
Abstract
A novel sulfonated carbon composite solid acid was successfully prepared by the pyrolysis of a polymer matrix impregnated with glucose followed by sulfonation. The title catalyst has higher acid site density, better esterification activity of both small and large free fatty acids (acetic acid and palmitic acid), and better reusability than the previously reported carbon-based catalyst prepared by sulfonating pyrolyzed sugar. This catalyst also exhibited higher esterification activity than tungstated zirconia (WZ) and Silica-Supported Nafion (Nafion®SAC-13). The higher activity of the sulfonated carbon composite solid acid catalyst was clearly due to the presence of a much higher acid site density than any of the other catalysts.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The transesterification of triglycerides (TGs) and esterification of free fatty acids (FFAs) are the two main reactions for converting vegetable oils and animal fats into biodiesel. Currently, most commercial processes used for biodiesel synthesis employ homogeneous catalysts to carry out these two reactions. Homogeneous catalysts, however, are corrosive, can be used only once and require energy intensive separation operations that lead to waste formation and environmental pollution. The use of heterogeneous catalysts to replace homogeneous ones could eliminate the problems associated with homogeneous catalysts and should allow the application of more environmentally friendly biodiesel synthesis protocols. In addition, using heterogeneous catalysts should enable the design of efficient continuous processes, improving the economics of biodiesel production [1–3]. In addition to the use in the esterification of FFAs in vegetable oils and animal fats, heterogeneous acid catalysts for biodiesel synthesis could in principle be employed to catalyze the simultaneous reactions of TGs and FFAs with low molecular alcohols in transesterification and esterification, respectively [3]. Among the heterogeneous acid catalysts studied to date for trans/esterification are zeolites [4, 5], MCM-41 [6, 7], tungstated zirconia [8–10], sulfated zirconia [11–14], Amberlyst-15 [15–17] and Nafion [18–22]. Some common problems with solid acid catalysts have been: low acid site concentrations, microporosity, hydrophilic character of catalyst surfaces, and active site leaching. High cost is also an obstacle for commercialization using many of the current heterogeneous acid catalysts being studied.
Recently, a new class of sulfonated carbons (C–SO3H) derived from the incomplete carbonization of simple sugars was reported to show excellent catalytic performance for the synthesis of biodiesel [23–27]. The sulfonated carbon catalysts showed better activities for trans/esterification than many of the other solid acids used for this purpose, including silica-supported Nafion, sulfonated zirconia (SZ), and niobic acid [23–26]. The sulfonated carbons have been suggested to be a flexible material composed of sulfonated polycyclic aromatic hydrocarbons. Depending on sulfonation conditions, the reported acid density of sulfonated carbons can be in the range of 0.48–1.74 mmol/g, a number higher than what has been reported for inorganic solid acids like SZ (0.1–0.5 mmol/g) [17], but lower than the acid density of commercially available strong acidic resins, such as Amberlyst-15 (4.7 mmol/g) [17]. Unlike sulfonic-acid resins, sulfonated polycyclic aromatic carbons should be more resistant to desulfonation [28, 29] as a result of the stabilization effect of the electron-withdrawing force that polycyclic aromatic hydrocarbons exert on bonded sulfonic groups. Hence, added stability is another appealing aspect of sulfonated carbon materials in addition to low cost.
We report the synthesis of a novel composite solid acid catalyst (P–C–SO3H), which shows higher acid site density, better fatty acid esterification activity, and better reusability than previously reported for sulfonated carbon catalysts (C–SO3H). Our main strategy for the synthesis of P–C–SO3H consisted of the sulfonation of a composite material formed through incomplete carbonization of hydrolyzed glucose supported in a porous copolymer. The as-prepared P–C–SO3H was characterized by elemental analysis, surface area analysis (BET), powder XRD, FTIR and TGA. Its catalytic activities were determined for the esterification of acetic acid and of palmitic acid with methanol, and were compared to the sulfonated carbon catalyst (C–SO3H), Nafion®SAC-13 (Nafion resin particles supported on a porous silica matrix [30]), and tungstated zirconia (WZ).
2 Experimental
Reagents including d-glucose (reagent grade, Aldrich), Amberlite XAD1180 (a porous polyaromatic styrene/divinylbenzene copolymer, Sigma–Aldrich), methanol (MeOH, 99.9%, Acros Organics), acetic acid (HAc, 99.7%, Aldrich), palmitic acid (HPa, hexadecanoic acid, Aldrich, 99%) were used without further purification. Commercial catalysts used in this work for comparison purposes, Nafion®SAC-13 and tungstated zirconia (WZ), were obtained from Sigma–Aldrich.
The composite catalyst P–C–SO3H was prepared by the pyrolysis of a polymer matrix impregnated with glucose and followed by sulfonation as shown in Scheme 1. The precursor of the composite catalyst P–C–SO3H was prepared by adding drop-wise an aqueous solution of glucose (1.2 g glucose, 3 mL deionized water) and a small amount of concentrated sulfuric acid (∼0.2 g) to pre-dried (100 °C air) Amberlite XAD1180 to incipient wetness. This mixture was dried at 100–120 °C overnight and then pyrolysized under dry N2 at 300 °C for 1 h.

Formation of C–SO3H and P–C–SO3H
The sugar catalyst (C–SO3H) precursor was obtained by the pyrolysis of glucose under dry N2 at 400 °C for 1 h [26]. All the precursors were then sulfonated using concentrated sulfuric acid (1 g solid/20 mL H2SO4) at 150–160 °C for 13 h under a dry N2 atmosphere. For comparison purposes, a resin-based catalyst (P–SO3H) prepared solely by the sulfonation of incompletely carbonized resin (pyrolysis of Amberlite XAD1180 under dry N2 at 300 °C for 1 h) without the presence of glucose was also synthesized. All the carbon-based catalysts were then washed with hot distilled water until no sulfate ions were detected in the wash water and dried at 100 °C. As-prepared P–C–SO3H and C–SO3H were black powders, while P–SO3H was a dark brownish powder.
The sulfur contents of the sulfonated carbon-based catalysts were determined by elemental analysis using ICP (Galbraith Laboratory, Knoxville, TN). BET surface areas were measured using N2 adsorption at −196 °C in a Micromeritics ASAP 2020. Prior to N2 adsorption, the catalyst samples were degassed for 3 h at 120 °C. IR spectra were recorded using a Nicolet Avatar 360 FTIR spectrometer equipped with a nitrogen-purged chamber and a DRIFTS attachment. Thermogravimetric analysis (TGA) was carried out in a TGA/SDTA 851 analyzer made by Mettler Toledo. A flow of air at 50 mL/min was employed and the temperature was ramped from room temperature to 900 °C at 5 °C/min for data collection. A Scintag XDS 2000 θ/θ powder X-ray diffractometer employing Cu Kα1/Kα2 (λ = 1.540592 and 1.544390 Å, respectively) radiation was used for the collection of X-ray diffraction patterns.
Liquid-phase esterification reactions were carried out under batch reaction conditions in a shaker reactor at an agitation rate of 225 rpm and 60 °C. Catalysts with the exception of WZ were dried at 100 °C for at least 2 h prior to reaction. WZ was calcined at 700 °C for 4 h prior to reaction. No pre-swelling was carried out before any of the reactions. Catalysts were added to the reaction vessels after reaction mixtures reached reaction temperature. A catalyst loading of 3 wt% was used for the esterification of acetic acid with methanol (the initial molar ratio of methanol-to-acid was 2). The esterification of a longer chain free fatty acid was carried out using an acid oil (usually contains 3–40% free fatty acid [31, 32]) by mixing 10 wt% palmitic acid and 90 wt% refined soybean oil. The initial molar ratio of methanol-to-oil was 6 and the catalyst loading was 2 wt% of the reaction mixture, following the parameters used by Marchetti for the esterification of oil with high amount of free fatty acids [29]. The transesterification of soybean oil was negligible under the experimental conditions used (60 °C and 1 h of reaction). Reaction conversions for the esterifications of acetic acid and palmitic acid were determined using 1H-NMR (JEOL ECX 300, 300.5 MHz) and acid titration [29, 33].
3 Results and Discussion
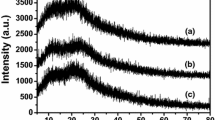
The exact structure of the composite material has not yet been precisely determined. Powder X-ray diffraction (PXRD) patterns (not shown) of P–C–SO3H and C–SO3H exhibited identical patterns with a broad peak centered around 2θ = 25°, characteristic of unordered carbon materials [34, 35]. As shown in Fig. 1, the diffuse reflectance FT-IR spectrum of the pre-dried (at 100 °C) polymer resin (P) exhibited characteristic peaks corresponding to aromatic C–H stretching (3,070–3,147 cm−1), aliphatic C–H stretching (2,931–2,998 cm−1), aromatic ring deformation (1,527–1,650 cm−1) and aromatic CH wag (755–971 cm−1). The composite material formed after pyrolysis but before sulfonation, P–C, showed a similar FT-IR spectrum with an additional peak centered around 1,700 cm−1 due to C=O stretching. Similar FTIR assignments have been made by other researchers for the analysis of chars of cellulose pyrolyzed at 450 °C [36], chlorogenic acid pyrolyzed at 250–750 °C [37], and pectin pyrolyzed at 250–550 °C [38]. From adsorption isotherms at −196 °C and BET calculations, it was estimated that dry (i.e., not swelled) P–C had a porous structure with about 70% of the surface area of the original resin matrix. P–C–SO3H, the composite material after sulfonation, had a broad band centered around 3,300 cm−1 due to OH stretching, with two shoulder peaks due to aromatic C–H stretching and aliphatic C–H stretching. The band around 1,666–1,882 cm−1 is stronger than that of its precursor, P–C, most likely due to the overlapping of C=O and S=O stretching bands. The IR spectrum of C–SO3H is similar to that of P–C–SO3H, except that stretching bands of aromatic CH and aliphatic CH3/CH2 groups are missing or overlapping with a broad band due to OH and adsorbed H2O from 2,600 to 3,500 cm−1. Both C–SO3H and P–C–SO3H had rather small surface areas in the dried form. Note that the low BET surface area in the dried form does not necessarily indicate limited access to active sites under reaction conditions as swelling of the catalysts can play a significant role in allowing full access of reagents to the interior sites in both materials.

FT-IR spectra of 100 °C pre-dried polymer matrix (P), composite material formed after pyrolysis (P–C), composite catalyst (P–C–SO3H) and sulfonated carbon catalyst (C–SO3H). BET surface area of each material was listed in the parentheses
The two pyrolyzed carbon-based catalysts showed similar thermal behaviors (Fig. 2) when heated in air except that C–SO3H lost more weight due to water desorption as the temperature increased from room temperature to 100 °C. For the temperature range of 100–240 °C, the TGA plot of P–C–SO3H exhibited a plateau with a much slower rate of weight loss, followed by a rapid weight loss up to 460 °C at which time no more mass was left.

TGA of C–SO3H and P–C–SO3H in air
The sulfonic acid site densities estimated by elemental analysis were used to compare the acid site densities of the carbon-based catalysts since only the –SO3H groups have sufficient acidity to contribute significantly to the reaction [26]. The sulfonic acid site density of P–C–SO3H calculated from elemental sulfur analysis (assuming that all S atoms in the catalyst are in the –SO3H form [23, 25]) was 2.42 mmol H+/g, about 3.6 times higher than that of C–SO3H, as shown in Table 1. Table 1 also compares the catalytic properties for the synthesis of biodiesel using esterification with methanol (MeOH) of a model free fatty acid (FFA) [acetic acid (HAc)] and a higher molecular weight FFA, palmitic acid (HPa, 10 wt% in soybean oil). To provide a better comparison of the catalytic performance of this catalyst, the conversions during 1-h reactions using two commercial acid catalysts, Nafion®SAC-13 and WZ, under the same reaction conditions are also shown in Table 1. It is obvious that P–C–SO3H exhibited the highest activity on a weight basis for both reactions. The 1-h conversions of HAc and HPa using P–C–SO3H as the catalyst were 43% and 68% higher, respectively, in relative terms than those obtained using C–SO3H, which exhibited superior catalytic performances compared to both Nafion®SAC-13 and WZ.
Multiple esterification reaction cycles were carried out to examine the deactivation of the composite catalyst. In these experiments, the catalyst samples were recovered after a particular cycle by simply decanting the reaction solution and drying the used catalysts at 100 °C for 2 h between cycles. Figure 3 shows that no significant deactivation was observed for P–C–SO3H during six consecutive esterification cycles of HAc or HPa with MeOH at 60 °C. Elemental analysis also showed negligible change in the sulfur content of P–C–SO3H. C–SO3H on the other hand, lost about 10% of the activity during three reaction cycles of acetic acid esterification with methanol (Fig. 4). A polymer-based catalyst prepared solely by the sulfonation of the incompletely carbonized resin (P–SO3H) without the presence of glucose showed comparable initial activity for the esterification of acetic acid at 60 °C but lost about 7% of its activity during each reaction cycle (Fig. 4). The deactivation characteristics of P–SO3H were similar to those of other acidic resins with sulfonic acid groups [28, 29], deactivating due to desulfonation. Thus, the stable catalytic performance for P–C–SO3H must be related to the integrated carbon-polymeric matrix precursor.

P–C–SO3H activity during six reaction cycles of the esterification at 60 °C of (a) HAc with MeOH, catalyst loading 3 wt%, 1 h reaction, CMeOH,0/CHAc,0 = 2; (b) HPa (in soybean oil, 10 wt%) with methanol, catalyst loading 2 wt%, 1 h reaction, CMeOH,0/Coil,0 = 6. The catalyst was retrieved between cycles by decanting the reaction mixtures

C–SO3H and P–SO3H (sulfonated pyrolyzed polymer matrix) activities during three reaction cycles of the esterification of HAc with MeOH at 60 °C (reaction conditions same as in Table 1 in text)
The deactivation mechanism for C–SO3H at 60 °C as a result of –SO3H loss has been discussed in another study [26]. Basically, the activity decrease of the C–SO3H catalysts in the initial reaction cycles is due to the leaching of sulfonated polycyclic aromatic hydrocarbons. Thus, P–C–SO3H exhibits higher catalytic activity and better stability than C–SO3H or P–SO3H at 60 °C.
Additional research is still required regarding the effect of each major preparation step (impregnation, carbonization, and sulfonation) on the structure and properties of the composite catalyst P–C–SO3H. It is, however, in light of the apparent similarities of both P–C–SO3H and C–SO3H, conceivable that the resultant composite catalyst may have a flexible local structure similar to that of C–SO3H [23–25]. This may be the reason why the original porous framework of the resin and its high surface area even after pyrolysis are no longer preserved after sulfonation and drying. The flexibility of the “soft carbon” (formed by the pyrolysis of sugar) on the surface and the high density of acid sites produced during sulfonation must favor a closed structure in the dry state as a result of strong hydrogen bonding that only unfolds upon swelling in a liquid. In our previous study of sulfonated catalysts (C–SO3H) [26], an induction period was observed during the first three consecutive reaction cycles when large reactants such as caprylic acid were used, indicating that catalyst swelling was playing a role in initial catalyst activity. In this study, no significant increase in conversion was observed during the consecutive cycling esterification reactions of palmitic acid with methanol using the composite catalyst, suggesting that the effect of swelling was minimized. Thus, P–C–SO3H, a flexible polymeric framework decorated with a layer of sulfonated polycyclic aromatic hydrocarbon, apparently can quickly swell in a liquid phase, enabling reactants, like palmitic acid, to access active sites freely. Because the polycyclic aromatic moieties may be more evenly spread along the resin framework, there should be more available sites for sulfonation, leading to the higher sulfonic acid density of P–C–SO3H compared to that of C–SO3H. Furthermore, several factors may have favored the stability of P–C–SO3H versus C–SO3H, e.g., the use of a structure-directing agent (the resin support) during its preparation, and the existence of favorable bonding interaction between the carbon moieties from the “soft carbon layer” and the polymer framework.
4 Conclusions
In conclusion, a novel solid acid catalyst showing high performance in liquid-phase esterification has been prepared by sulfonating a composite material formed by incomplete carbonization of hydrolyzed glucose supported on a porous polymeric resin. The resulting strong solid acid likely consists of a flexible carbon-based framework decorated with highly dispersed polycyclic aromatic hydrocarbons containing sulfonic acid groups. Choosing a suitable initial matrix is key in preparing catalysts with high performance. For instance, our investigations have shown severe leaching of polycyclic polyaromatic hydrocarbons following reactions using porous silica materials as matrices for sulfonated-carbon catalyst preparation. We anticipate that the activity and stability of the composite catalyst could be further improved by optimizing catalyst composition and preparation.
References
Huber GW, Iborra S, Corma A (2006) Chem Rev 106:4044
Lotero E, Goodwin JG Jr, Bruce DA, Suwannakarn K, Liu Y, Lopez DE (2006) Catalysis 19:41
Lotero E, Liu Y, Lopez DE, Suwannakarn K, Bruce DA, Goodwin JG Jr (2005) Ind Eng Chem Res 44:5353
Corma A (1997) Chem Rev 97:2373
Machado MD, Perez-Pariente J, Sastre E, Cardoso D, de Guerenu AM (2000) Appl Catal A-Gen 203:321
Perez-Pariente J, Diaz I, Mohino F, Sastre E (2003) Appl Catal A-Gen 254:173
Barrault J, Bancquart S, Pouilloux Y (2004) C R Chimie 7:593
Furuta S, Matsuhashi H, Arata K (2004) Catal Commun 5:721
Ramu S, Lingaiah N, Devi BLAP, Prasad RBN, Suryanarayana I, Prasad PSS (2004) Appl Catal A-Gen 276:163
López DE, Suwannakarn K, Goodwin JG Jr, Bruce DA (2007) J Catal 247:43
Yadav GD, Murkute AD (2004) J Catal 224:218
Omota F, Dimian AC, Bliek A (2003) Chem Eng Sci 58:3175
Corma A, Garcia H (1997) Catal Today 38:257
Jitputti J, Kitiyanan B, Rangsunvigit P, Bunyakiat K, Attanatho L, Jenvanitpanjakul P (2006) Chem Eng J 116:61
Bozek-Winkler E, Gmehling J (2006) Ind Eng Chem Res 45:6648
Stroehlein G, Assuncao Y, Dube N, Bardow A, Mazzotti M, Morbidelli M (2006) Chem Eng Sci 61:5296
López DE, Goodwin JG Jr, Bruce DA, Lotero E (2005) Appl Catal A-Gen 295:97
Heidekum A, Harmer MA, Hoelderich WF (1999) J Catal 181:217
Alvaro M, Corma A, Das D, Fornes V, Garcia H (2005) J Catal 231:48
Liu Y, Lotero E, Goodwin JG Jr (2006) J Catal 242:278
Liu Y, Lotero E, Goodwin JG Jr (2006) J Catal 243:221
López DE, Goodwin JG Jr, Bruce DA (2007) J Catal 245:379
Toda M, Takagaki A, Okamura M, Kondo JN, Hayashi S, Domen K, Hara M (2005) Nature 438:178
Takagaki A, Toda M, Okamura M, Kondo JN, Hayashi S, Domen K, Hara M (2006) Catal Today 116:157
Okamura M, Takagaki A, Toda M, Kondo JN, Domen K, Tatsumi T, Hara M, Hayashi S (2006) Chem Mater 18:3039
Mo X, López D, Suwannakarn K, Liu Y, Lotero E, Goodwin JG Jr, Lu C (2008) J Catal 254:332
Zong M-H, Duan Z-Q, Lou W-Y, Smith TJ, Wu H (2007) Green Chem 9:434
Petrus L, Stamhuis EJ, Joosten GEH (1981) Ind Eng Chem Prod Res Dev 20:366
Marchetti JM, Miguel VU, Errazu AF (2007) Fuel 86:906
Harmer MA, Farneth WE, Sun Q (1996) J Am Chem Soc 118:7708
Srivastava A, Prasad R (2000) Renew Sust Energy Rev 4:111
Miao XL, Wu QY (2006) Bioresour Technol 97:841
Morgenstern M, Cline J, Meyer S, Cataldo S (2006) Energy Fuels 20:1350
Manocha LM, Bhatt H, Manocha SM (1996) Carbon 34:841
Tsubouchi N, Xu C, Ohtsuka Y (2003) Energy Fuels 17:1119
Mok WSL, Antal MJ Jr, Szabo P, Varhegyi G, Zelei B (1992) Ind Eng Chem Res 31:1162
Sharma RK, Hajaligol MR, Smith PAM, Wooten JB, Baliga V (2000) Energy Fuels 14:1083
Sharma RK, Wooten JB, Baliga VL, Hajaligol MR (2001) Fuel 80:1825
Acknowledgments
This research was funded by the US Department of Agriculture (Award No 68-3A75-3-147). The authors would like to thank Prof. S.-J. Hwu and Prof. D. A. Bruce for their assistance in the catalyst preparation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mo, X., Lotero, E., Lu, C. et al. A Novel Sulfonated Carbon Composite Solid Acid Catalyst for Biodiesel Synthesis. Catal Lett 123, 1–6 (2008). https://doi.org/10.1007/s10562-008-9456-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-008-9456-y