
Abstract
Background
In recent studies, granulocyte-colony stimulating factor (G-CSF) was shown to improve cardiac function in myocardial infarction and non-ischemic cardiomyopathies. The mechanisms of these beneficial effects of G-CSF in diabetic cardiomyopathy are not yet fully understood. Therefore, we investigated the mechanisms of action of G-CSF on diabetic cardiomyopathy in a rat model of type 2 diabetes.
Methods
Seventeen-week-old OLETF (Otsuka Long Evans Tokushima Fatty) diabetic rats and LETO (Long Evans Tokushima Otuska) rats were randomized to treatment with 5 days of G-CSF (100 μg/kg/day) or with saline. Cardiac function was evaluated by serial echocardiography performed before and 4 weeks after treatment. We measured expression of the G-CSF receptor (GCSFR) and Bcl-2, as well as the extent of apoptosis in the myocardium.
Results
G-CSF treatment significantly improved cardiac diastolic function in the serial echocardiography assessments. Expression of G-CSFR was down-regulated in the diabetic myocardium (0.03 ± 0.12 % vs. 1 ± 0.15 %, p < 0.05), and its expression was stimulated by G-CSF treatment (0.03 ± 0.12 % vs. 0.42 ± 0.06 %, p < 0.05). In addition, G-CSF treatment increased the expression of Bcl-2 in the diabetic myocardium (0.69 ± 0.06 % vs. 0.26 ± 0.11 %, p < 0.05), consistent with the reduced cardiomyocyte apoptosis (9.38 ± 0.67 % vs. 17.28 ± 2.16 %, p < 0.05).
Conclusions
Our results suggest that G-CSF might have a cardioprotective effect in diabetic cardiomyopathy through up-regulation of G-CSFR, attenuation of apoptosis by up-regulation of Bcl-2 expression, and glucose-lowering effect. Our findings support the therapeutic potential of G-CSF in diabetic cardiomyopathy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic patients frequently develop a cardiomyopathy that is characterized by ventricular hypertrophy and diastolic dysfunction. It may be precipitated by hyperglycemia, an independent risk factor for cardiac damage [1, 2]. Although, the mechanisms of diabetic cardiomyopathy are not fully understood, recent studies have shown that massive loss of ventricular myocytes followed by compensatory hypertrophy of the remaining myocytes, and reproductive fibrosis, play key roles in its development in rodents [3] and humans [4]. Moreover, deleterious effects of diabetes mellitus on cardiac performance due to fibrosis, increased apoptosis, and microvascular and endothelial dysfunction, have been demonstrated in isolated heart preparations and isolated cardiomyocytes [2, 5].
In a number of recent studies, granulocyte-colony stimulating factor (G-CSF) was shown to reduce the size of infarcts and induce myocardial regeneration and the recovery of cardiac function after myocardial infarction [6, 7]. Beneficial effects of G-CSF were also observed in patients suffering from congestive heart failure due to ischemic or dilated cardiomyopathy [7–9]. Similarly, in a rat model, G-CSF was shown to improve cardiac function in adriamycin-induced dilated cardiomyopathy, suggesting that its beneficial effects may extend to non-ischemic heart failure [10]. Previously we reported that G-CSF treatment of diabetic rats ameliorated cardiac diastolic dysfunction and morphological damage, especially fibrosis of the myocardium, but we did not provide any insights into the mechanisms of these therapeutic effects [11].
The aim of this study was to explore the mechanisms behind the beneficial effects of G-CSF on diabetic cardiomyopathy. To this end, we studied its impact on G-CSF receptor (G-CSFR) and on markers of the apoptosis in the cardiac tissue of diabetic animals.
Methods
Experimental Animals
In this study, we followed the ARRIVE guidelines on animal research [12]. All protocols were approved by the Hanyang University Institutional Animal Care and Use Committee. Our experimental subjects were twelve male Otsuka Long-Evans Tokushima Fatty (OLETF) rats with spontaneous long-term hyperglycemia and type 2 diabetes [13], and our controls were twelve male Long-Evans Tokushima Otsuka (LETO) rats, developed from the same colony by selective mating but without diabetes. The OLEFT and LETO rats were supplied by the Tokushima Research Institute (Otsuka Pharmaceutical, Tokushima, Japan). All rats were kept in a dedicated, pathogen-free facility at the Hanyang University Medical School Animal Experiment Center, at controlled temperature (23 ± 2 °C) and humidity (55 ± 5 %) with a 12-h artificial light and dark cycle.
Experimental Design
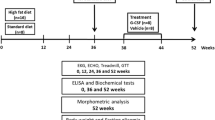
The experimental design, which had developed previously [11], is outlined in Fig. 1. The study began with rats at the age of 7 weeks, given ad libitum access to standard laboratory chow. Because diastolic dysfunction was more pronounced in the sucrose-fed OLTETF rats in our previous study, the OLETF rats were given free access to water containing 30 % sucrose to facilitate the development of diabetic cardiomyopathy, while the LETO rats were given free access to tap water. At 17 weeks of age (after 10 weeks), each group was randomly divided into two sub-groups, that were injected intraperitoneally either with saline or with 100 μg/kg/day recombinant human G-CSF (Leucostim®, Dong-A Pharmacological, Seoul, Korea) for five days.

Schematic description of the experimental protocol. OLETF, Otsuka Long-Evans Tokushima Fatty rats; LETO, Long-Evans Tokushima Otsuka rats; G-CSF, granulocyte-colony stimulating factor
Body weight, fasting blood glucose, total cholesterol (TC) and triglyceride (TG) levels were measured. Blood samples were collected from tail veins after 8 h of fasting, and the levels of serum glucose, TC, and TG were measured using an Olympus AU400 auto analyzer (Olympus GmbH, Hamburg, Germany) [14]. Doppler echocardiography was performed twice: at 17 weeks (before treatment) and at 22 weeks (4 weeks after the saline and G-CSF injections).
Echocardiography and Measurements of Cardiac Function
The rats were anesthetized with 50 mg/kg ketamine and 5.8 mg/kg xylazine HCl. The left side of the chest was shaved to obtain a clear image. Serial echocardiographic examinations (Philips iE33, Philips Medical System with an S8-3 probe) were performed by a single sonographer, with the rats in the left lateral decubitus position. The measurements included left ventricular ejection fraction (LVEF), peak velocity (E), the deceleration time (DT) of the early diastolic filling wave, and early mitral annulus velocity (E’). All measurements were based on the mean of five consecutive cardiac cycles, and mean values were used in analyses [11].
Histopathology
The rats were anesthetized with an intra-peritoneal injection of 50 mg/kg ketamine and 5.8 mg/kg xylazine HCl. They were weighed, and their hearts were removed and divided into two halves along the anterior longitudinal middle line. One half of each heart was fixed in formalin, embedded in paraffin, and cut into 4 μm thick sections in preparation for terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end-labeling (TUNEL) assays. The other half was frozen in liquid nitrogen and stored at −80 °C for the qPCR and western blot analyses.
Detection of myocardial apoptosis by TUNEL assay
The TUNEL assay was performed to detect apoptotic cells in paraffin-embedded cardiac sections. Fragmented double-stranded DNA was identified with an in situ cell detection kit (Roche Diagnostics, Indianapolis, IN, USA). Briefly, endogenous peroxidase was inhibited with 3 % H2O2 in methanol for 10 min, and the sections were permeabilized with 0.5 % Triton-X-100 for 10 min. Proteinase K was applied to induce proteolysis for 15 min at 37 °C, and blocked with 10 % normal goat serum in PBS for 60 min at 37 °C. The slides were incubated with the TUNEL reaction mixture (enzyme and labeling solution in a 1:10 dilution) for 90 min at 37 °C, and incubated with converted-POD for 30 min at 37 °C. Levels of myocardial apoptosis were detected using a DAB kit (Vector Laboratories, Burlingame, CA, USA); apoptotic nuclei were counted to calculate the apoptotic index (number of labeled nuclei/number of total nuclei).
Immunohistochemical Staining for G-CSFR
To identify the presence of G-CSFR in the cardiomyocyte of each heart section, we performed immunohistochemical staining. Tissue sections were incubated for with a mouse monoclonal anti-G-CSFR antibody as the primary antibody (1:50 dilution; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) at 4 °C overnight. The sections were washed and then incubated with a horseradish peroxidase-conjugated donkey anti-rabbit secondary antibody (1:300 dilution; Jackson ImmunoResearch, West Grove, PA) for 90 min. After rinsing with PBS, immunoreactivity was visualized using DAB (Vector Laboratories, Burlingame, CA, USA). Images were obtained on an ECLIPSE 80i microscope equipped with an iAi progressive scan camera (Nikon, Tokyo, Japan) and cytoVision system software (Applied Imaging, Newcastle, UK).
Quantitative Real-Time PCR Analysis of G-CSFR and Bcl-2 Expression
Quantitative real-time polymerase chain reactions (qPCR) were performed using a SYBR Green qPCR Mix (Toyobo, Tokyo, Japan) and analyzed on a LightCycler 1.5 (Roche Diagnostics, Indianapolis, IN, USA). The genes of interest included those for G-CSFR and Bcl-2 (Table 1). qPCR amplification was performed with incubation for 10 min at 95 °C followed by 45 cycles of 10 s at 95 °C, 10 s at 60 °C, and 10 s at 72 °C, and a final dissociation step at 65 °C for 15 s. The crossing point of each sample was automatically determined by the LightCycler, and the relative change ratio was determined using the ratio of mRNA for the selected gene that for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [15]. PCR was performed in duplicate and the transcript levels were normalized against those of GAPDH.
Western Blot Analysis for Bcl-2
Proteins were extracted from fresh-frozen LV myocardium. The samples were homogenized on ice with homogenization buffer (Pro-preb; iNtRON, Seongnam, South Korea). Then, samples containing 50 μg protein were transferred into sample buffer, separated by 10 % sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (0.45 μm pore size, Bio-Rad, Hercules, CA, USA). After blocking in 5 % skim milk solution for 60 min, the membranes were incubated with primary anti-Bcl-2 antibody (dilution 1:2,000; Cell Signaling Technology, Boston, MA, USA), or GADPH (1:1,000, Cell Signaling Technology, Boston, MA, USA) followed by HRP-conjugated anti-rabbit antibody (1:1,000, Jackson Immunoresearch, West Grove, PA, USA). GAPDH was used as a protein loading control. Positive protein bands were visualized using an ECL kit (GenDEPOT, Barker, TX, USA), and the results were quantified with an image analyzer (Image lab 3.0, Bio-Rad, Hercules, CA, USA).
Statistical Analysis
Statistical analyses were performed using SPSS software version 18.0 (SPSS Inc., Chicago, IL, USA). All data are presented as mean ± standard error of the mean (SEM). Differences in measured echocardiographic values between the groups were determined using two-way repeated measures ANOVA with Bonferroni’s post-hoc test for multiple comparisons. The remaining data were analyzed using t-tests (for single comparisons) or one-way ANOVAs (for multiple comparisons) with Tukey’s post hoc test (equal variances assumed). P values < 0.05 were considered statistically significant.
Results
Effects of G-CSF on Body Weight and Biochemical Markers
Table 2 presents measurements of body weight and biochemical markers before and after treatment with G-CSF. The body weight of the OLETF rats (in both sub-groups) was greater than that of the LETO rats. In addition, both OLETF groups had higher fasting glucose, TC, and TG levels than the LETO groups at 17 weeks of age. Regardless of treatment group, there was a significant increase in body weight and fasting glucose levels from 17 to 22 weeks of age in the OLETF rats compared to the LETO rats (p < 0.05). After treatment, the body weight, blood level of TC and TG levels did not differ between the two OLETF groups; however, fasting glucose was significantly lower in the G-CSF treated OLETF sub-group compared to saline treated group (p < 0.05).
Effects of G-CSF Treatment on Cardiac Diastolic Dysfunction
Figure 2 compares the echocardiographic parameters at 17 and 22 weeks of age in the four groups. At 17 weeks, LVEF (i.e. indicator of LV systolic function) was preserved, but the Doppler indices of mitral valve flow (i.e. indicators of LV diastolic function) showed that the E and E’ velocities were lower and the E/E’ ratio was higher in the two OLETF groups than in the LETO groups, suggesting that the OLETF rats had developed diastolic dysfunction. Echocardiography performed at 22 weeks (after the completion of treatment) revealed that, while LVEF did not vary significantly, E’ velocity was higher in the OLTETF rats treated with G-CSF than in the OLETF saline control rats (4.08 ± 0.19 cm/s vs. 2.83 ± 0.21 cm/s, p < 0.05). In addition, there was a significant rise in E’ (3.32 ± 0.36 cm/s vs. 4.08 ± 0.19 cm/s, p < 0.05) and decline in E/E’ (22.07 ± 2.58 vs. 17.95 ± 2.42, p < 0.05) in the G-CSF treated OLETF rats compared to before treatment (Fig. 2). These results show that LV diastolic dysfunction in the OLETF rats was clearly improved by G-CSF treatment.

Effects of G-CSF on cardiac function. EF, ejection fraction; E, peak velocity of the early diastolic filling wave; A, peak velocity of late diastolic filling wave; E’, early mitral annulus velocity during the diastolic phase. LS, LETO rats saline control; LG, LETO rats treated with G-CSF; OS, OLETF rats saline control; OG, OLETF rats treated with G-CSF. All data are mean ± SEM. * p < 0.05 vs. OLETF saline control rats; † p < 0.05 vs. LETO saline control rats
Effects of G-CSF on G-CSFR Expression
To look for evidence of potential direct effects of G-CSF on myocardial tissue, we assessed the expression of G-CSFR in the heart. Immunohistochemistry showed that G-CSFR-positive cells were distributed throughout the cardiomyocytes of LETO rats and there was no significant difference in the pattern of positive staining among LETO rats. However, G-CSFR-positive cells were stained more strongly in G-CSF treated OLETF rats compared to control OLETF rats (Fig. 3a). qPCR showed that the level of G-CSFR mRNA was lower in OLETF rats than in the control LETO rats (0.03 ± 0.12 % vs. 1 ± 0.15 %, p < 0.05). Compared to control OLETF rats, G-CSF mRNA level was significantly increased in G-CSF treated OLETF rats (0.03 ± 0.12 % vs. 0.42 ± 0.06 %, p < 0.05). As expected, G-CSFR expression was very low in the diabetic hearts, and a significant increase was observed when G-CSF was administered (Fig. 3b).

Effects of G-CSF on G-CSF receptor (G-CSFR) expression. a. Immunohistochemical staining for G-CSFR. b. Quantitative PCR for G-CSFR mRNA. LS, LETO rats saline control; LG, LETO rats treated with G-CSF; OS, OLETF rats saline control; OG; OLETF rats treated with G-CSF. All data are mean ± SEM. * p < 0.05 vs. OLETF saline control rats; † p < 0.05 vs. LETO saline control rats
Effects of G-CSF on Bcl-2 Expression and Apoptosis
The apoptotic index was significantly higher in the control OLETF rats than in the control LETO rats (17.28 ± 2.16 % vs. 5.89 ± 2.78 %, p < 0.05), and G-CSF treatment reduced apoptosis in the OLETF rats (9.38 ± 0.67 % vs. 17.28 ± 2.16 %, p < 0.05) (Fig. 4a and b). To understand the molecular basis of the increased apoptosis in the hearts of the diabetic rats, we examined the expression of Bcl-2, an anti-apoptotic protein, and it’s mRNA. Bcl-2 mRNA levels were significantly higher in the G-CSF treated OLETF rats than in the control OLETF rats (1.20 ± 0.19 % vs. 0.58 ± 0.08 %, p < 0.05) (Fig. 4c), and Bcl-2 protein was also significantly higher in the G-CSF treated OELTF rats (0.69 ± 0.06 % vs. 0.26 ± 0.11 %, p < 0.05) (Fig. 4d). These results suggest that the protective effect of G-CSF as at least partly due to up-regulation of Bcl-2 expression in the diabetic myocardium.

Effects of G-CSF on apoptosis and Bcl-2 expression. a. Apoptotic cells in each group. Apoptotic nuclei were stained brown by the TUNEL assay and non-apoptotic nuclei were blue. b. Apoptotic index of cardiomyocytes in each group. c. Quantitative PCR for Bcl-2 mRNA. d. Western blotting for Bcl-2 proteins. LS, LETO rats saline control; LG, LETO rats treated with G-CSF; OS, OLETF rats saline control; OG, OLETF rats treated with G-CSF. All data are mean ± SEM. * p < 0.05 vs. OLETF saline control rats; † p < 0.05 vs. LETO saline control rats
Discussion
In the present study, we examined the effect of G-CSF treatment on relatively early diabetic cardiomyopathy in a rat model. We showed that G-CSF treatment improved diastolic dysfunction and attenuated apoptosis. Moreover, it induced up-regulation of Bcl-2, which provides a molecular explanation for the beneficial effects of G-CSF on diabetic cardiomyopathy. We also found the glucose-lowering effect induced G-CSF particularly noteworthy, as it is likely the critical mechanism contributing to the beneficial effects of this cytokine on improvement in diastolic function and reduction of apoptosis. Expression of the G-CSFR was found to be down-regulated in the diabetic myocardium and up-regulated by G-CSF treatment, which suggests that there is sensitization of the myocardium to direct effects of G-CSF. Together, our results provide insight into the key mechanisms that contribute to the beneficial effects of this cytokine on diabetic cardiomyopathy in OLETF rats.
The interest in G-CSF in the context of ischemic heart disease stemmed from its ability to mobilize bone marrow-derived stem cells into peripheral blood. In particular, it was thought that G-CSF might contribute to tissue regeneration [16]. Much research has been devoted to the role of G-CSF in ischemic and non-ischemic cardiomyopathies, and various mechanisms of its therapeutic actions have been proposed [6–8, 10, 17–19]. Among them are trans-differentiation of bone marrow-derived cells into cardiomyocytes, increased homing and fusion of bone marrow-derived cells with resident cells in the myocardium, direct protective effects on cardiomyocytes through G-CSFR, and stimulation of the production of growth factors and chemokines associated with neovascularization and angiogenesis by paracrine mechanism [6, 20–23]. However, it is not clear which of these mechanisms is most likely responsible for the positive effects of G-CSF treatment. In particular, since previous studies reported that endogenous bone marrow-derived stem cells contribute only to a small proportion of the regenerated myocardium in the acute infarction model [24, 25], it is doubtful that the improvement in cardiac function can be explained solely by bone marrow mobilization.
In this study, we have identified several factors that may contribute to the cardioprotective effect of G-CSF in diabetic cardiomyopathy. The first is up-regulation of G-CSFR expression in the myocardium. We found that G-CSFR was down-regulated in the diabetic myocardium, and its expression could be regulated by a positive feedback mechanism dependent on stimulation by G-CSF itself [26]. Thus there may be a direct protective effect of G-CSF on diabetic cardiomyopathy through the G-CSFR. The enhanced expression of this receptor after G-CSF treatment in cardiomyocytes suggests a sensitization of the heart to direct influences of this cytokine. This is in line with previous studies demonstrating a direct cardioprotective effect of G-CSF in ischemic and non-ischemic cardiomyopathy [6, 9]. However, the expression of G-CSFR, as a mediator of the cardioprotective effect of G-CSF treatment in diabetic cardiomyopathy, is merely speculative, since precise mechanisms of G-CSF’s actions mediated through the G-CSFR and downstream signals of G-CSFR evoked by G-CSF have not been investigated in this study.
A second important finding of the present study is that G-CSF treatment reduces apoptosis. Specifically, G-CSF treatment enhanced the expression of cardiac Bcl-2 protein in diabetic rats, which can be interpreted as inhibition of apoptosis. The ability of G-CSF to inhibit apoptotic death of cardiomyocytes has been demonstrated before in a number of studies [6, 10, 17, 27]. Furthermore, the accumulated evidence suggests that apoptotic cell death is a key element in the pathogenesis and progression of various cardiac diseases, including diabetic cardiomyopathy [5, 10, 17, 28]. Apoptosis can cause a loss of contractile tissue, compensatory hypertrophy of myocardial cells, and reparative fibrosis [29]. Recent studies have shown that the incidence of apoptosis increases in the hearts of patients with diabetes and in streptozotocin-induced diabetic animals, and inhibition of apoptosis improves cardiac function in diabetic cardiomyopathy [3, 4, 30]. Apoptotic proteins in the diabetic myocardium appears to be regulated by a pathway that up-regulates pro-apoptotic proteins and down-regulates anti-apoptotic proteins, which may induce apoptosis. In previous experimental studies, G-CSF treatment reduced cardiomyocyte apoptosis by modulating these apoptosis-related proteins. The groups of Harada et al. and Baldo MP et al. reported a similar anti-apoptotic effect of G-CSF, which was associated with up-regulation of anti-apoptotic proteins such as Bcl-2 and Bcl-xL [6, 17]. Hou et al. showed that G-CSF inhibited the expression of Fas, a pro-apoptotic protein in adriamycin-induced dilated cardiomyopathy in rat [10]. However, while the anti-apoptotic effect of G-CSF has been demonstrated in ischemic cardiomyopathy and dilated cardiomyopathy, its influence on apoptosis in vivo in diabetic cardiomyopathy model remains unknown. In this study, G-CSF treatment significantly increased expression of Bcl-2 (both mRNA and protein), suggesting that G-CSF-mediated cytoprotection may be, at least in part, via an action on apoptosis in diabetic cardiomyopathy.
Another important finding obtained from the present study is that G-CSF treatment reduces plasma glucose concentration. Multiple mechanisms have been described whereby hyperglycemia contributes to the pathological cardiac remodeling, such as direct effects of elevated glucose on cells, oxidative stress, nonenzymatic glycation, and apoptosis [31–33]. Since hyperglycemia presents the most important link for the development of diabetic cardiomyopathy, adequate metabolic control, especially improvement of glycoregulation is the basic strategy in its prevention. Previous studies have shown the protective effect of G-CSF from diabetes. Serum glucose level decreased in healthy peripheral blood stem cell transplantation donors after G-CSF treatment [34]. In addition, other studies have shown that G-CSF treatment prevents spontaneous autoimmune type 1 diabetes and insulitis in the NOD mouse [35], and transplantation of G-CSF-mobilized peripheral blood mononuclear cells may also improve blood glucose metabolism [36]. In this study, G-CSF treatment significantly decreased blood glucose, suggesting that glucose-lowering effect of G-CSF may be another cardioprotective action of G-CSF in diabetic rats, as well as may account for the improvement of diastolic dysfunction and reduction of apoptosis.
This study has several limitations. First, we cannot rule out the possibility that the improved cardiac performance, especially diastolic function, after G-CSF treatment is associated with one of the other, previously postulated, mechanisms, i.e. systemic effect or mobilization or homing of bone marrow stem cells or other paracrine effects, such as fibrosis, vascularization, oxidative stress or structural changes involving the extracellular matrix proliferation, the change of collagen content, and the myocellular hypertrophy. These effects are worth exploring in further studies. Second, this study did not investigate any potential effects of the downstream pathways activated by the binding of G-CSF to its receptor, such as JAK/Stat, PI3K/Akt, MAPK/ERK [6, 26]. And we also could not investigate the more precise mechanisms whether anti-apoptotic actions of G-CSF in the diabetic heart were associated with to the G-CSFR downstream signals. Further intensive studies are also needed to address the question whether the improvements in diastolic dysfunction are mediated through a direct action of G-CSF or through the G-CSFR-mediated signaling pathway or through its improvement of the metabolic milieu. Third, we have found the glucose-lowering effect of G-CSF in early diabetic cardiomyopathy rat model, but we could not investigate the mechanisms how G-CSF reduced the blood glucose level in OLETF rats and any potential causal relationship between glucose reduction and antiapoptotic effect. Further studies about the potential mechanism of this effect linked to modulation of insulin sensitivity, oxidative stress and free fatty acid β-oxidation, as well as the function or histology of pancreas and spleen would be desirable. Finally, our study included only a small number of subjects, and the treatment regimens, such as dosage, timing and duration of G-CSF, were very limited. Before G-CSF can be considered for pharmacological use in the treatment of diabetic cardiomyopathy, the optimal treatment regimen must be determined.
Conclusion
Our results disclose that G-CSF might have a cardioprotective effect in diabetic cardiomyopathy through up-regulation of G-CSFR, attenuation of apoptosis by enhancement of Bcl-2 protein expression, and glucose-lowering effect. Our findings support the therapeutic potential of G-CSF in diabetic cardiomyopathy.
References
Battiprolu PK, Gillette TG, Wang ZV, Lavandero S, Hill JA. Diabetic Cardiomyopathy: Mechanisms and Therapeutic Targets. Drug Discov Today Dis Mech. 2010;7(2):e135–e43. doi:10.1016/j.ddmec.2010.08.001.
Falcao-Pires I, Leite-Moreira AF. Diabetic cardiomyopathy: understanding the molecular and cellular basis to progress in diagnosis and treatment. Heart Fail Rev. 2012;17(3):325–44. doi:10.1007/s10741-011-9257-z.
Fiordaliso F, Li B, Latini R, Sonnenblick EH, Anversa P, Leri A, et al. Myocyte death in streptozotocin-induced diabetes in rats in angiotensin II- dependent. Lab Investig. 2000;80(4):513–27.
Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, et al. Myocardial cell death in human diabetes. Circ Res. 2000;87(12):1123–32.
Yoon YS, Uchida S, Masuo O, Cejna M, Park JS, Gwon HC, et al. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation. 2005;111(16):2073–85. doi:10.1161/01.cir.0000162472.52990.36.
Harada M, Qin Y, Takano H, Minamino T, Zou Y, Toko H, et al. G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat Med. 2005;11(3):305–11. doi:10.1038/nm1199.
Minatoguchi S, Takemura G, Chen XH, Wang N, Uno Y, Koda M, et al. Acceleration of the healing process and myocardial regeneration may be important as a mechanism of improvement of cardiac function and remodeling by postinfarction granulocyte colony-stimulating factor treatment. Circulation. 2004;109(21):2572–80. doi:10.1161/01.cir.0000129770.93985.3e.
Huttmann A, Duhrsen U, Stypmann J, Noppeney R, Nuckel H, Neumann T, et al. Granulocyte colony-stimulating factor-induced blood stem cell mobilisation in patients with chronic heart failure–Feasibility, safety and effects on exercise tolerance and cardiac function. Basic Res Cardiol. 2006;101(1):78–86. doi:10.1007/s00395-005-0556-1.
Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, et al. Granulocyte colony-stimulating factor improves left ventricular function of doxorubicin-induced cardiomyopathy. Lab Investig. 2007;87(5):440–55. doi:10.1038/labinvest.3700530.
Hou XW, Son J, Wang Y, Ru YX, Lian Q, Majiti W, et al. Granulocyte colony-stimulating factor reduces cardiomyocyte apoptosis and improves cardiac function in adriamycin-induced cardiomyopathy in rats. Cardiovasc Drugs Ther. 2006;20(2):85–91. doi:10.1007/s10557-006-7652-9.
Lim YH, Joe JH, Jang KS, Song YS, So BI, Fang CH, et al. Effects of granulocyte-colony stimulating factor (G-CSF) on diabetic cardiomyopathy in Otsuka Long-Evans Tokushima fatty rats. Cardiovasc Diabetol. 2011;10:92. doi:10.1186/1475-2840-10-92.
Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412. doi:10.1371/journal.pbio.1000412.
Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes. 1992;41(11):1422–8.
Blom D, Yamin TT, Champy MF, Selloum M, Bedu E, Carballo-Jane E, et al. Altered lipoprotein metabolism in P2Y (13) knockout mice. Biochim Biophys Acta. 2010;1801(12):1349–60. doi:10.1016/j.bbalip.2010.08.013.
Yi-Sun S, Cheng-Hu F, Byung-Im S, Jun-Young P, Dae Won J, Kyung-Soo K. Therapeutic effects of granulocyte-colony stimulating factor on non-alcoholic hepatic steatosis in the rat. Ann Hepatol. 2013;12(1):115–22.
Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood. 1991;78(11):2791–808.
Baldo MP, Davel AP, Damas-Souza DM, Nicoletti-Carvalho JE, Bordin S, Carvalho HF, et al. The antiapoptotic effect of granulocyte colony-stimulating factor reduces infarct size and prevents heart failure development in rats. Cell Physiol Biochem. 2011;28(1):33–40. doi:10.1159/000331711.
Sato T, Suzuki H, Kusuyama T, Omori Y, Soda T, Tsunoda F, et al. G-CSF after myocardial infarction accelerates angiogenesis and reduces fibrosis in swine. Int J Cardiol. 2008;127(2):166–73. doi:10.1016/j.ijcard.2007.05.007.
Li JM, Yao ZF, Zou YZ, Ge JB, Guan AL, Wu J. The therapeutic potential of G-CSF in pressure overload induced ventricular reconstruction and heart failure in mice. Mol Biol Rep. 2012;39(1):5–12. doi:10.1007/s11033-011-0703-8.
Balsam LB, Wagers AJ, Christensen JL, Kofidis T, Weissman IL, Robbins RC. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature. 2004;428(6983):668–73. doi:10.1038/nature02460.
Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, et al. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature. 2004;428(6983):664–8. doi:10.1038/nature02446.
Norol F, Merlet P, Isnard R, Sebillon P, Bonnet N, Cailliot C, et al. Influence of mobilized stem cells on myocardial infarct repair in a nonhuman primate model. Blood. 2003;102(13):4361–8. doi:10.1182/blood-2003-03-0685.
Gnecchi M, He H, Liang OD, Melo LG, Morello F, Mu H, et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med. 2005;11(4):367–8. doi:10.1038/nm0405-367.
Hamamoto M, Tomita S, Nakatani T, Yutani C, Yamashiro S, Sueda T, et al. Granulocyte-colony stimulating factor directly enhances proliferation of human troponin I-positive cells derived from idiopathic dilated cardiomyopathy through specific receptors. J Heart Lung Transplant. 2004;23(12):1430–7. doi:10.1016/j.healun.2003.09.031.
Fukuhara S, Tomita S, Nakatani T, Yutani C, Kitamura S. Endogenous bone-marrow-derived stem cells contribute only a small proportion of regenerated myocardium in the acute infarction model. J Heart Lung Transplant. 2005;24(1):67–72. doi:10.1016/j.healun.2003.09.032.
Li Y, Takemura G, Okada H, Miyata S, Esaki M, Maruyama R, et al. Treatment with granulocyte colony-stimulating factor ameliorates chronic heart failure. Lab Investig. 2006;86(1):32–44. doi:10.1038/labinvest.3700367.
Dai Y, Ashraf M, Zuo S, Uemura R, Dai YS, Wang Y, et al. Mobilized bone marrow progenitor cells serve as donors of cytoprotective genes for cardiac repair. J Mol Cell Cardiol. 2008;44(3):607–17. doi:10.1016/j.yjmcc.2007.11.011.
Baraka A, AbdelGawad H. Targeting apoptosis in the heart of streptozotocin-induced diabetic rats. J Cardiovasc Pharmacol Ther. 2010;15(2):175–81. doi:10.1177/1074248409356557.
Engel D, Peshock R, Armstong RC, Sivasubramanian N, Mann DL. Cardiac myocyte apoptosis provokes adverse cardiac remodeling in transgenic mice with targeted TNF overexpression. Am J Physiol Heart Circ Physiol. 2004;287(3):H1303–11. doi:10.1152/ajpheart.00053.2004.
Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, et al. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes. 2001;50(6):1414–24.
Young ME, McNulty P, Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation. 2002;105(15):1861–70.
King GL, Wakasaki H. Theoretical mechanisms by which hyperglycemia and insulin resistance could cause cardiovascular diseases in diabetes. Diabetes Care. 1999;22 Suppl 3:C31–7.
Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. 2002;51(6):1938–48.
Stroncek DF, Clay ME, Petzoldt ML, Smith J, Jaszcz W, Oldham FB, et al. Treatment of normal individuals with granulocyte-colony-stimulating factor: donor experiences and the effects on peripheral blood CD34+ cell counts and on the collection of peripheral blood stem cells. Transfusion. 1996;36(7):601–10.
Kared H, Masson A, Adle-Biassette H, Bach JF, Chatenoud L, Zavala F. Treatment with granulocyte colony-stimulating factor prevents diabetes in NOD mice by recruiting plasmacytoid dendritic cells and functional CD4 (+) CD25 (+) regulatory T-cells. Diabetes. 2005;54(1):78–84.
Huang P, Li S, Han M, Xiao Z, Yang R, Han ZC. Autologous transplantation of granulocyte colony-stimulating factor-mobilized peripheral blood mononuclear cells improves critical limb ischemia in diabetes. Diabetes Care. 2005;28(9):2155–60.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shin, J.H., Lim, YH., Song, YS. et al. Granulocyte-Colony Stimulating Factor Reduces Cardiomyocyte Apoptosis and Ameliorates Diastolic Dysfunction in Otsuka Long-Evans Tokushima Fatty Rats. Cardiovasc Drugs Ther 28, 211–220 (2014). https://doi.org/10.1007/s10557-014-6519-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-014-6519-8