
Abstract
Purpose
Systemic hypertension may induce adverse hypertrophy of the left cardiac ventricle. Pathological cardiac hypertrophy is a common cause of heart failure. We investigated the significance of ferroptosis repressor xCT in hypertrophic cardiomyopathy.
Methods
xCT expression in angiotensin II (Ang II)-treated mouse hearts and rat cardiomyocytes was determined using qRT-PCR and Western blotting. Cardiac hypertrophy was induced by Ang II infusion in xCT knockout mice and their wildtype counterparts. Blood pressure, cardiac pump function, and pathological changes of cardiac remodeling were analyzed in these mice. Cell death, oxidative stress, and xCT-mediated ferroptosis were examined in Ang II-treated rat cardiomyocytes.
Results
After Ang II infusion, xCT was downregulated at day 1 but upregulated at day 14 at both mRNA and protein levels. It was also decreased in Ang II-treated cardiomyocytes, but not in cardiofibroblasts. Inhibition of xCT exacerbated cardiomyocyte hypertrophy and boosted the levels of ferroptosis biomarkers Ptgs2, malondialdehyde, and reactive oxygen species induced by Ang II, while overexpression of xCT opposed these detrimental effects. Furthermore, knockout of xCT aggravated Ang II-mediated mouse cardiac fibrosis, hypertrophy, and dysfunction. Ferrostatin-1, a ferroptosis inhibitor, alleviated the exacerbation of cardiomyocyte hypertrophy caused by inhibiting xCT in cultured rat cells or ablating xCT in mice.
Conclusion
xCT acts as a suppressor in Ang II-mediated cardiac hypertrophy by blocking ferroptosis. Positive modulation of xCT may therefore represent a novel therapeutic approach against cardiac hypertrophic diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypertension is a major risk fact for cardiovascular disease and remains a main cause of morbidity and mortality [1]. It induces adaptive cardiac remodeling (cardiac hypertrophy and fibrosis), disturbs cardiac contractility, and eventually leads to heart failure [2,3,4]. Angiotensin II (Ang II), an important component of the rennin–angiotensin–aldosterone system (RAS), is involved in the pathogenesis of hypertension and cardiac hypertrophy. Neurohormonal blocking drugs have been used to treat cardiac hypertrophy [5].
A physiological range of reactive oxygen species (ROS) abundance is required for normal function of human beings. ROS play dual roles in physiological and pathophysiological processes [6]. Their bimodal actions are both beneficial and detrimental in a context-dependent manner. ROS-related disease can be either due to a lack of ROS (e.g., chronic granulomatous disease, certain autoimmune disorders) or a surplus of ROS (e.g., cardiovascular and neurodegenerative diseases) [7]. ROS involvement has been indicated in the pathogenesis of cardiac hypertrophy and heart failure [8]. Ang II-induced excessive ROS aggravates cardiac hypertrophy by activating various hypertrophic signaling kinases and transcription factors [9]. Distinct from apoptosis, necroptosis, and autophagy, ferroptosis is an iron-dependent and ROS-reliant cell death [10]. Slc7a11 gene-encoded plasma membrane cystine/glutamate antiporter xCT promotes cystine uptake. Cystine is reduced to cysteine to increase the synthesis of glutathione (GSH), which is an ROS scavenger [11]. Recent publications suggest that xCT prevents transverse aortic constriction (TAC)-induced cardiac remodeling and iron overload cardiomyopathy by suppressing ferroptosis [12, 13]. However, the significance of xCT and ferroptosis in Ang II-induced pathological cardiac hypertrophy remains largely unknown.
In this study, we analyzed the expression of xCT in Ang II-induced hypertrophic cardiomyocytes and hearts in vitro and in vivo. We then checked the role of xCT in cardiac hypertrophy. Lastly, we elucidated the mechanism underlying potential xCT involvement in cardiac hypertrophy.
Methods
Antibody and Reagents
The rabbit xCT antibody (12691S, 1:1000) and GAPDH antibody (2118, 1:10000) for Western blot assay were from Cell Signaling Technology (Danvers, MA, USA). All of the HRP-labeled secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). For immunofluorescent staining, the α-actinin antibody (A5044, 1:100) was from Sigma (Saint Louis, MO, USA) and the fluorescence-conjugated Alexa Fluor 488 secondary antibody (A32723, 1:200) was from Invitrogen (Carlsbad, CA, USA). Sulfasalazine (SASP) (S0883) was from Sigma. Ferrostatin-1 (Fer-1) (S7243) was from Selleck (Houston, TX, USA). N-acetyl-L-cysteine (NAC) (S0077) was from Beyotime Institute of Biotechnology (Shanghai, China). DAPI (abs9235) was from Absin Bioscience (Shanghai, China).
Cell Culture
Cardiomyocytes (NRCMs) and cardiofibroblasts (NRCFs) were isolated from 1 to 2 day-old neonatal rat hearts using a conventional method as described [14]. Briefly, heart tissues were digested by trypsin and collagenase II. The dissociated cells in DMEM/F-12 medium with 10% FBS and 1% penicillin and streptomycin were plated to obtain adhered NRCFs after 90 min in an incubator (5% CO2). NRCMs in suspension were transferred to the dishes coated with laminin and were grown in DMEM/F12 supplemented with 15% FBS, 1% penicillin/streptomycin, and 1% 5-bromodeoxyuridine for 24 h. NRCFs were grown in DMEM/F12 supplemented with 10% FBS, 1% penicillin/streptomycin. Both adhered NRCFs and NRCMs were cultured in serum-free DMEM/F-12 for 12 h before experiments.
Measurement of Cell Surface Area by Immunofluorescence
NRCMs were seeded onto coverslips at a concentration of 5 × 105 cells/ml for 24 h. After treatment, cultured NRCMs were fixed with acetone for 15 min, permeabilized with pre-cooled methanol for 10 min, blocked with 5% BSA solution for 1 h, and incubated with α-actinin antibody at 4 °C overnight. Then, the cells were washed and stained with fluorescence-conjugated Alexa Fluor 488 secondary antibody for 1 h. Finally, the cells were counter-stained with DAPI. Immunofluorescence was captured by Laser Confocal Microscopy (Leica TCS-NT SP8) and the surface areas were measured using Image J.
Generation of xCT Knockout Mice
Mice carrying floxed xCT alleles (xCTfl/fl) were constructed by Cyagen Biotechnology (Guangzhou, China). Mice expressing EIIa-cre recombinase (B6. FVB-Tg (EIIa-cre) C5379Lmgd/J) were obtained from Jackson Laboratory (Bar Harbor, ME, USA). To generate xCT−/− mice, we cross-mated xCTfl/fl mice with EIIa-cre mice to delete the 2nd exon of slc7a11 globally using the Cre-loxP-system.
Cardiac Hypertrophy Mouse Model
Male C57BL/6 mice were from Vital River Laboratory Animal Technology Company (Beijing, China). All animal experiments were approved by the Institutional Animal Care and Use Committee of Capital Medical University. Hypertensive cardiac hypertrophy was induced in 8–10 week-old male mice by chronic subcutaneous infusion of angiotensin II (Ang II, Sigma-Aldrich, A9525) at a dose of 1000 ng/kg/min using the ALZET® Osmotic Pumps (Model 1002D) for 14 days. The control mice were infused with saline for 14 days. For Fer-1 treatment, mice were randomized to be treated with either vehicle or Fer-1 at a dose of 1 mg/kg intraperitoneal daily injection [15]. All mice were analyzed by echocardiography for structure and hemodynamic measurements at 14 days.
Echocardiography
Cardiac function was evaluated by a high-resolution Micro-Ultrasound system (Vevo 770, VisualSonics, Toronto, Ontario, Canada). Briefly, cardiac contractile function and structure were evaluated by M-mode echocardiography. Ventricular parameters including diastolic left ventricular anterior wall (LVAW; d) and systolic left ventricular anterior wall (LVAW; s) were measured. All the measurements were obtained from more than three beats and averaged. The fractional shortening (FS%) and ejection fraction (EF%) were calculated to judge cardiac systolic function.
Measurement of Blood Pressure
Mice were acclimatized for 7 days prior to experimentation. Systolic blood pressure (SBP) and diastolic blood pressure (DBP) were recorded under conscious condition using the tail-cuff method with the CODA noninvasive BP system (Kent Scientific Corporation, Torrington, CT, USA).
Histopathology Analysis
For histological analysis, hearts were fixed, embedded, and sectioned into 5 μm thick slices. Sections were used to stain wheat germ agglutinin (WGA) to assess cellular hypertrophy. Sections were stained with Masson’s trichrome (collagen, blue; cytoplasm, red/pink) for collagen deposition analysis. Immunohistochemistry images were captured with Pannoramic SCAN II (3Dhistech Ltd., Budapest, Hungary) and analyzed by a person blinded to treatment using Image J.
Quantitative RT-PCR (qRT-PCR) Analysis
Total RNA was extracted from heart tissues, NRCMs or NRCFs cells using the Trizol method (Sigma-Aldrich, T9424). The first strand cDNA was synthesized from 1 μg of total RNA by HIScript-II qRT SuperMix for qRT-PCR from Vazyme Biotechnology (Nanjing, China). qRT-PCR was performed utilizing SYBR Green Master Mix from Transgen Biotechnology (Beijing, China) with an iCycler IQ system (Bio-Rad Laboratories, Hercules, CA, USA) at 94 °C for 30 s, 94 °C for 5 s, and 60 °C for 1 min for 45 cycles. The expression of mRNA was normalized with endogenous GAPDH. All samples were run in duplicate. The following of primers for genes:
Mouse:
GAPDH: 5’-GGTTGTCTCCTGCGACTTCA-3′; 5’-GGTGGTCCAGGGTTTCTTACTC-3′;
xCT: 5’-AAGTCTAATGGGGTTGCCCT-3′; 5’-TGATAGCCATGGAGATGCAG-3′;
ANF: 5’-CACAGATCTGATGGATTTCAAGA-3′; 5’-CCTCATCTTCTACCGGCATC-3′;
BNP: 5’-GAAGGTGCTGTCCCAGATGA-3′; 5’-CCAGCAGCTGCATCTTGAAT-3′;
Collagen I: 5’-GAGTACTGGATCGACCCTAACCA-3′; 5’-ACGGCTGAGTAGGGAACACA-3′;
Collagen III: 5’-TCCCCTGGAATCTGTGAATC-3′; 5’-TGAGTCGAATTGGGGAGAAT-3′;
Rat:
GAPDH: 5’-CCCCCAATGTATCCGTTGTG-3′; 5’-TAGCCCAGGATGCCCTTTAGT-3′;
xCT: 5’-TCACGGGCACATAGGA-3′; 5’-GCATAATGAGTAACTGGGTCT-3′;
ANF: 5’-GAAGATGCCGGTAGAAGATGAG-3′; 5’-AGAGCCCTCAGTTTGCTTTTC-3′;
BNP: 5’-GGTGCTGCCCCAGATGATT-3′; 5’-CTGGAGACTGGCTAGGACTTC-3′;
Myh7: 5′- GCCCCAAATGCAGCCAT-3′; 5′- CGCTCAGTCATGGCGGAT-3′;
Ptgs2: 5’-ATGTTCGCATTCTTTGCCCAG-3′; 5’-TACACCTCTCCACCGATGAC-3′.
Western Blotting
Total proteins were extracted from hearts or cells in RIPA lysis buffer. Equal protein samples were loaded, separated by SDS-PAGE, and transferred to a membrane from Millipore (Billerica, MA, USA). The membrane was blocked and incubated with a primary antibody at 4 °C overnight, and then with secondary antibodies for 1 h at room temperature. The enhanced chemiluminescence system (Millipore) was used for detection, and signal intensities were analyzed with Image J.
Adenovirus and Cell Infection
The pAdEasy-EF1-MCS-3flag-CMV-EGFP vector was used to build an adenoviral vector encoding rat xCT gene (Ad-xCT). Ad-xCT and Ad-GFP viruses were produced at HanBio Biotechnology (Shanghai, China). NRCMs were infected with Ad-xCT or Ad-GFP viruses at a multiplicity of infection of 50 MOI and then treated with Ang II. Expression of xCT was evaluated by Western blotting.
siRNA Transfection
The siRNAs used to knockdown xCT in NRCMs were synthesized by HanBio Biotechnology. The sequence of xCT siRNA was 5’-CTTGCAATATGTATATCCA-3′ and the sequence of control siRNA was 5’-UUCUCCGAACGUGUCACGU-3′. The siRNA knockdowns were performed using Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Briefly, cells were seeded in 6-well plates to obtain 30–50% confluence. DMEM/F-12 with mixture of siRNA (20pM/well) and transfection reagents (5ul/well) were added to the cells for 6 h and then replaced with fresh complete culture medium.
Trypan Blue Assay
NRCMs were harvested and stained with 0.4% Trypan Blue. Positive and negative cells were counted under a light microscope (Olympus, CKX-41, Japan).
Cell Counting Assay
A 10 μl Cell Counting Kit-8 (CCK-8) solution (YEASEN, Shanghai, China) was added to each well and incubated at 37 °C for 2 h, then the OD value for each well was read at wavelength 450 nm with a microplate reader (Multiskan, Thermo Fisher Scientific). The cell viability was calculated as follows:
Cell Oxidative Stress Assays
ROS levels were determined by flow cytometry and NRCMs were stained with 2′, 7′-dichlorodihydrofluorescein diacetate (DCFH-DA) according to manufacturer’s instructions. Briefly, 5 × 105 NRCMs were pretreated with 0.15 mM SASP or vehicle for 8 h and then treated with Ang II for 12 h. The cells were washed twice with PBS and then incubated with 10 μM DCFH-DA for 20 min at 37 °C. After washing with PBS, cells were harvested and analyzed for the fluorescence of DCFH by flowcytometry (BD Accuri C6, Frankin Lakes, NJ, USA). Malondialdehyde (MDA) levels in cells were measured using the MDA Assay Kit (Beyotime) according to the manufacturer’s instructions, and MDA concentration was calculated in μM per gram of protein.
Statistical Analysis
All data were expressed as mean ± standard error of the mean (SEM) and were analyzed using GraphPad Prism 7.0 (GraphPad Soft-ware, San Diego, CA, USA) or SPSS Version 21 software. Comparison between two groups was conducted using two-tailed, unpaired Student’s t test. In the result with more than two groups, analysis of variance (ANOVA) was applied to analyze the difference. Two-way ANOVA moderated by Bonferroni post-hoc test was used to detect variation between three or more groups. Statistical significance was defined as P < 0.05.
Results
xCT Expression Is Fluctuated during Progression of Cardiac Hypertrophy
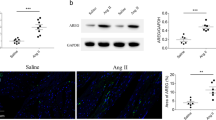
To investigate the potential involvement of xCT during cardiac hypertrophy development, we checked the expression of xCT in Ang II-induced acute hypertrophic mouse hearts. After Ang II infusion, xCT was downregulated at day 1 (Fig. 1a and b) but upregulated at day 14 at both mRNA and protein levels (Supplementary Fig. 1). It was also decreased in Ang II-treated NRCMs but not in NRCFs (Fig. 1c and d). Because xCT expression fluctuates during the process of compensatory cardiac hypertrophy induced by Ang II, it may play a role in cardiac remodeling.

xCT expression is reduced in hypertrophic heart. Mice were infused with Ang II (1000 ng/kg/min) for 1 day (a and b). (a) qRT-PCR analyses of xCT mRNA (n = 5). (b) Western blot and quantification of xCT (n = 3). (c and d) NRCMs and NRCFs were treated with Ang II (100 nM) or saline (n = 6). qRT-PCR and Western blot analyses of xCT levels. *P<0.05,**P<0.01
Suppression of xCT Aggravates Ang II–Induced Cardiomyocyte Hypertrophy
To elucidate whether xCT exerts a pro- or anti-hypertrophic effect in cardiomyocyte hypertrophy induced by Ang II, we inhibited xCT in NRCMs with xCT inhibitor, SASP. Compared with control, SASP further enlarged Ang II-induced cardiomyocytes and potentiated the expression of hypertrophic marker genes, such as arterial natriuretic polypeptide (ANP), brain natriuretic peptide (BNP), and myosin heavy chain 7 (Myh7) (Fig. 2a and b). In addition, we assessed NRCFs’ function after Ang II and SASP incubation. SASP did not alter the upregulation of collagen I/III expression induced by Ang II (Supplementary Fig. 2). Consistently, knocking down xCT by siRNA also increased the expression of ANF and BNP (Supplementary Fig. 3b). To ascertain xCT repression of Ang II–induced cardiomyocyte hypertrophy, we overexpressed xCT by infection of NRCMs with xCT expressing adenoviruses (Ad-xCT) compared with Ad-GFP viruses (Supplementary Fig. 4a). Augmented xCT decreased Ang II-mediated enlargement of cardiomyocytes and overexpression of ANF, BNP, and Myh7 in NRCMs (Fig. 2c and d). Therefore, xCT suppresses Ang II–induced cardiomyocyte hypertrophy.

Inhibition of xCT aggravates Ang II–induced cardiomyocyte hypertrophy. NRCMs were treated with saline or Ang II (100 nM) for 48 h in the presence of SASP (150 μM) or DMSO (a and b). (a) α-Actinin staining was performed to determine cell size. Representative images (Left) and quantification of cell size of total 30 NRCMs (Right) in each group are shown (scale bar = 20 μm). (b) qRT-PCR analyses (n = 6). NRCMs were first infected with Ad-GFP or Ad-xCT and then treated with/without Ang II for 48 h (c and d). (c) α-Actinin staining was performed to determine cell size. Representative images (Left) and quantification of cell size of total 30 NRCMs (Right) in each group are shown (scale bar = 20 μm). (d) qRT-PCR analyses of mRNA (n = 6). **P < 0.01
Knockout of xCT Exasperates Mouse Cardiac Hypertrophy
To ascertain the protective role of xCT in cardiac hypertrophy, we generated xCT−/− mice (Fig. 3a). Systolic and diastolic blood pressures boosted by Ang II treatment were not affected in the absence of xCT (Fig. 3b). Induced by Ang II for 14 days, xCT−/− mice exhibited reduced cardiac pump function in terms of left ventricular ejection fraction (EF%) and fractional shortening (FS%) (Fig. 3c). However, the reduction of cardiac output (20.62%) was not as drastic as that of EF% (33.14%) in Ang II-treated xCT KO mice (Supplementary Table 1). HW/BW and HW/TL ratios were increased in Ang II-infusion mice (Fig. 3d). Ang II increased cardiomyocyte size, fibrosis, and hypertrophic and fibrosis genes ANP, BNP, collagen I and III in WT mice (Fig. 3e–h). The hypertrophic effect of Ang II was markedly amplified in xCT−/− mice (Fig. 3c–f). Moreover, xCT knockout aggravated fibrosis in Ang II-induced hearts (Fig. 3g and h). Hence, knockout of xCT significantly exaggerates cardiac hypertrophy in mice.

Knockout of xCT aggravates cardiac hypertrophy in mice. WT (xCT+/+) and xCT knockout (xCT−/−) mice were treated with saline or Ang II (1000 ng/kg/min) for 14 days. (a) Western blot of xCT in cardiac tissues. (b) Systolic and diastolic blood pressures. (c) Representative M-mode echocardiography of the left ventricle (left). Measurement of ejection fraction (EF%) and fractional shortening (FS%) (right, n = 5). (d) Representative images of heart size photographed (left). HW/BW and HW/TL ratios (right, n = 5). (e) TRITC-labeled wheat germ agglutinin (WGA) staining of hearts. Scale bar, 50 μm (left). Quantification of the relative myocyte cross-sectional area (n = 6, right). (f) Masson’s trichrome staining. Scale bar, 50 μm (left). Quantification of the relative fibrosis (n = 6, right). (g and h) qRT-PCR analysis of mRNA expression (n = 5). *P < 0.05, **P < 0.01
xCT Reduces Cardiomyocyte Death and Hypertrophy by Blocking Ferroptosis
xCT is a key plasma membrane exchanger for cystine uptake in maintaining intracellular redox homeostasis to block ferroptosis. SASP aggravated Ang II-induced cardiomyocytes death in vitro (Fig. 4a and b). Meanwhile, lipid peroxidation (manifested as fluorescence intensity of DCFH-DA) and MDA, markers of oxidative stress, and ptgs2 mRNA, marker of ferroptosis [16] were further increased by SASP in Ang II-treated NRCMs (Fig. 4c–e). Consistently, si-xCT exaggerated Ang II-induced cell death, ptgs2, and ROS overproduction (Supplementary Fig. 3c–f). In contrast, ferroptosis was reversed by overexpression of xCT (Supplementary Fig. 4). We then tested whether blocking ferroptosis ameliorated SASP-exacerbated cardiomyocyte death, hypertrophy, and oxidative stress. Fer-1, an inhibitor of ferroptosis, reversed the detrimental effect of SASP, illustrated by reversing cardiomyocyte death, decreased ANF, BNP and Myh7 expression and reduced ROS, MDA, and Ptgs2 overproduction. Because ferroptosis is oxidative stress-dependent cell death, the antioxidant agent NAC also overturned the negative effect of SASP (Fig. 4f–i and Supplementary Fig. 5). These results demonstrate that xCT reverses Ang II-medicated cardiomyocyte death and hypertrophy by blocking ferroptosis.

xCT abrogates cardiomyocyte death and hypertrophy by blocking ferroptosis. (a and b) NRCMs were incubated with SASP (150 μM) or DMSO for 8 h and saline or Ang II was then added for 24 h. (a) Trypan blue assay was used to determine the cell death (n = 6). (b) CCK-8 assay was used to examined the cell viability (n = 6). (c–e) NRCMs were incubated with SASP or DMSO for 8 h and saline or Ang II was then added for 12 h. (c) ROS levels were detected with DCFH-DA staining by flow cytometry (n = 6). (d) MDA levels were measured by MDA Assay Kit (n = 6). (e) qRT-PCR analysis (n = 6). (f–i) NRCMs were first treated with SASP for 6 h, then with DMSO, Fer-1, or NAC for 2 h and lastly with Ang II for 24 (f and g) or 48 h (h and i). (f) Trypan blue assay (n = 6). (G) CCK-8 assay (n = 6). (h and i) qRT-PCR analyses (n = 6). *P < 0.05, **P < 0.01
Aggravation of Cardiac Hypertrophy by xCT Knockout Is Reversed by Inhibition of Ferroptosis
Recently, we have established that inhibition of mTOR-xCT signaling cascade induces ferroptosis [17]. Because Fer-1 has no influence on wildtype mouse hearts [15], we used Fer-1 to suppress ferroptosis in vivo. Accordingly, Fer-1 reduced cardiac hypertrophy/fibrosis and improved cardiac function of xCT−/− mice with Ang II infusion. In contrast, Fer-1 exacerbated the cardiomyopathy of WT mice (Fig. 5 and Supplementary Table 2). These results indicate that xCT−/−-mediated aggravation of cardiac hypertrophy can be alleviated by suppression of ferroptosis.

The aggravation of cardiac hypertrophy induced by xCT knockout is abolished by ferroptosis inhibition. 15% DMSO saline as solvent or Fer-1 was given to WT and xCT−/− mice for 1 day and then the treatment was combined with Ang II fusion for 14 days. (a) Diagrammatic illustration of experimental schemes and timelines. (b) Systolic and diastolic blood pressures. (c) Representative M-mode echocardiography of the left ventricle (left). Measurement of ejection fraction (EF%) and fractional shortening (FS%) (right, n = 5). (d) Representative hearts (left). HW/BW and HW/TL ratios (right, n = 5). (e) TRITC-labeled wheat germ agglutinin (WGA) staining of heart sections. Scale bar, 50 μm (left). Quantification of the relative myocyte cross-sectional area (n = 5, right). (f) Masson’s staining of cardiac sections. Scale bar, 50 μm (left). Quantification of the relative fibrosis (n = 5, right). (g and h) qRT-PCR analysis of mRNA expression (n = 5,). *P < 0.05, **P < 0.01
Discussion
xCT suppresses ferroptosis by disposing ROS [18, 19]. In this study, we identified xCT as a cardioprotective factor in pathological cardiac hypertrophy. xCT decreased in mouse hearts immediately in response to Ang II infusion. Overexpression of xCT attenuated cardiomyocyte hypertrophy. Pharmacological inhibition or knockout of xCT aggravated cardiac fibrosis, hypertrophy, and dysfunction. Blockage of xCT induced ferroptosis of cardiomyocytes. Inhibitor of ferroptosis alleviated the exacerbation of xCT deficiency on Ang II-induced pathological cardiac remodeling. Therefore, xCT plays a protective role in cardiac hypertrophic diseases (Fig. 6).

xCT prevents cardiac hypertrophy by inhibiting ferroptosis. Diagrammatic illustration of xCT in prevention of cardiac hypertrophy
Cardiac hypertrophy is an early compensatory change of heart in response to many stresses, especially hypertension, myocardial infarction, and pressure overload [20,21,22]. The heart is capable of significant remodeling and hypertrophic growth as means of adapting pathological stimuli [23, 24]. However, progressive cardiac hypertrophy could lead to severe heart failure and sudden death [25]. The primary function of xCT is to increase biosynthetic and bioenergetic needs to maintain redox homeostasis and protect cells from ferroptosis [26]. Recent studies indicated that ferroptosis contributed to pathological process in a variety of diseases and conditions, including cerebral ischemia reperfusion, ischemia reperfusion-triggered acute renal failure, sepsis-induced acute lung injury, TAC-induced cardiac remodeling, and iron overload cardiomyopathy [27,28,29,30,31, 12, 13]. Consistent with TAC-induced cardiac hypertrophy [12], xCT was decreased in mouse hearts after Ang II infusion for 1 day. It is known that p53 can be induced by Ang II [32]. We speculate that Ang II downregulated xCT through induction of p53 because p53 can enhance ferroptosis by inhibiting the expression of xCT [33]. However, xCT was elevated in Ang II-treated mouse hearts by 14 days. xCT may be upregulated by Ang II-induced oxidative stress, which is an inducer of xCT [34], leading to enhanced GSH production and maintaining intracellular redox balance at late stage. Inhibition of xCT aggravated cardiomyocyte hypertrophy and overexpression of xCT attenuated this detrimental effect. Consistently, depletion of xCT augmented cardiac hypertrophy in mice. Thus, xCT is an anti-hypertrophic regulator in cardiac hypertrophy.
Improving cardiac remodeling due to cardiac hypertrophy and fibrosis should be a cardioprotective strategy. Although ROS is a common denominator during cardiac remodeling, the failure of clinical trials with antioxidant compounds has underscored the need for a better understanding of the sources and contribution of ROS in these diseases [35]. Depletion of GSH causes accumulation of ROS, which induces lipid peroxidation and consequently ferroptosis [36]. Iron-dependent accumulation of lipid hydroperoxides triggers ferroptosis, which contributes to pathological process in a variety of diseases, including cardiomyopathy [15, 37,38,39]. xCT plays a major role in maintaining intracellular cysteine balance and GSH biosynthesis. We speculate that mice have somehow adapted to xCT deficiency by reprograming cystine/cysteine metabolism from xCT-dependent to independent pathways not only at the cellular level but also at tissue, organ, and systemic levels. Although there are no phenotypical changes under physiological conditions, mice failed to compensate faulty xCT and exhibited defects when they were challenged under stress conditions. Thus, xCT is essential for redox homeostasis, and its inhibition causes ferroptosis. In this study, we revealed that Ang II markedly induced ferroptosis, which was exacerbated by pretreatment of xCT inhibitor SASP in NRCMs. In contrast, the ferroptosis was reversed by overexpression of xCT. As a ROS scavenger, Fer-1 ameliorated Ang II-induced cardiac hypertrophy/fibrosis and improved cardiac function presumably through depleting excessive ROS and consequently inhibiting ROS-dependent ferroptosis in xCT knockout mice. However, depletion of ROS by Fer-1 might be harmful to WT mice whose ROS could be less abundant. In analogy to the observations in mice, SASP-treated culture cells phenocopied the alteration of xCT knockout mice. Both Fer-1 and NAC were presumably harmful to Ang II-treated cardiomyocytes with limited ROS but ameliorated Ang II-induced cardiomyocyte hypertrophic changes through depleting excessive ROS in SASP-treated cardiomyocytes.
In summary, we have found xCT to be a suppressor of pathological hypertrophy by blocking Ang II-induced ferroptosis. We propose positive modulation of xCT as a potential therapeutic strategy for the treatment of cardiac hypertrophic diseases.
Data Availability
All data involved in this study are available from the corresponding author upon reasonable request.
References
Weber KT, Sun Y, Gerling IC, Guntaka RV. Regression of established cardiac fibrosis in hypertensive heart disease. Am J Hypertens. 2017;30(11):1049–52.
Gjesdal O, Bluemke DA, Lima JA. Cardiac remodeling at the population level--risk factors, screening, and outcomes. Nat Rev Cardiol. 2011;8(12):673–85.
Devereux RB, Roman MJ. Left ventricular hypertrophy in hypertension: stimuli, patterns, and consequences. Hypertens Res. 1999;22(1):1–9.
Tang X, Chen XF, Wang NY, et al. SIRT2 acts as a Cardioprotective deacetylase in pathological cardiac hypertrophy. Circulation. 2017;136(21):2051–67.
Schiattarella GG, Hill JA. Inhibition of hypertrophy is a good therapeutic strategy in ventricular pressure overload. Circulation. 2015;131(16):1435–47.
D'Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8(10):813–24.
Brieger K, Schiavone S, Miller FJ Jr, Krause KH. Reactive oxygen species: from health to disease. Swiss Med Wkly. 2012;142:w13659.
Rababa'h AM, Guillory AN, Mustafa R, Hijjawi T. Oxidative stress and cardiac remodeling: An updated edge. Curr Cardiol Rev. 2018;14(1):53–9.
Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49(2):241.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–76.
Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (−): cystine supplier and beyond. Amino Acids. 2012;42(1):231–46.
Wang JY, Deng B, Liu Q, et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. 2020;11(7):574.
Fang X, Cai Z, Wang H, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res. 2020;127(4):486–501.
Bi HL, Zhang XL, Zhang YL, et al. The deubiquitinase UCHL1 regulates cardiac hypertrophy by stabilizing epidermal growth factor receptor. Sci Adv 2020;6(16):eaax4826.
Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–80.
Wang H, An P, Xie E, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66(2):449–65.
Li C, Chen H, Lan Z, et al. mTOR-dependent upregulation of xCT blocks melanin synthesis and promotes tumorigenesis. Cell Death Differ. 2019;26(10):2015–28.
Sehm T, Rauh M, Wiendieck K, Buchfelder M, Eyupoglu IY, Savaskan NE. Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget. 2016;7(46):74630–47.
Daher B, Parks SK, Durivault J, et al. Genetic ablation of the Cystine transporter xCT in PDAC cells inhibits mTORC1, growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res. 2019;79(15):3877–90.
Xie X, Bi HL, Lai S, et al. The immunoproteasome catalytic beta5i subunit regulates cardiac hypertrophy by targeting the autophagy protein ATG5 for degradation. Sci Adv 2019; 5(5): eaau0495.
Simko F, Pechanova O. Remodelling of the heart and vessels in experimental hypertension: advances in protection. J Hypertens. 2010;28(Suppl 1):S1–6.
Gibb AA, Hill BG. Metabolic coordination of physiological and pathological cardiac remodeling. Circ Res. 2018;123(1):107–28.
Li Y, Li Z, Zhang C, et al. Cardiac fibroblast-specific activating transcription factor 3 protects against heart failure by suppressing MAP2K3-p38 signaling. Circulation. 2017;135(21):2041–57.
Sun SJ, Yao JL, Xu LB, et al. Cardiac structural remodeling in hypertensive cardiomyopathy. Hypertens Res. 2017;40(5):450–6.
Shenasa M, Shenasa H. Hypertension, left ventricular hypertrophy, and sudden cardiac death. Int J Cardiol. 2017;237:60–3.
Shin CS, Mishra P, Watrous JD, et al. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat Commun. 2017;8:15074.
Lo M, Wang YZ, Gout PW. The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol. 2008;215(3):593–602.
Lewerenz J, Hewett SJ, Huang Y, et al. The cystine/glutamate antiporter system x(c)(−) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18(5):522–55.
Ottestad-Hansen S, Hu QX, Follin-Arbelet VV, et al. The cystine-glutamate exchanger (xCT, Slc7a11) is expressed in significant concentrations in a subpopulation of astrocytes in the mouse brain. Glia. 2018;66(5):951–70.
Qian M, Lou Y, Wang Y, et al. PICK1 deficiency exacerbates sepsis-associated acute lung injury and impairs glutathione synthesis via reduction of xCT. Free Radic Biol Med. 2018;118:23–34.
Shibasaki T, Iuchi Y, Okada F, et al. Aggravation of ischemia-reperfusion-triggered acute renal failure in xCT-deficient mice. Arch Biochem Biophys. 2009;490(1):63–9.
Liu Q, Wang G, Zhou G, et al. Angiotensin II-induced p53-dependent cardiac apoptotic cell death: its prevention by metallothionein. Toxicol Lett. 2009;191(2–3):314–20.
Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the Ferroptosis network. Free Radic Biol Med. 2019;133:163–8.
Lim J, Delaidelli A, Minaker SW, et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc Natl Acad Sci U S A. 2019;166(19):9433–42.
Schiattarella GG, Hill JA. Metabolic control and oxidative stress in pathological cardiac remodelling. Eur Heart J. 2017;38(18):1399–401.
Liang D, Deng L, Jiang X. A new checkpoint against ferroptosis. Cell Res. 2020;30(1):3–4.
Baba Y, Higa JK, Shimada BK, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2018;314(3):H659–H68.
Park TJ, Park JH, Lee GS, et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019;10(11):835.
Weiland A, Wang Y, Wu W, et al. Ferroptosis and its role in diverse brain diseases. Mol Neurobiol. 2019;56(7):4880–93.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81670380, 81770466), the CAMS Initiative for Innovative Medicine at Chinese Academy of Medical Sciences (2017-I2M-1-008), the State Key Laboratory Special Fund 2060204, and the National Key R&D Program of China (2017YFC1308000).
Author information
Authors and Affiliations
Contributions
Xiyu Zhang and Cuiting Zheng contributed to the collection of data, data analysis and interpretation, and article writing; Zhenqiang Gao, Hongyu Chen, Kai Li, Lingling Wang, Chunjia Li, and Yuanyuan Zheng contributed to the collection of data, as well as data analysis and interpretation; Hongjia Zhang and Ming Gong provided financial support; Hongbing Zhang and Yan Meng provided financial support and contributed to conception and design, and manuscript writing. Yan Meng provided administrative support and final approval of the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, X., Zheng, C., Gao, Z. et al. SLC7A11/xCT Prevents Cardiac Hypertrophy by Inhibiting Ferroptosis. Cardiovasc Drugs Ther 36, 437–447 (2022). https://doi.org/10.1007/s10557-021-07220-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-021-07220-z