grown by chemical vapor transport [11]. While the exposed plane is well-controlled, these wafers are limited by very small surface areas (tens of square millimeters) and therefore very few active sites are available. To minimize ambient contamination on these limited active sites, catalytic tests on single crystal wafers are conducted under ultrahigh vacuum (UHV) conditions which lend themselves to single turnover experiments. The temperature programmed desorption experiments conducted by Hyman et al. [7] demonstrated that CO was more weakly bound to the Pd/ZnO (0001) surface than to Pd/ZnO \((10\bar{1}0)\) and that the production of CO by MeOH decomposition was likewise greater on Pd/ZnO (0001). The authors suggested that this could be due to more facile PdZn alloy formation on the ZnO (0001) surface rather than the \((10\bar{1}0)\) surface. Such studies offer useful mechanistic insights into the behavior of catalysts, but these samples do not lend themselves to steady state reactivity measurements. Therefore, a high surface area ZnO (0001) powder is needed to take advantage of the molecular level insights into catalytic behavior provided by these surface science studies.
Many techniques exist to synthesize highly faceted ZnO powders, but the platelet surface area is low \((<15 \frac{m^2}{g})\) [12, 13] and thermal stability has not been demonstrated. Some examples of successful routes to powders exposing the (0001) plane include the research efforts of Xu et al. [14] when they prepared ZnO (0001) platelets from a vapor phase transport technique. While this was effective to produce ZnO (0001), it was not easily scalable to appropriately evaluate the catalytic behavior. Wang et al. [15] have reported an electrodeposition method for the production of ZnO (0001) films on ITO glass, but these tended to pit upon annealing. Such behavior is not unexpected as work by Staemmler et al. [16] has suggested that the (0001) and \((000\bar{1})\) surfaces reconstruct or balance the charge via hydrogenation of the surface.
Charge balance by ligands in solution is an alternate approach to stabilize the polar facets and solution techniques are amenable to large scale synthesis. Of the solution routes available, Yu et al. [17] have demonstrated a technique wherein a range of concentrations of zinc acetate [Zn(CH3COO)2·H2O, noted as Zn(OAc)2] and hexamethylenetetramine [C6H12N4, termed HMT] produced nanorods or prisms. The prism morphology was improved upon by Wang et al. [18], who produced highly faceted powders composed of low aspect ratio (0.33) (0001) plates using a mixture of water and ethanol as the solvent. A different technique by Tian et al. [19] used a sodium citrate [HOC(COONa)(CH2COONa)2·2H2O, denoted as Na3Cit] solution at room temperature to produce powders with (0001) facet exposure. The amount of Na3Cit used was very small in comparison to the amount of ZnO precursor, which suggested that the Na3Cit was a very aggressive capping agent and only required in small amounts. While these studies did not report the surface area, it is clear from the size of the prisms presented that the area was minimal (on the order of a few square microns).
This report details the preparation of high surface area (75 m2/g) ZnO(0001) powders capped with Na3Cit in the presence of HMT as a buffering agent. Commercial ZnO powders have surface areas of a few square meters per gram, but they do not expose the polar plane. Hence this work represents a significant improvement in the surface area of ZnO powders that predominately expose the polar plane.
2 Experimental
All chemicals used were ACS reagent grade obtained from Sigma-Aldrich. ZnO platelets were prepared by mixing 3.3 g (15 mmol) Zn(OAc)2 with 2.1 g (15 mmol) HMT and either 1 mg (0.0034 mmol), 10 mg (0.034 mmol) or 100 mg (0.34 mmol) of Na3Cit in 30 mL of distilled water in a three neck round bottom flask fitted with a reflux condenser open to the atmosphere on the benchtop. Prepared samples are described as ZnO–Na3Cit-1, -10 or -100 throughout this manuscript. An additional batch without Na3Cit was prepared using otherwise similar conditions and is referred to as ZnO–HMT. The solution was stirred for 5 min or until all reagents were completely dissolved and then heated to reflux temperature (∼90 °C) for 5 h. After cooling, the resultant precipitate was separated from the mother liquor by centrifugation and rinsed with ∼50 mL EtOH. The powder was allowed to air dry prior to characterization. Platelet shape was analyzed using images collected in a scanning electron microscope (SEM, Hitachi S5200) operated at 2 kV accelerating voltage. Transmission electron micrographs were acquired on a JEOL 2010F field emission gun transmission electron microscope (TEM) operated at 200 kV. Selected area diffractograms (SAED) were likewise collected on the JEOL 2010F using a Gatan ES500W camera. Powder X-ray diffractograms (XRD) were collected on a Scintag Pad V diffractometer with DataScan 4 software from MDI, Inc. for system automation and data collection. Cu–Kα radiation (40 kV, 35 mA) was used with a Bicron Scintillation detector with a pyrolitic graphite curved crystal monochromator. The scan settings used a 0.02 step size and a 10 s dwell time. Whole-pattern Reitveld refinement [20] was conducted on the collected patterns using GSAS [21] and EXPGUI [22]. Reference values for the ZnO profile were obtained from the American Mineralogist Crystal Structure Database [23]. Fourier-transform infrared spectroscopy (FT-IR) was conducted using an attenuated total reflectance (ATR, Smart Orbit) attachment on a Nicolet 6700 using 256 scans at a resolution of 2 per cm. Thermogravimetric analysis and differential thermal analysis (TGA/DTA) were performed on a TA SQT Q600 under flowing N2 at a ramp rate of 20 °C/min to 700 °C. The surface area was determined by the BET method [24] with a Micromeretics Gemini 2360 after outgassing at 120 °C in flowing N2 overnight. Additional pretreatments were conducted at 250 and 340 °C in flowing N2 to evaluate the stability of the plates at and above a commonly used temperature for methanol steam reforming [3].
3 Results and Discussion
3.1 Role of HMT on ZnO Morphology
Following the synthetic procedure outlined in Sect. 2, ZnO powders were synthesized in the presence of HMT to produce ZnO–HMT regular hexagonal prisms, as shown in Fig. 1. The exposed area of the polar (0001)/\((000\bar{1})\) planes was comparable to that of the \((10\bar{1}0)\) facet as determined by a visual inspection of the SEM images. The surface area was estimated at 1 m2/g using the average crystallite size found by inspection of the SEM images. BET analysis (Table 1) was unable to provide a reliable measurement as the value was below the minimum range of the instrument and could not be accurately determined. Since the surface area was undetectable at the lowest (120 °C) outgas temperature, further pretreatment was not undertaken.

Hexagonal prisms of the ZnO–HMT sample did not display any substantial preference towards the exposure of the (0001) facet
3.2 Role of Na3Cit on ZnO Morphology
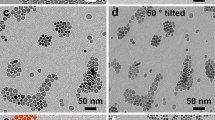
ZnO powders were also synthesized in the presence of HMT with 1, 10 or 100 mg of Na3Cit to produce ZnO–Na3Cit-1, -10 and -100 platelets. These samples had a plate-like morphology, and the plates became thinner with increasing Na3Cit content. The dependence upon Na3Cit concentration can be seen in Fig. 2, where the lowest concentration (ZnO–Na3Cit-1, Fig. 2a) produced a combination of thin plates and prisms. Increasing the concentration further (ZnO–Na3Cit-10, Fig. 2b and c) yielded plates which tended to stack regularly in the [0001] direction like a ream of paper, while the ZnO–Cit-100 plates were much more disordered (Fig. 2d). This was suspected to be a result of the differing degrees of surface charge compensated for by the capping agent. TGA and FTIR analysis of the Na3Cit plates revealed that the surfaces were capped in Na3Cit with no detectable HMT present. TGA/DTA analysis (Fig. 3) indicated that the HMT volatilized completely between 200 and 300 °C. In contrast, both the Na3Cit and the ZnO–Na3Cit-100 exhibited similar peaks near 150 and 315 °C. The close match suggests that the ZnO–Na3Cit-100 was coated with Na3Cit, thereby causing similar peaks to occur during TGA/DTA analysis. In addition, FT-IR spectroscopy (Fig. 4) confirmed the presence of the Na3Cit on the prepared powder. Combined, these results suggested that the Na3Cit was strongly bound to the ZnO surface with no evidence to indicate the presence of HMT. This behavior suggested that the HMT acted as a buffer in solution, while the Na3Cit was actively bound to the ionic surfaces, consistent with reports in the literature [17, 25]. Therefore the Na3Cit acted as a charge stabilizer and was observed in the product, while the HMT was not retained on the surface. As a result, the platelets synthesized with Na3Cit and HMT had a more thoroughly charge compensated surface and were able to grow much thinner (Fig. 2c and d) than the prisms produced with HMT alone (Fig. 2a).

SEM images of samples synthesized with Na3Cit (a) ZnO– Na3Cit-1, (b) ZnO–Na3Cit-10 (c)higher magnification view of image (b) to illustrate the stacking of ZnO sheets and (d) ZnO–Na3Cit-100 which shows the formation of increasingly thin platelets

TGA–DTA analysis showing thermal stability of pure HMT (top), Na3Cit (middle) and ZnO–Na3Cit-100 (bottom) to 400 °C

FTIR spectra showing similarity between the ZnO–Na3Cit-100 and pure solid Na3Cit, in contrast to the pure solid HMT
3.3 Characterization of ZnO Platelets
The above data indicated the Na3Cit concentration had a strong influence on the stacking of the platelets as well as the morphology. In order to determine the exposed facets, we studied the powders via XRD and electron diffraction in the TEM. Reitveld analysis of the XRD pattern (Fig. 5) indicated a substantial anisotropic broadening of the (0002) peak, which is a higher order reflection of the forbidden (0001) reflection. This peak broadening also substantially reduced the (0002) peak height. An additional set of peaks was identified as {[Zn2(C2H2N3O)(OH)3]·2H2O}n based upon a match to a similar copper compound in the ICSD database (card # 260453, [26]). This phase was found to comprise ∼20 wt% of the crystalline fraction of the sample by Reitveld analysis, which suggested that the material existed as a bulk contaminant in the powder rather than a monolayer adsorbed on the surface and did not contribute to the layered structure. The average plate thickness was found by XRD to be 25 nm, which is at the high end of the range (10–25 nm) measured from SEM images (Fig. 2c). Since the XRD average determined by the Scherrer equation is a volume average, the calculated value was weighted towards thicker plates. SEM measurements were also imprecise due to the error induced by measuring images of a tilted surface. The platelet thickness calculated from the BET surface area, as described in Eq. 1, was 7 nm.

XRD diffraction pattern showing ZnO–Na3Cit-10 (Open diamonds) with corresponding Reitveld refinement curve shown as a solid line. Major peaks corresponding to ZnO (Wurtzite, [23]) are noted by filled diamonds. The other set of peaks (filled triangles) fit the pattern for an organometallic phase (ICSD card # 260453)
While it was not possible to determine an exact aspect ratio due to the difficulty in measuring the width of the platelets, we estimate it to be on the order of 0.01 when considering the stacks of micron-sized platelets with 7 nm thickness.
In contrast, the ZnO–Na3Cit-100 sample showed randomly oriented platelets which did not form layered structures. Inspection of individual ZnO–Na3Cit-100 platelets via selected area electron diffraction (Fig. 6) confirmed exposure of the (0001) facet. The stacking of platelets in each sample strongly influenced the surface area and thermal stability. The surface area measurements (Table 1) for each sample correlated to the concentration of Na3Cit in the synthesis step. The ZnO–Na3Cit-10 sample demonstrated a surface area of 50 m2 per gram, and the surface area of the ZnO–Na3Cit-100 sample was nearly doubled (96 m2/g) (Fig. 7). The weight loss (by TGA, Fig. 8) of the ZnO–Na3Cit-100 sample was double that of the ZnO–Na3Cit-10.

SAED (inset) of an individual platelet of the ZnO–Na3Cit-100 sample after heating to 150 °C exposing the (0001) facet. Images processed with ImageJ [27]
3.4 Thermal Stability of ZnO Plates
Since these platelets were developed as novel supports for steam reforming catalysts, the thermal stability was tested by heating the samples to 250 °C. When heated, the surface area of the ZnO–Na3Cit-10 decreased, which is believed to be due to the stacked orientation of the platelets. The layered structure observed by SEM (Fig. 2c) placed each crystallite in close proximity to a neighboring platelet. Upon heating, the energetically unstable \((0001)/(000\bar{1})\) facets were in close contact and fused easily (Fig. 9a). Similar stacking behavior has been reported by Cao et al. [28] when growing ZnO platelets on Si substrates. In contrast, the ZnO–Na3Cit-100 sample was very disordered, and thus a greater void space existed between each platelet (Fig. 9b). Since the polar facets were not in immediate contact, the platelets could not condense like the ZnO–Na3Cit-10 sample. As a result, the sample retained a high surface area (75 m2/g) when heated to methanol steam reforming temperatures. The plates developed a mottled texture, as seen in Fig. 9. TEM images show that the plate had broken into individual crystallites, but these retained their orientation as seen from electron diffraction patterns in Fig. 7. These small crystallites also exhibited (0001) faces as shown by SAED (Fig. 7, inset). Reitveld analysis indicated that the average thickness after heating to 250 °C was 11 nm, which along with the BET (5 nm) and SEM measurements (20–30 nm) demonstrated that the platelets could survive moderate heat treatment. As noted previously, determination of plate thickness via SEM was difficult since it was not easy to find plates that were sitting edge-on. Further pretreatment at 340 °C caused additional loss in surface area. The presence of Na3Cit was found to have no effect on the platelet thermal stability. FTIR scans (Fig. 10) of post-treated ZnO–Na3Cit-100 samples indicated that some Na3Cit remained on the surface at 340 °C and thus removal of the ligand was not considered to be a factor in the break up of the plates into smaller crystallites. As such, the ligand is not necessary to maintain platelet stability and can be removed to avoid interference with subsequent catalytic activity.

Both the ZnO–Na3Cit-10 (a) and ZnO–Na3Cit-100 (b) samples maintained the (0001) facet upon heating to 250 °C, as shown by the SAED images (insets)

TGA–DTA analysis of ZnO–Na3Cit-10 and -100 samples show a significant difference in weight loss due to ligand removal

After heating to 250 °C, the ZnO–Na3Cit-10 (a) and ZnO–Na3Cit-100 (b) plates remained intact but showed signs of thermal damage

FTIR spectra of the ZnO–Na3Cit-100 sample as a function of pretreatment show that the sample retained some Na3Cit at each pretreatment temperature
4 Conclusion
We have demonstrated a simple solution-based synthetic technique that produced high surface area ZnO platelets with (0001) faces. The growth mechanisms via synthesis using the amine-based competing ion technique and citrate based poisoning method show that the competing ion effect provided only a weak interaction, while the poisoning method was a far more robust process. FTIR and TGA/DTA results indicated the HMT ligand was not bound to the ZnO after synthesis, thereby implying that it provided a weak interaction in solution to moderately favor (0001) facet growth. In contrast, the presence of Na3Cit produced a much thinner platelet. The thermal stability of the sample was found to be dependent on the relative proximity of the individual platelets. Stacked platelets tended to condense upon removal of the citrate ligand by heat treatment whereas plates that were separated from each other retained a high surface after thermal treatment. The stacked platelets lost surface area when they sintered to form thicker crystallites. In contrast, the disordered platelets did not have the nearest neighbor contact that the stacked platelets did, and thus remained distinct. Many of the deformed plates continued to expose the (0001) facet, which suggests that moderate heat treatments were not detrimental to the overall exposed facet. Studies to investigate the activity of supported catalysts using these polar powders are underway.
References
Vesborg PCK, Chorkendorff I, Knudsen I, Balmes O, Nerlov J, Molenbroek AM, Clausen BS, Helveg S (2009) J Catal 262(1):65. doi:10.1016/j.jcat.2008.11.028
Iwasa N, Takezawa N (2003). Top Catal V22(3):215. doi:10.1023/A:1023571819211
Karim AM, Conant T, Datye AK (2008) Phys Chem Chem Phys 10:5584. doi:10.1039/b800009c
Cheng WH, Akhter S, Kung HH (1983) J Catal 82(2):341. doi:10.1016/0021-9517(83)90200-2
Wilmer H, Kurtz M, Klementiev KV, Tkachenko OP, Grünert W, Hinrichsen O, Birkner A, Rabe S, Merz K, Driess M, Wöll C, Muhler M (2003) Phys Chem Chem Phys 5:4736. doi:10.1039/b304425d
Vohs J, Barteau M (1986) Surf Sci 176(1–2):91. doi:10.1016/0039-6028(86)90165-2
Hyman MP, Lebarbier VM, Wang Y, Datye AK, Vohs JM (2009) J Phys Chem C 113(17):7251. doi:10.1021/jp809934f
Bowker M, Houghton H, Waugh KC (1981) J Chem Soc Faraday Trans 1: Phys Chem Condens Phases 77:3023. doi:10.1039/F19817703023
Halevi B, Vohs JM (2005) J Phys Chem B 109(50):23976
Diebold U, Koplitz LV, Dulub O (2004) Appl Surf Sci 237(1–4):336
Dulub O, Boatner LA, Diebold U (2002) Surf Sci 519(3):201
Ada K, Gökgöz M, Önal M, Sarıkaya Y (2008) Powder Technol 181(3):285. doi:10.1016/j.powtec.2007.05.015
Li GR, Hu T, Pan GL, Yan TY, Gao XP, Zhu HY (2008) J Phys Chem C 112(31):11859. doi:10.1021/jp8038626
Xu CX, Sun XW, Dong ZL, Yu MB (2004) Appl Phys Lett 85(17):3878. doi:10.1063/1.1811380
Wang F, Liu R, Pan A, Cao L, Cheng K, Xue B, Wang G, Meng Q, Li J, Li Q, Wang Y, Wang T, Zou B (2007) Mater Lett 61(10):2000. doi:10.1016/j.matlet.2006.08.007
Staemmler V, Fink K, Meyer B, Marx D, Kunat M, Girol SG, Burghaus U, Wöll C (2003) Phys Rev Lett 90(10):106102/1
Yu Q, Yu C, Yang H, Fu W, Chang L, Xu J, Wei R, Li H, Zhu H, Li M, Zou G, Wang G, Shao C, Liu Y (2007) Inorg Chem 46(15):6204. doi:10.1021/ic070008a
Wang M, Hahn SH, Kim JS, Chung JS, Kim EJ, Koo KK (2008) J Cryst Growth 310(6):1213. doi:10.1016/j.jcrysgro.2008.01.001
Tian ZR, Voigt JA, Liu J, McKenzie B, McDermott MJ, Rodriguez MA, Konishi H, Xu H (2003) Nat Mater 2(12):821. doi:10.1038/nmat1014
Rietveld HM (1969) J Appl Crystallogr 2(2):65. doi:10.1107/S0021889869006558
Larson AC, Dreele RBV (2004) General structure analysis system (GSAS). Technical report, Los Alamos National Laboratory Report LAUR 86-748
Toby BH (2001) J Appl Crystallogr 34(2):210 doi:10.1107/S0021889801002242
Downs RT, Hall-Wallace M (2003) . Am Mineral 88:247
Brunauer S, Emmett PH, Teller E (1938) J Am Chem Soc 60(2):309
Cho S, Jang JW, Jung SH, Lee BR, Oh E, Lee KH (2009) Langmuir 25(6):3825. doi:10.1021/la804009g
Chippindale AM, Hibble SJ, Bilbé EJ (2009) Acta Crystallogr Sect C 65(7):i39. doi:10.1107/S0108270109020885. http://dx.doi.org/10.1107/S0108270109020885
Abràmoff MD, Magalhães PJ, Ram SJ (2004) Biophotonics Int 11(7):36
Cao X, Zeng H, Wang M, Xu X, Fang M, Ji S, Zhang L (2008) J Phys Chem C 112(14):5267. doi:10.1021/jp800499r
Acknowledgements
This work has been supported by the United States Department of Energy Office of Basic Energy Science, Division of Chemical Sciences under contract number DE-FG02-05ER15712 (University of New Mexico), and DE-AC0494AL85000 (Sandia National Laboratories), Division of Material Science. Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company for the United States Department of Energy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Burton, P.D., Peterson, E.J., Boyle, T.J. et al. Synthesis of High Surface Area ZnO(0001) Plates as Novel Oxide Supports for Heterogeneous Catalysts. Catal Lett 139, 26–32 (2010). https://doi.org/10.1007/s10562-010-0405-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-010-0405-1