
Abstract
Mesenchymal stem cells (MSCs) are an accepted candidate for cell-based therapy of multiple diseases. The interest in MSCs and their possible application in cell therapy have resulted in a better understanding of the basic biology of these cells. Recently, like aggregation and transforming growth factor beta (TGFβ) delivery, hypoxia has been indicated as crucial for complete chondrogenesis. The aim of this study was to test different culture conditions for directing stem cell differentiation into the chondrogenic lineage in vitro by testing different TGFβ superfamily members into the culture media under normoxic conditions. All chondrogenic culture conditions used allowed the differentiation of bone marrow-MSCs (BM-MSCs) into chondrogenic lineage. Chondrogenic induction capacity depended on the growth factor added to the culture media. In particular, the chondrogenic culture condition that better induced chondrogenesis was the medium that included the combination of three growth factors: bone morphogenetic protein-2 (BMP-2), BMP-7 and TGFβ-3. In this culture media, differentiated cells showed the highest levels expression of two markers of chondrogenesis, SOX9 and COL2A1, compared to the control points (p < 0.05, two-tailed t test). In our experimental conditions, the combination of BMP-2, BMP-7 and TGFβ-3 was the most effective in promoting chondrogenesis of BM-MSCs. These results underline the importance of determining in each experimental design the best protocol for in vitro directing stem cell differentiation into the chondrogenic lineage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stem cells (MSCs) are undifferentiated, nonhematopoietic, multipotent adult stem cells that have the capability to renew themselves and differentiate into different cell types (fat, bone, cartilage and hematopoietic supporting stroma) as described abundantly in literature (Ryden et al. 2003; Pittenger, 2008; Hombach-Klonisch et al. 2008; Diaz-Prado et al. 2011).
The most commonly used tissue source for the isolation of MSCs is the bone marrow as discussed by different authors (Conget and Minguell, 1999; Pittenger et al. 1999). Three minimal criteria, established by the International Society for Cellular Therapy in 2006 (Dominici et al. 2006), defines bone marrow-MSCs (BM-MSCs): adherence to plastic, specific surface antigen expression and multipotent differentiation potential. BM-MSCs represent a promising cell resource for tissue engineering and cell-based therapies.
The use of autologous or allogenic stem cells is suggested by Jung et al. (2009) and coauthors as an alternative therapeutic approach for treatment of cartilage defects . Cartilage tissue engineering has the potential to provide cartilaginous constructs capable of restoring the normal function of articular cartilage as described by Buckwalter and Mankin (1998). To improve the repair of cartilage by tissue engineering, several cell treatments have been developed with the aim of forming a repair tissue with structural, biochemical, and functional characteristics equivalent to those of natural articular cartilage as described by Díaz Prado et al. (2011).
The use of differentiated chondrogenic progenitors has to be chosen on the use of undifferentiated stem cells. Chondrogenesis is the process by which MSCs differentiate towards chondrocyte, as resumed by Goldring et al. (2006). Different protocols allow the induction of BM-MSCs chondrogenic differentiation. To mimic mesenchymal condensation and cell–cell interactions, MSCs are cultured in high density aggregates as described in different papers (Cicione et al. 2010; DeLise et al. 2000; Johnstone et al. 1998). Derfoul reported the necessity of culture MSCs in medium containing dexamethasone to induce chondrogenic differentiation (Derfoul et al. 2006) and Tuli the necessity to add growth factors belonging to the TGFβ superfamily that constitute the earliest signals in chondrogenic condensation (Tuli et al. 2003).
Several growth factors are known to influence the anabolic and catabolic processes of chondrocytes. Therefore, a number of these growth factors have been used in cartilage tissue engineering studies in vitro to promote the chondrogenic phenotype, to stimulate extracellular matrix production and to promote chondrogenesis of MSCs, as reviewed by Kock recently (Kock et al. 2012). Among others, TGFβ-3, BMP-2 and osteogenic protein 1 (OP-1 or BMP-7) stimulate the synthesis of proteoglycans and collagens matrix components as described by Spagnoli (2008) and are necessary for different steps during chondrogenesis as shown by Pizette and Niswander (2000).
A number of studies (Blaney Davidson et al. 2007; Bobick et al. 2009; Yoon and Lyons, 2004) have demonstrated that TGFβ superfamily polypeptides (TGFβs and BMPs) induce the expression of chondrocyte specific genes, such as Collagen type 2 and SOX9, suggesting their critical role of signaling in chondrogenesis.
In the last years, many studies have been focused on finding better culture conditions to differentiate MSCs towards mature chondrocytes. The use of chondrocyte-conditioned medium proposed by Hwang et al. (2008) or the co-culture of MSCs with chondrocytes described by Chen et al. (2009) have been described to be able to drive MSCs chondrogenic differentiation.
In this study we examined the capacity of three different growth factors (BMP-2, BMP-7 and TGFβ-3), alone or in combinations, to induce chondrogenic differentiation in normoxic conditions. We defined the optimal conditions, between the stimuli used, to obtain chondrogenic differentiation of BM-MSCs aggregate cultures by the use of histological, immunohistochemical analysis and real-time (qPCR) technique. We determined the expression levels of different genes involved in chondrogenesis corresponding to the mature chondrocyte phenotype (COL2A1, SOX9 and aggrecan).
Materials and methods
Isolation and culture of MSCs
The BM samples used to isolate MSCs were obtained from three patients (mean age 69 years, range 55–82 years) undergoing total hip replacement. The donors were not selected; the samples submitted were processed as they arrived at the laboratory. This study was approved by the institutional review board, and informed consent was obtained from all subjects in the study.
BM-derived mesenchymal stem cells (BM-MSCs) were isolated by washing the BM with 50 mL of Dulbecco’s modified Eagle’s medium (DMEM), 10 % fetal bovine serum (FBS) with a gauge needle (BD Microbalance™ 3, Oxford, Ireland) and a 20 mL syringe (Kendall Monoject Syringe, Istambul, Turkey) as previously described (Hermida-Gomez et al. 2011). The resulting cell suspension was filtered through a sterile 40 µm cell strainer and centrifuged at 300xg for 8 min. Isolated BM cells were cultured in monolayer in DMEM, 20 % FBS, 1 % penicillin and streptomycin (P/S) (all from Sigma-Aldrich, St. Louis, MO, USA) until 80 % confluent. Pre-plating for 15 min in the first two passages eliminated any fibroblasts remaining in the culture as suggested by Richler and Yaffe (1970).
The cells were cultured and expanded in monolayer until the beginning of the differentiations experiments at 37 °C in a humidified atmosphere containing 5 % CO2. At this point, when cells reached 80 % of confluence, they were trypsinized, washed and submitted both to phenotypical analysis and to differentiation experiments.
Phenotypic characterization using flow cytometry
At the 3rd passage, after culture expansion, the cells were trypsinized, washed and analyzed by flow cytometry for hematopoietic and MSCs markers as described before (Cicione et al. 2013). The antibodies listed in supplementary Table 1 were used for these experiments. A minimum of 25,000 cell events per assay were acquired on a FACsCalibur flow cytometer (BD Biosciences, Madrid, Spain). Data were analyzed using Cell Quest software (BD Biosciences) and the results were expressed as positive percentage.
Multipotential characterization
At 3rd passage the cells were differentiated towards three different lineages (adipocyte, osteoblast and chondrocyte) in humidified atmosphere with 5 % CO2. The culture conditions used to induce each differentiation are described in the following paragraphs.
Adipogenic and osteogenic differentiation
At 3rd passage BM-MSCs were detached using trypsin–EDTA (Sigma-Aldrich, St. Louis, MO, USA), seeded at 1.5 × 105 cells/cm2 in a chamber slide (BD Falcon, France) and cultured in growth medium until confluence in normoxic conditions. Adipogenesis was induced by culturing for 3 weeks in hMSC Commercial Adipogenic Differentiation Medium (Lonza, Biowhittaker, Belgium), following the manufacturer’s instructions. Osteogenesis was induced by culture for 3 weeks in normoxic conditions in hMSC Commercial Osteogenic Differentiation Medium (Lonza, Biowhittaker, Belgium), following the manufacturer’s instructions. Each differentiation point was compared with a control point that corresponded to cells cultured for the same period of time with DMEM and 20 % FBS. Differentiation was assessed trough histological and real-time PCR (qPCR) techniques.
Chondrogenic differentiation
Chondrogenesis was assessed using the micropellet formation (2.5 × 105 cells) technique first described by Johnstone et al. (1998), with some modifications. BM-derived cells from the third passage were detached using trypsin–EDTA and centrifuged at 300g for 10 min. The resulting pellet was treated for 14 days to induce chondrogenic differentiation. The medium used for the first 2 days contained DMEM with 15 % FBS, supplemented with 5 mg/mL ascorbic acid (AA), 1/1,000 monotioglycerol (MTG), and 1 % P/S to promote chondroprogenitor condensation. This medium was replaced by DMEM with 15 % knockout (KO) serum (Gibco, Madrid, Spain), 1 % P/S and supplemented with 5 mg/mL AA, 10 µM dexamethasone, 6 µg/mL transferrin, 1 × 10−7 M retinoic acid, and the use of three different growth factors alone or in combination: TGFβ-3 1 ng/mL (Prospec-Tany Technogene Ltd., Rehovot, Israel); TGFβ-3 10 ng/mL; BMP-2 10 ng/mL (Prospec-Tany Technogene Ltd., Rehovot, Israel); BMP-7 10 ng/mL (Prospec-Tany Technogene Ltd., Rehovot, Israel); TGFβ-3 1 ng/mL + BMP-2 10 ng/mL + BMP-7 10 ng/mL. All the differentiation points were compared to cells grown in DMEM with 15 % KO serum. The culture medium was changed every 3–4 days. After 14 days, cell aggregates were directly frozen for RNA extraction or embedded in Tissue-Tech OCT compound (Sakura Finetek) and frozen for histological and immunohistochemical techniques. The presence of hyaline cartilage-characteristic molecules, such as type II collagen and proteoglycans, were detected by histological, immunohistochemical and qPCR techniques as described below.
Histologic analyses
For adipogenesis evaluation, differentiation was confirmed by detection of cytoplasmic lipid droplets by Oil Red O staining after cell fixation in 4 % paraformaldehyde. Quantification of Oil Red O-positive cells was performed using analiSIS1 software (version D; Olympus, Germany).
For osteogenesis evaluation, differentiation was analyzed by Alizarin Red staining after cell fixation in 4 % paraformaldehyde, to assess the presence of calcium deposits. Quantification of Alizarin Red-positive cells was performed using analiSIS1 software (version D; Olympus, Germany).
For chondrogenesis evaluation, 4 µm-thick frozen sections of aggregates were stained with hematoxylin and eosin (HE), Masson´s trichrome (MT), toluidine blue (TB) and safranin O (SaO) for proteoglycans and collagens.
Immunohistochemical analyses
For chondrogenesis evaluation, 4 µm-thick frozen sections were incubated with primary antibodies to detect the presence of types I (Abcam, Cambridge, UK), II (Chemicon, Australia) and X (Sigma Aldrich Quimica, Spain) collagens and metalloproteinase 13 (Thermo Fisher Scientific, Spain) (supplementary table 2). The peroxidase/DAB ChemMateTM DAKO EnVision™ detection kit (Dako, Barcelona, Spain) was used to determine antigen–antibody interactions. Negative staining controls were achieved by omitting the primary monoclonal antibody. Samples were visualized using an optical microscope. Quantification of the immunostaining for Col II was performed using analiSIS1 software (version D; Olympus, Germany).
RNA isolation, cDNA (complementary DNA) synthesis and reverse transcription-PCR analysis (qPCR)
Isolation of total RNA from cell cultures was accomplished using Trizol Reagent (Invitrogen, Barcelona, Spain), following the manufacturer´s protocol as described before (Cicione et al. 2013). RNA was assessed for quantity at 260 nm using a NanoDrop™ spectrophotometer (Thermo Scientific, Madrid, Spain). The A260/A280 ratio was calculated to assess quality and purity. Total RNA (1 µg) was further processed or stored at -80°C until used (Table 1).
Total RNA underwent DNase digestion (Fermentas, Spain) for complete removal of DNA contamination. Subsequently, the reverse-transcription reaction was performed on 1 µg of total RNA using SuperScript™ First-Strand Synthesis System for RT-PCR (Invitrogen™, Spain), following the manufacturer instructions as described before (Cicione et al. 2010). Samples were stored at −20 °C before the amplification of target cDNAs. Positive and negative controls were included in each experiment.
Real-time PCR analysis (qPCR) was performed on a LightCycler® 480 Instrument (Roche, Mannheim, Germany) to study mRNA expression of the genes implicated in chondrogenesis (SOX9, aggrecan, COL1A1 and COL2A1), osteogenesis (ALP, OP) and adipogenesis (FABP4, APM1, and LPL). For this purpose the primers shown in Table 1 were used.
The primers were purchased from Roche (Mannheim, Germany). The TATA box binding protein gene (TBP) was used as the internal control housekeeping gene to normalize the amount of target cDNA.
Data analysis was performed using LightCycler 480 Relative Quantification software (Roche). Relative levels of expression were calculated by the 2−ΔΔCt method. Each assay was done at least in triplicate and included marker-positive and marker-negative controls and reagent with no template controls. Each data was normalized against the housekeeping gene. For each gene expression, we assigned the value 1 to the lowest level of expression and the values were measured as relative expression levels (REL).
RNA extraction, reverse transcription-PCR assay setup and post reverse transcription-PCR product analysis were carried out in separate dedicated rooms to prevent cross-contamination.
Statistical analysis
Each experiment was repeated at least three times. The statistical significance of the differences between mean values was determined using a two-tailed t test; p < 0.05 was considered significant. Results are expressed as the mean ± standard error (mean ± SE) and mean ± standard deviation (mean ± SD)
Results
Isolation of BM-MSCs and characterization of cultured expanded cells
Spindle-shaped bipolar cells attached to the flask were observed at the first medium change at 48 h of culture. We expanded, differentiated and analyzed the surface antigens expression and multipotentiality of the cells isolated from three donors.
Phenotypic analysis
At the third passage, at the beginning of the differentiation experiments, the BM-MSCs were characterized according to the expression of surface markers using flow cytometry. Table 2 shows data for age, gender and surface marker expressions obtained doing the mean value between the three BM-MSCs donors. The cells displayed very similar expression of surface antigen expression and were negative for the expression of CD34, CD45 and STRO-1. In the table, each antigen expression value is expressed as positive percentage and the range of its expression is reported. CD73 and CD44 antigens were co-expressed at 94 % ± 0.05. CD105 was expressed at 96 % ± 0.12 when alone and co-expressed with CD106 at 24 % ± 0.14. CD166 was expressed at 92 % ± 0.06. The percentage of CD90 expression was around 95 % ± 0.02 and of CD29 was around 94 % ± 0.03. Our cultured cells expressed CD117 at 23 % ± 0.38 and SSEA4 at 83 % ± 0.09.
Multi-differentiation potential
We induced adipogenic, osteogenic and chondrogenic differentiations under normoxic conditions, as described in the materials and methods part, in order to confirm the multi-potentiality of our cultured expanded cells.
Adipogenic and osteogenic differentiation
After 21 days of culture with the appropriate differentiation medium or with DMEM plus 20 % FBS serum for the control points, the cells were stained with Oil Red O for lipid droplets to evaluate adipogenic differentiation and with Alizarin Red for calcium deposits to evaluate osteogenic differentiation. All the controls resulted nearly almost negative for staining, showing the absence of lipid droplets and calcium deposits. The cells grown in differentiation medium were positively stained (Fig. 1a), indicating differentiation. In the adipogenic and in the osteogenic differentiation, the percentage of cells with positive Oil Red O staining and Alizarin Red staining was respectively more than 40 and 60 times higher than in the control medium (p value <0.05, two-tailed t test) (Fig. 1b). To confirm this result, total RNA was extracted from cells treated in the same way, reverse transcribed into cDNA and the resulting cDNA was used as template for subsequent qPCR amplification. We measured the expression levels of LPL, APM1 and FABP4 marker genes for adipogenic differentiation and of OP and ALP marker genes for osteogenic differentiation (Fig. 1c). In each experiment, each gene expression level of cells treated with the differentiation medium was compared to cells cultured in DMEM with 20 % FBS (control points). Cells treated with adipogenic medium showed higher levels of LPL, APM1 and FABP4 expressions compared to the control cells (4534.31; 3404.75 and 13369.58 times, respectively) and were statistically significant (p value <0.005, two-tailed t test). Cells treated with osteogenic medium showed higher levels of OP and ALP expressions compared to the control cells (27.67 and 57.28 times, respectively) and were statistically significant (p value <0.05, two-tailed t test). These results confirmed the histological stainings.

Adipogenic and osteogenic differentiation of BM-MSCs grown in adipogenic (ADM) and osteogenic (ODM) differentiation medium with their respective control grown in DMEM with 20 % FBS (CTRL) (a). The percentage of cells positive for Oil Red O (adipogenic differentiation) and for Alizarin Red (osteogenic differentiation) stain is expressed as the mean ± SE; *p < 0.05 and **p < 0.005 (b). The adipogenic and osteogenic differentiation potential was confirmed by qPCR, comparing results to the corresponding control. Data are expressed as mean ± SE of the REL (c)
Chondrogenic differentiation
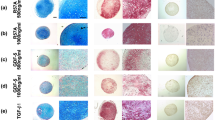
After 14 days of culture with the diverse chondrogenic differentiation media or with DMEM plus 15 % KO serum for the control cells, the aggregates were analyzed by histochemistry using HE, TB, MT and SaO staining, and immunohistochemistry for type I, II and X collagens, and metalloproteinase 13. These techniques confirmed chondrogenic differentiation under our experimental conditions (Fig. 2a,b). Specifically, TB and SaO showed the presence of extracellular matrix, stained respectively in violet-blue and orange-red, mainly in the points corresponding to cells treated with TGFβ-3 10 ng/mL and when combination of the three growth factors. By means of histochemistry, the presence of type II collagen and proteoglycans were higher in the different chondrogenic differentiation media than in the corresponding controls. Immunohistochemistry results for Col1A1, Col10A1 and MMP13 were nearly almost negative (data not shown). To confirm this result, total RNA was extracted from cells treated in the same way, reverse transcribed into cDNA and the resulting cDNA was used as template for subsequent qPCR amplification (Fig. 3). In each experiment, each gene expression level of cells treated with each of the chondrogenic differentiation medium was compared to cells cultured in DMEM plus 15 % KO serum (control points). We determined the expression levels of different genes involved in chondrogenesis corresponding to the mature chondrocyte phenotype (COL2A1, SOX9 and aggrecan) and also COL1A1 (corresponding to hypertrophic chondrocytes). Treated cells showed higher levels of SOX9 expression compared to the control points, obtaining the higher values when the combination TGFβ-3 1 ng/mL + BMP-2 10 ng/mL + BMP-7 10 ng/mL and also BMP-2 10 ng/mL alone were added as growth factors to the culture media (5.13 and 2.51 times, respectively) and were statistically significant (p < 0.05, two-tailed t test) when using the combination TGFβ-3 1 ng/mL + BMP-2 10 ng/mL + BMP-7 10 ng/mL. However aggrecan and COL1A1 expression levels were only higher than the control points when TGFβ-3 10 ng/mL was added to the culture media (1.76 times for aggrecan and 1.94 for COL1A1), resulting to be statistically significant (p < 0.05, two-tailed t test) for aggrecan. The COL2A1 gene was detected (Ct = 27) only when the following combination of growth factors was added: TGFβ-3 1 ng/mL + BMP-2 10 ng/mL + BMP-7 10 ng/mL. The results of the expression of COL2A1 cannot be shown due to the absence of its detection in the other conditions analyzed.

Histologic and immunohistochemical analysis of the BM-MSCs chondrogenic differentiation in normoxic conditions. The pictures show the results obtained after 14 days of pellet culture with chondrogenic medium plus three different growth factors alone or in combination. TGFβ3-1 means 1 ng/mL TGFβ-3; TGFβ3-10 means 10 ng/mL TGFβ-3; BMP2 means 10 ng/mL BMP-2; BMP7 means 10 ng/mL BMP-7; TGF + BMPs means 1 ng/mL TGFβ-3 + 10 ng/mL BMP-2 + 10 ng/mL BMP-7. All the chondrogenic differentiation points were compared to cells grown in DMEM with 15 % KO serum (CTRL). HE, Hematoxylin-eosin; TB, toluidine blue; MT, Masson´s trichrome; SaO, safranin O; and Col2A1, type II collagen stainings (a). Percentage of cells positive for Col2A1 staining is expressed as the mean ± SE (b)

Chondrogenic differentiation potential was analyzed by qRT-PCR, comparing the different chondrogenic culture media (TGFβ3-1, TGFβ3-10, BMP-2, BMP-7 and TGF + BMPs) with the control grown in DMEM with 15 % KO serum (CTRL). Expression levels of SOX9, aggrecan (AGG) and COL1A1 at day 14 of culture in normoxic conditions; *p < 0.05. Data are expressed as mean ± SE of the RELs
Discussion
Chondrogenic differentiation of MSCs leads to the expression of two chondrocyte hypertrophy-associated genes, collagen X and MMP13 as shown by Sekiya et al. (2002) and by Pelttari et al. (2006). Due to this, it is imperative to establish the best protocol for in vitro directing stem cell differentiation into the chondrogenic lineage in each experimental design. In this study we examined the in vitro chondrogenic induction of BM-MSCs by three different growth factors (BMP-2, BMP-7 and TGFβ-3), alone or in combination, in normoxic conditions. In all the experiments performed, BM-MSCs from the three donors were able to differentiate toward adipogenic, osteogenic and chondrogenic lineage. In addition, qPCR analysis of the marker genes for each specific lineage confirmed these results, since it was observed an up-regulation of the genes analyzed in the treated cells compared to the corresponding controls.
In the last years, many studies have been focused on finding better culture conditions capable to differentiate MSCs towards mature chondrocytes (Heng et al. 2004). For this purpose different growth and differentiation factors were studied in detail by Sekiya et al. (2002) and by Kim et al. (2005). In particular, the ability of BMPs to promote chondrogenesis of MSCs and to increase matrix production by these multipotent cells have been studied extensively over the past two decades (Kurth et al. 2007; Nawata et al. 2005; Kaps et al. 2002; Keller et al. 2011). BMPs are members of the TGFβ superfamily of growth and differentiation factors. These proteins, originally identified by Toh as inducers of bone and cartilage formation in ectopic sites (Toh et al. 2007), act as autocrine and/or paracrine factors regulating bone growth, development and remodeling. It is well-known that bone and cartilaginous cells produce BMPs. BMPs induce differentiation of pluripotent mesenchymal cell lines into chondrocytes when grown at high density, since high-density culture mimics the condensation step that precedes the chondrogenesis in vivo. Kameda reported that BMP-2, -5, and -6 maintain and promote later stages of chondrocyte differentiation rather than initiation of maturation (Kameda et al. 2000) while Haaijman that BMP-7, which is expressed in proliferating chondrocytes, promoted chondrocyte proliferation and inhibited terminal differentiation (Haaijman et al. 2000). BMP-2 regulates the chondrogenic differentiation, the maturation of mesenchymal progenitors and stimulates the synthesis of chondrocyte matrix components, as described by Toh et al. (2007). For this reason BMP-2 has been proposed as a tool for cartilage repair and as an inducer of chondrogenesis. In healthy cartilage, Blaney assessed that BMP-2 is hardly present, whereas it is highly expressed during osteoarthritis (Blaney Davidson et al. 2007). In our study, and regarding chondrogenic differentiation, we obtained the better chondrogenic induction when the combination TGFβ-3, BMP-2 and BMP-7 of growth factors was added to the culture medium, since it was the unique culture condition that showed COL2A1 gene expression and that revealed the highest SOX9 expression level. Likewise, chondrogenic culture medium containing only BMP2 also showed high SOX9 mRNA expression levels. This result is in agreement with those published by Nawata et al. (2005) and Toh et al. (2007). The former generated cartilage tissue by inducing the in vivo differentiation of rat muscle-derived cells into the chondrocytic lineage with recombinant human bone morphogenetic protein 2 (rHuBMP-2). The latter demonstrated that the high-density 3D micromass culture condition, in combination with BMP-2, resulted in an enhancement and enrichment of chondrogenic differentiation.
Combinations of growth factors have also been used in the past to promote chondrogenic differentiation of MSCs. Xiang et al. (2007) induced the chondrogenesis of MSCs using the combination of IGF-1 (insulin-like growth factor-1) and TGFβ-1; whereas Kim and Im (2009) demonstrated that administration of a combination of TGFβ-2 and BMP-7 was the most effective in promoting chondrogenesis.
Both COL2A1, that encodes a key cartilage-specific extracellular matrix protein, and SOX9, that encodes a nuclear transcription factor, are two of the earlier markers expressed during chondrogenesis. SOX9 is required for the differentiation of chondroprogenitors towards mature chondrocytes and for the deposition of cartilage matrix containing collagens II, IX, XI and aggrecan, as described by Lefebvre et al. (1997). This transcription factor is expressed continuously in chondrocytes up to the hypertrophic stage and is required not only to maintain the chondrocyte phenotype but also for BMP induced chondrogenesis, as described by Yoon and Lyons (2004). In all the chondrogenic media tested we observed, by immunohistochemistry, the presence of type II collagen immunoreactivity in the extracellular matrix, however mRNA transcript level of collagen II was only detected when the combination TGFβ-3, BMP-2 and BMP-7 was added. This absence of type II collagen mRNA expression in the other culture conditions analyzed may be due to the destabilization during the translation of some mRNAs with AU-rich elements as described by Lian et al. (2007).
Aggrecan is one of the more abundant proteoglycans in the hyaline articular cartilage, as reported by Heinegard (2009). This large proteoglycan consists of a 200 kDa core protein to which keratin sulphate and chondroitin sulphate glycosaminoglycan side-chains are attached. During chondrogenesis, agreccan mRNA begins to accumulate at the onset of cellular condensation and continues to be expressed throughout differentiation. In our study, we observed the highest aggrecan mRNA expression level when TGFβ-3 10 ng/mL alone or the combination TGFβ-3, BMP-2 and BMP-7 of growth factors were added to the chondrogenic culture medium. Regarding the use of TGFβ-3 alone, the work by Kim (2005) demonstrated that cells derived from human embryonic germ cells differentiate toward chondrogenic lineage when they are culture in the presence of this transforming growth factor. These authors demonstrated that accumulation of proteoglycans, not only in cell pellets but also in culture medium, was increased by TGFβ-3 treatment. This proteoglycan release was maintained higher in cells treated with TGFβ-3 than in the controls without treatment, suggesting chondrogenic differentiation. However, after 21 days of culture control-cell pellets without treatment and cell pellets treated with BMP-2 alone or co-treated with BMP-2 and TGFβ-3, showed a similar proteoglycan release. On the other hand, it has been demonstrated that BMP-2 and BMP-7 promote cartilage differentiation and matrix maturation in artificial engineered cartilage tissues in vitro. In this sense, Kaps et al. (2002) demonstrated that BMP-7-expressing chondrocytes are able to promote and maintain the chondrocyte phenotype by inverting the collagen expression profile from type I collagen to type II collagen during prolonged cultivation in vitro. On the contrary, BMP-2-expressing chondrocytes showed no alteration in collagen gene expression compared with control-transfected chondrocytes still displaying a de-differentiated phenotype, surprisingly in these cells the expression of the large cartilage proteoglycan aggrecan was slightly down-regulated.
Conclusions
In our experimental conditions the three growth factors used, alone or in combination, were capable to induce the beginning of chondrogenesis in normoxia environment. Nevertheless, the chondrogenic culture condition that better induced BM-MSCs chondrogenesis was the one that included the combination of the three growth factors tested followed by the medium with the higher concentration of TGFβ-3. In our experimental conditions, we have determined the best method for in vitro studying BM-MSCs efficient chondrogenesis in normoxia conditions. This method could provide a powerful tool for study the contribution of specific genes to the process of chondrogenesis.
Abbreviations
- AA:
-
Ascorbic acid
- AGG:
-
Aggrecan
- ALP:
-
Alkaline phosphatase
- APM1:
-
Adiponectin
- BM-MSCs:
-
Bone marrow-mesenchymal stem cells
- BMP-2:
-
Bone morphogenetic protein-2
- BMP-7:
-
Bone morphogenetic protein-7
- cDNA:
-
Complementary deoxyribonucleic acid
- COL10A1:
-
Collagen type XA1
- COL1A1:
-
Collagen type IA1
- COL2A1:
-
Collagen type IIA1
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- FABP4:
-
Fatty acid-binding protein 4
- FBS:
-
Fetal bovine serum
- HE:
-
Hematoxylin–eosin
- IGF-1:
-
Insulin-like growth factor-1
- KO:
-
Knockout serum
- LPL:
-
Lipoprotein lipase
- MMP13:
-
Matrix metalloproteinase 13
- MSCs:
-
Mesenchymal stem cells
- MT:
-
Masson’s trichrome
- MTG:
-
Monotioglycerol
- OP:
-
Osteoprotegerin
- OP-1:
-
Osteogenic protein 1
- P/S:
-
Penicillin and streptomycin
- PCR:
-
Polymerase chain reaction
- qPCR:
-
Real-Time PCR
- REL:
-
Relative expression levels
- rHuBMP-2:
-
Recombinant human bone morphogenetic protein 2
- RNA:
-
Ribonucleic acid
- SaO:
-
Safranin O
- SOX9:
-
Sex determining region Y-box 9
- TB:
-
Toluidine blue
- TBP:
-
TATA box binding protein
- TGFβ:
-
Transforming growth factor beta
- TGFβ-3:
-
Transforming growth factor beta-3
References
Blaney Davidson EN, Vitters EL, van Lent PL, van de Loo FA, van den Berg WB, van der Kraan PM (2007) Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis res Ther 9:R102
Bobick BE, Chen FH, Le AM, Tuan RS (2009) Regulation of the chondrogenic phenotype in culture. Birth Defects Res C Embryo Today 87:351–371
Buckwalter JA, Mankin HJ (1998) Articular cartilage: degeneration and osteoarthritis, repair, regeneration, and transplantation. Instr Course Lect 47:487–504
Chen WH, Lai MT, Wu AT, Wu CC, Gelovani JG, Lin CT, Hung SC, Chiu WT, Deng WP (2009) In vitro stage-specific chondrogenesis of mesenchymal stem cells committed to chondrocytes. Arthritis Rheum 60:450–459
Cicione C, Diaz-Prado S, Muinos-Lopez E, Hermida-Gomez T, Blanco FJ (2010) Molecular profile and cellular characterization of human bone marrow mesenchymal stem cells: donor influence on chondrogenesis. Differentiation 80:155–165
Cicione,C., Muinos-Lopez,E., Hermida-Gomez,T., Fuentes-Boquete,I., Diaz-Prado,S., and Blanco,F.J. (2013). Effects of severe hypoxia on bone marrow mesenchymal stem cells differentiation potential. Stem Cells Int. 2013:232896. doi:10.1155/2013/232896
Conget PA, Minguell JJ (1999) Phenotypical and functional properties of human bone marrow mesenchymal progenitor cells. J Cell Physiol 181:67–73
DeLise AM, Stringa E, Woodward WA, Mello MA, Tuan RS (2000) Embryonic limb mesenchyme micromass culture as an in vitro model for chondrogenesis and cartilage maturation. Methods Mol Biol 137:359–375
Derfoul A, Perkins GL, Hall DJ, Tuan RS (2006) Glucocorticoids promote chondrogenic differentiation of adult human mesenchymal stem cells by enhancing expression of cartilage extracellular matrix genes. Stem Cells 24:1487–1495
Díaz Prado S, Fuentes Boquete I, Blanco FJ Cell therapy and tisular engineering to regenerate articular cartilage. In: Malgorzata Komorowska A, Olsztynska-Janus S (eds) Biomedical engineering, trends, researches and technologies. Intech Editorial 2011, p 193-216. http://www.intechopen.com/articles/show/title/cell-therapy-and-tissular-engeenering-to-regenerate-articular-cartilage
Diaz-Prado S, Muinos-Lopez E, Hermida-Gomez T, Cicione C, Rendal-Vazquez ME, Fuentes-Boquete I, de Toro FJ, Blanco FJ (2011) Human amniotic membrane as an alternative source of stem cells for regenerative medicine. Differentiation 81:162–171
Dominici M, Le BK, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317
Goldring MB, Tsuchimochi K, Ijiri K (2006) The control of chondrogenesis. J Cell Biochem 97:33–44
Haaijman A, Burger EH, Goei SW, Nelles L, Dijke PT, Huylebroeck D, Bronckers ALJJ (2000) Correlation between ALK-6 (BMPR-IB) distribution and responsiveness to osteogenic protein-1 (BMP-7) in embryonic mouse bone rudiments. Growth Factors 17(3):177–192
Heinegard D (2009) Proteoglycans and more—from molecules to biology. Int J Exp Pathol 90:575–586
Heng BC, Cao T, Lee EH (2004) Directing stem cell differentiation into the chondrogenic lineage in vitro. Stem Cells 22:1152–1167
Hermida-Gomez T, Fuentes-Boquete I, Gimeno-Longas MJ, Muinos-Lopez E, Diaz-Prado S, de Toro FJ, Blanco FJ (2011) Bone marrow cells immunomagnetically selected for CD271+ antigen promote in vitro the repair of articular cartilage defects. Tissue Eng Part A 17:1169–1179
Hombach-Klonisch S, Panigrahi S, Rashedi I, Seifert A, Alberti E, Pocar P, Kurpisz M, Schulze-Osthoff K, Mackiewicz A, Los M (2008) Adult stem cells and their trans-differentiation potential–perspectives and therapeutic applications. J Mol Med (Berl) 86:1301–1314
Hwang NS, Varghese S, Lee HJ, Zhang Z, Ye Z, Bae J, Cheng L, Elisseeff J (2008) In vivo commitment and functional tissue regeneration using human embryonic stem cell-derived mesenchymal cells. Proc Natl Acad Sci U.S.A 105:20641–20646
Johnstone B, Hering TM, Caplan AI, Goldberg VM, Yoo JU (1998) In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells. Exp Cell Res 238:265–272
Jung DI, Ha J, Kang BT, Kim JW, Quan FS, Lee JH, Woo EJ, Park HM (2009) A comparison of autologous and allogenic bone marrow-derived mesenchymal stem cell transplantation in canine spinal cord injury. J Neurol Sci 285:67–77
Kameda T, Koike C, Saitoh K, Kuroiwa A, Iba H (2000) Analysis of cartilage maturation using micromass cultures of primary chondrocytes. Dev Growth Differ 42:229–236
Kaps C, Bramlage C, Smolian H, Haisch A, Ungethum U, Burmester GR, Sittinger M, Gross G, Haupl T (2002) Bone morphogenetic proteins promote cartilage differentiation and protect engineered artificial cartilage from fibroblast invasion and destruction. Arthritis Rheum 46:149–162
Keller B, Yang T, Chen Y, Munivez E, Bertin T, Zabel B, Lee B (2011) Interaction of TGFbeta and BMP signaling pathways during chondrogenesis. PLoS ONE 6:e16421
Kim HJ, Im GI (2009) Combination of transforming growth factor-beta2 and bone morphogenetic protein 7 enhances chondrogenesis from adipose tissue-derived mesenchymal stem cells. Tissue Eng Part A 15:1543–1551
Kim MS, Hwang NS, Lee J, Kim TK, Leong K, Shamblott MJ, Gearhart J, Elisseeff J (2005) Musculoskeletal differentiation of cells derived from human embryonic germ cells. Stem Cells 23:113–123
Kock L, van Donkelaar CC, Ito K (2012) Tissue engineering of functional articular cartilage: the current status. Cell Tissue Res 347:613–627
Kurth T, Hedbom E, Shintani N, Sugimoto M, Chen FH, Haspl M, Martinovic S, Hunziker EB (2007) Chondrogenic potential of human synovial mesenchymal stem cells in alginate. Osteoarthritis Cartilage 15:1178–1189
Lefebvre V, Huang W, Harley VR, Goodfellow PN, de Crombrugghe B (1997) SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol 17:2336–2346
Lian Q, Lye E, Suan YK, Khia Way TE, Salto-Tellez M, Liu TM, Palanisamy N, El Oakley RM, Lee EH, Lim B, Lim SK (2007) Derivation of clinically compliant MSCs from CD105+. Stem Cells 25:425–436
Nawata M, Wakitani S, Nakaya H, Tanigami A, Seki T, Nakamura Y, Saito N, Sano K, Hidaka E, Takaoka K (2005) Use of bone morphogenetic protein 2 and diffusion chambers to engineer cartilage tissue for the repair of defects in articular cartilage. Arthritis Rheum 52:155–163
Pelttari K, Winter A, Steck E, Goetzke K, Hennig T, Ochs BG, Aigner T, Richter W (2006) Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum 54:3254–3266
Pittenger MF (2008) Mesenchymal stem cells from adult bone marrow. Methods Mol Biol 449:27–44
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147
Pizette S, Niswander L (2000) BMPs are required at two steps of limb chondrogenesis: formation of prechondrogenic condensations and their differentiation into chondrocytes. Dev Biol 219:237–249
Richler C, Yaffe D (1970) The in vitro cultivation and differentiation capacities of myogenic cell lines. Dev Biol 23:1–22
Ryden M, Dicker A, Gotherstrom C, Astrom G, Tammik C, Arner P, Le BK (2003) Functional characterization of human mesenchymal stem cell-derived adipocytes. Biochem Biophys Res Commun 311:391–397
Sekiya I, Vuoristo JT, Larson BL, Prockop DJ (2002) In vitro cartilage formation by human adult stem cells from bone marrow stroma defines the sequence of cellular and molecular events during chondrogenesis. Proc Natl Acad Sci U.S.A 99:4397–4402
Spagnoli A (2008) Mesenchymal stem cells and fracture healing. Orthopedics 31:855–856
Toh WS, Yang Z, Liu H, Heng BC, Lee EH, Cao T (2007) Effects of culture conditions and bone morphogenetic protein 2 on extent of chondrogenesis from human embryonic stem cells. Stem Cells 25:950–960
Tuli R, Tuli S, Nandi S, Huang X, Manner PA, Hozack WJ, Danielson KG, Hall DJ, Tuan RS (2003) Transforming growth factor-beta-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J Biol Chem 278:41227–41236
Xiang Y, Zheng Q, Jia BB, Huang GP, Xu YL, Wang JF, Pan ZJ (2007) Ex vivo expansion and pluripotential differentiation of cryopreserved human bone marrow mesenchymal stem cells. J Zhejiang Univ Sci B 8:136–146
Yoon BS, Lyons KM (2004) Multiple functions of BMPs in chondrogenesis. J Cell Biochem 93:93–103
Acknowledgments
This study was supported by grants: Servizo Galego de Saúde, Xunta de Galicia (PS07/84), Cátedra Bioiberica de la Universidade da Coruña and Instituto de Salud Carlos III CIBER BBN; Ministerio Ciencia e Innovacion PLE2009-0144; Fondo Investigacion Sanitaria-PI 08/2028 with participation of funds from FEDER (European Community), Tamara Hermida-Gómez is the beneficiary of a contract from Fondo de Investigación Sanitaria (2008), Spain. We would like to thank P.Filgueira and M.J.Sánchez for technical assistance.
Conflict of interest
The authors declare that no competing financial interest exists.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cicione, C., Muiños-López, E., Hermida-Gómez, T. et al. Alternative protocols to induce chondrogenic differentiation: transforming growth factor-β superfamily. Cell Tissue Bank 16, 195–207 (2015). https://doi.org/10.1007/s10561-014-9472-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10561-014-9472-7