
Abstract
Purpose
Tolvaptan may reduce the signs of volume overload in heart failure (HF) patients who experience volume overload despite using conventional diuretics. In this study, we evaluated the dose-response effects of tolvaptan on weight loss, urine volume and electrolyte excretion in furosemide-treated Japanese HF patients exhibiting volume overload.
Methods
In the study, 117 HF patients with volume overload on stable doses of furosemide (≥40 mg/day) were treated with tolvaptan (15, 30 or 45 mg) or placebo once-daily for 7 days.
Results
The decrease in body weight from baseline to the day after the final dose with 15, 30 or 45 mg tolvaptan (–1.62 ± 1.55, –1.35 ± 1.54 and –1.85 ± 1.10 kg, respectively), was significantly greater compared with that in the placebo group (–0.53 ± 0.96 kg) (p < 0.05). However, the decrease in body weight with tolvaptan was not significantly dose-dependent. Signs of volume overload improved at all doses of tolvaptan. Tolvaptan elicited a dose-dependent increase in urine volume and a decrease in urine osmolality, but did not affect urinary sodium or potassium excretion. Adverse reactions associated with diuresis were most frequently observed at the higher doses of tolvaptan.
Conclusions
Once-daily tolvaptan (15, 30 or 45 mg) was effective and tolerable as an add-on treatment to furosemide therapy in Japanese HF patients with volume overload.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The clinical symptoms of heart failure (HF) can generally be divided into volume overload-related and hypoperfusion-related symptoms [1]. In patients with HF, a volume increase can be seen in the vascular and extravascular spaces, resulting in symptoms such as peripheral edema, pulmonary congestion and jugular venous distention. Progression of volume overload leads to decreased quality-of-life and increased risk of hospitalization.
Diuretics are widely used to reduce volume overload associated with HF but can lead to several undesirable clinical effects [2]. In particular, electrolyte imbalances are often associated with the use of diuretics, making volume overload difficult to control. Increased sodium (Na+) and potassium (K+) excretion associated with most diuretics can lead to hyponatremia and hypokalemia, which also worsen quality-of-life and increase patient mortality as a result of neurological symptoms and fatal arrhythmias.
The use of the loop diuretic furosemide is associated with increased activation of the renin–angiotensin system and the sympathetic nervous system [3]. Additionally, the majority of diuretics are also potent antihypertensive agents, meaning hypotension may become a problem. Thus, because of the adverse effects associated with conventional diuretics, diuretic agents targeting different pathways are needed.
Tolvaptan is a selective arginine vasopressin (AVP) V2 receptor antagonist that increases free water clearance by inhibiting V2 receptor-mediated water reabsorption in the renal collecting tubule. Accordingly, tolvaptan is expected to improve volume overload in patients in whom conventional diuretics are ineffective or decrease serum electrolyte concentrations. In one phase II study conducted in the United States (US), Gheorghiade et al. [4] reported that 30, 45 and 60 mg tolvaptan reduced body weight and improved lower limb edema in furosemide-refractory (40–240 mg/day) Caucasian HF patients with volume overload.
In this study, we evaluated the dose-response effects of tolvaptan on weight loss, urine volume and electrolyte excretion in Japanese HF patients with volume overload and compared the results with the study by Gheorghiade et al. [4].
Methods
Study population
HF patients (aged 20–80 years) with signs of volume overload assessed by the investigator, such as edema, jugular venous distention, hepatomegaly, pulmonary congestion, or third cardiac sound at the study screening were eligible for the study. All patients were required to be treated with oral furosemide at a stable dose (≥40 mg/day) for at least 3 days before enrollment. Patients with a body weight change of ±1.0 kg in the last 2 days of a 3-day observation period were selected to enter the study treatment period. The main exclusion criteria included unstable state with acute HF or acute worsening of chronic HF, the presence of a ventricular assist device, open heart surgery or a pacemaker implanted within 60 days before screening, heart surgery scheduled within 30 days of enrolment, reduced circulating plasma volume, anuria, acute myocardial infarction or sustained ventricular tachycardia or ventricular fibrillation within 30 days of enrolment, hypertrophic cardiomyopathy (except in the dilated phase), valvular disease with significant valve stenosis, untreated thyroid disease, severe neurogenic disease, diabetes with poor glycemic control, or cerebral infarction. Patients with any of the following abnormal laboratory findings were also excluded: serum Na+ <120 mEq/L or >147 mEq/L, body mass index ≥35 kg/m2, supine systolic blood pressure <90 mmHg, hemoglobin <9 g/L, total bilirubin >3 mg/dL, serum potassium >5.5 mEq/L).
Study design
This was a multicenter, randomized, double-blind, placebo-controlled, parallel-group study. The objective of this study was to investigate the dose-response effects of once-daily oral tolvaptan for 7 days on weight loss, urine volume and electrolyte excretion in Japanese HF patients with furosemide-refractory volume overload.
The primary endpoint was the change in body weight from baseline to after the final dose. The final dose was the day when the study drug or placebo was last administered; being Day 7 for patients who completed the study, or Day 1–6 for patients who withdrew from the study. Secondary endpoints included signs of volume overload, including lower limb edema, jugular venous distention, hepatomegaly, pulmonary rales, third cardiac sound, and pulmonary congestion. The study was approved by the institutional review board at each participating site. Before screening, written informed consent was obtained from all patients involved.
The study consisted of a screening evaluation, a 3-day observation period, a 7-day treatment period, an end-of-study examination conducted on the day after the final dose, and a post-study examination 1 week later. During the treatment period, all eligible patients continued their stable furosemide dose (≥40 mg/day) and were randomly assigned to receive tolvaptan (15, 30 or 45 mg) or placebo once-daily for 7 days. Furosemide and the study drug were administered after breakfast. Patients were hospitalized from the observation period until the end-of-study examination. The use of loop diuretics other than furosemide, human atrial natriuretic peptides, phosphodiesterase III inhibitors, catecholamines, CYP3A1 inhibitors, or CYP3A4-inducing agents was prohibited. All patients on salt restriction before the study, or those receiving drugs affecting volume status or the underlying disease were instructed to continue these interventions until the last assessment.
Two of the tolvaptan doses (30 and 45 mg/day) used in this study were chosen with reference to the dose-finding study conducted in the US [4]. We also included a low-dose of 15 mg/day. To examine the efficacy of tolvaptan as add-on treatment to current therapy, a placebo group was also included. A total of 30 patients were randomly assigned to each group. The number of patients was determined by weighting the results from the US study [4], and a two-tailed significance test (p ≤ 0.05) with an 80% power of detection and adjustment for multiple comparisons across four groups.
Study endpoints
Body weight was measured after urination and before breakfast to the nearest 0.1 kg from the start of the observation period until the day after final dosing. Weight fluctuations due to defecation or clothing were minimized by measuring body weight at the same time of day and in similar light clothing.
Patients were evaluated daily, 6–8 hours after furosemide administration, for lower limb edema, jugular venous distention, hepatomegaly, pulmonary rales, third cardiac sound, and pulmonary congestion. Patients were also examined for pulmonary congestion once during the 3-day observation period, and during the end-of-study examination.
Lower limb edema was analyzed by palpation of the tibial border or the dorsum of the foot and its severity was evaluated on a four-point scale. Jugular venous distention was evaluated in a semi-supine position by measuring the height (in cm) from the sternal angle to the highest level of jugular venous pulsation. The liver was examined for palpability and hepatomegaly was evaluated as the length (in cm) on the right breast line from the costal arch. Pulmonary rales and third cardiac sounds were examined by auscultation. Pulmonary congestion was evaluated by chest X-rays.
A 24-hour urine sample was collected daily during the observation period and the treatment period to determine urine volume, urine osmolality and urine electrolyte (Na+ and K+) excretion. Other parameters examined included serum osmolality, serum electrolyte (Na+ and K+) concentrations; plasma AVP, brain natriuretic peptide (BNP) and atrial natriuretic peptide (ANP) concentrations; and laboratory readings, vital signs and 12-lead electrocardiograms. Changes in plasma AVP, BNP and ANP were measured using blood samples taken before dosing on the first day of the treatment period and at the end-of-study examination.
Statistical analysis
In analyses of changes in body weight, jugular venous distention, hepatomegaly, rate of improvement of lower limb edema, and the resolution rate of rales and third cardiac sound after final dosing, missing data were supplemented using the last-observation-carried forward method. To evaluate changes in body weight, jugular venous distention and hepatomegaly, comparisons were made between each tolvaptan dose group and the placebo group using Dunnett’s test with a two-tailed significance level of 0.05. Fisher’s exact test was used to compare the rates of improvement of lower limb edema and pulmonary congestion, and the resolution rate of pulmonary rales and third cardiac sound with a two-tailed significance level of 0.0167, after adjustment by the Bonferroni method. Descriptive statistics were also calculated for changes from baseline to all postdose time-points. Changes in body weight and pharmacodynamic characteristics at all postdose time-points were compared between each tolvaptan group and the placebo group using Dunnett’s test at a two-tailed significance level of 0.05.
Patients who showed no abnormalities during the examination period in lower limb edema, pulmonary congestion, pulmonary rales and third cardiac sound prior to dosing were excluded from these analyses to calculate the rates of improvement and/or resolution.
The Kruskal-Wallis rank sum test for age, height, body weight, New York Heart Association class and furosemide fixed-dose range, and Fisher’s exact test or χ 2 test for other demographic characteristics, were used to confirm that any differences in baseline characteristics among the four treatment groups, including significant differences, would not affect the study results.
Results
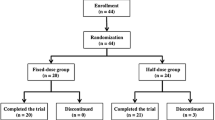
The trial was conducted at 46 centers throughout Japan from August 27, 2004 to January 31, 2006. Patient enrolment and disposition are summarized in Table 1. Of the 122 participants, 118 were eligible for the safety analyses (29, 28, 33 and 28 patients from the placebo, 15, 30, and 45 mg tolvaptan groups, respectively), and 117 were eligible for efficacy analyses (28, 28, 33 and 28 patients, respectively).
Patient characteristics
The characteristics of the patients who were eligible for efficacy analyses (n = 117) are summarized in Table 2. Sex, height, body weight and furosemide fixed-dose range used were unevenly distributed among patients eligible for the efficacy analyses at baseline (p < 0.15).
Efficacy outcomes
The effects of tolvaptan on changes in body weight are shown in Fig. 1. The mean ± standard deviation change in body weight from baseline to after the final dose was −0.53 ± 0.96 kg in the placebo group and −1.62 ± 1.55, −1.35 ± 1.54 and −1.85 ± 1.10 kg, in the 15, 30 and 45 mg tolvaptan groups, respectively, (Fig. 1a). Reductions in body weight were observed on the first day of tolvaptan treatment at all doses and were maintained throughout the study. The mean change in body weight from baseline on Day 1 was −0.20 ± 0.47 kg in the placebo group and −0.74 ± 0.70, −0.90 ± 0.53 and −1.17 ± 0.67 kg in the 15, 30 and 45 mg tolvaptan groups, respectively, indicating a dose-dependent decrease at this time (Fig. 1b). Overall, the three tolvaptan doses elicited comparable reductions in body weight that were significantly greater compared with placebo, although there was no apparent dose-dependency.

Changes in body weight from baseline to the day after the final tolvaptan dose (a) and over time (b) during the 7-day treatment period. Values are means ± standard deviation. *p < 0.05, **p < 0.01
All three doses of tolvaptan resulted in a general improvement in signs of volume overload, including pulmonary congestion, jugular venous distention, hepatomegaly, and lower limb edema. These improvements were not dose-dependent for any symptom (Table 3).
Pharmacodynamic outcomes
As shown in Fig. 2a, tolvaptan dose-dependently increased urine volume from baseline. In contrast, urine osmolality decreased in a nearly dose-dependent manner in the tolvaptan groups from baseline (Fig. 2b). Mean changes from baseline in urinary excretion of Na+ and K+ were not noticeably different between the tolvaptan groups and the placebo group (Fig. 2c, d).

Time-course of changes from baseline in daily urine volume (a), urine osmolality (b), and urinary sodium (c) and potassium (d) excretion during the 7-day treatment period. Values are means ± standard deviation. *p < 0.05
The serum Na+ concentration and serum osmolality on Days 3 and 8 were higher in the tolvaptan groups than in the placebo group (Fig. 3a, b). There was no notable difference in either the time-course or the magnitude of changes in serum K+ concentrations between the tolvaptan groups and the placebo group (Fig. 3c).

Time-course of changes from baseline in serum osmolality (a), and serum sodium (b) and potassium (c) concentrations. Values are means ± standard deviation. *p < 0.05
The changes in plasma neurohormonal peptide levels are shown in Table 4. The plasma AVP concentration was dose-dependently increased by tolvaptan. In contrast, ANP and BNP tended to decrease in the tolvaptan and placebo groups compared with baseline. However, no significant differences in ANP or BNP were observed between groups.
Safety
The incidence of adverse events was 62.1% (n = 18/29, 55 episodes) with placebo and 85.7% (n = 24/28, 72 episodes), 97.0% (n = 32/33, 92 episodes), and 82.1% (n = 23/28, 63 episodes) with 15, 30 and 45 mg tolvaptan, respectively. The most common adverse events pooled from all tolvaptan groups were thirst, elevated uric acid, and dehydration, in order of descending prevalence. Thirst and dehydration appeared to be related to aquaresis—the pharmacological effect of tolvaptan. The number of subjects presenting with thirst and dehydration increased in a tolvaptan dose-dependent manner (Table 5). Most of the adverse events were transient and either improved or resolved over time.
Serious adverse events occurred in seven patients. Of these, four were in the placebo group, and included two deaths (sudden death, cardiopulmonary arrest), congestive heart failure in one patient and umbilical hernia in one patient. Among patients given tolvaptan, nasal bleeding occurred in one patient given 30 mg tolvaptan, effort dyspnea in one patient given 45 mg tolvaptan, and transient ischemic attack in one patient given 45 mg tolvaptan. Only effort dyspnea could not be excluded as being related to tolvaptan use.
Discussion
Diuretics are widely recommended in various guidelines, including the Guidelines for the Treatment of Chronic Heart Failure (2005 Revised Edition) from the Japanese Circulation Society [5–7]. However, correction of volume overload in HF remains a challenging clinical task. Furthermore, the use of conventional diuretics in HF patients is associated with electrolyte abnormalities, hypotension, prerenal azotemia and upregulation of the renin–angiotensin system, and there are currently few therapeutic options to optimize fluid volume [8,9]. Based on the current situation, the concomitant use of an AVP V2 receptor antagonist with a loop diuretic or thiazide may offer an effective approach to control fluid volume in patients with HF.
Tolvaptan is a selective AVP V2 receptor antagonist. In the US, tolvaptan has been reported to improve volume overload in patients with HF [4]. Tolvaptan also has been reported to improve dyspnea, edema, and hyponatremia in decompensated HF patients [10]. In Europe and the US, tolvaptan has been approved for the treatment of hyponatremia [11].
Improvement in long-term prognosis and volume overload are the objectives of any new medications for patients with HF. In this study, we investigated the short-term efficacy of tolvaptan as add-on to furosemide in terms of improvements in volume overload estimated by body weight and congestive signs in Japanese patients with HF. In the earlier US study that evaluated 30, 45 and 60 mg tolvaptan, a similar reduction in body weight was observed [4]. Therefore, in our study, we also evaluated the effects of a lower dose of tolvaptan (15 mg) in Japanese patients.
Efficacy
The primary endpoint of the study was the change in body weight from baseline, since this parameter closely reflects hypervolemic status [12] and is a convenient and accurate measurement. Secondary endpoints were changes in signs and symptoms associated with hypervolemic status.
In this study, all three doses of tolvaptan elicited significant reductions in body weight over 7 days of 1.0–1.5 kg compared with placebo. Based on prior estimates of a 2.0–3.0-kg weight gain associated with worsening of HF [5], this amounts to an approximately 50% reduction in hypervolemia. This finding is agrees with those of a previous study in which tolvaptan given as add-on to furosemide reduced body weight by 1.0–1.5 kg compared with placebo [4]. Furosemide has been shown to reduce body weight by <1 kg in HF patients with volume overload [13]. Thus, the 1.0–1.5 kg reduction in body weight with tolvaptan given as add-on treatment with furosemide appears to be clinically meaningful in terms of the magnitude of its effect.
A previous report demonstrated that in addition to Na+ retention, water retention is also implicated in volume overload-induced HF [14]. The results of this study showing an improvement in volume overload with tolvaptan indicate that water retention is involved in volume overload in HF. These results suggest a benefit in using an aquaretic to promote excretion of excess water in HF patients with volume overload.
Analysis of the secondary endpoints analysis showed that all three tolvaptan doses improved the signs of volume overload such as lower limb edema, pulmonary congestion, jugular venous distention, and hepatomegaly, but these improvements were not dose-dependent. In the earlier US study, no dose-response relationships in terms of body weight reduction or ankle edema were observed for 30–60 mg tolvaptan [4].
Tolvaptan exhibited a dose-dependent relationship in terms of its aquaretic effects, but not for changes in body weight or signs of volume overload. This could be explained by the fact that tolvaptan-induced aquaresis led to an increased sense of thirst and water intake by patients. However, water intake was not measured in this study and future studies will need to examine the relationship between water balance and body weight. In this study, a reduction in body weight was observed, even with unrestricted water intake, which likely led to a negative water balance and an improvement in volume overload.
Of particular note is that tolvaptan did not decrease serum electrolyte concentrations, unlike conventional diuretics. In this study, we found that none of the patients exhibited an acute increase in serum Na+ level >10 mEq/24 h, while one patient given 15 mg, two patients given 30 mg and four patients given 45 mg tolvaptan exhibited an increase in the serum Na+ level beyond the normal range. These results suggest a therapeutic role for tolvaptan in patients with impaired water excretion, and may prevent electrolyte imbalances that may lead to fatal arrhythmias.
An increase in the plasma AVP level was seen in patients given tolvaptan. A likely explanation for this is that tolvaptan increased the serum osmolality, thereby stimulating AVP secretion. An increase in AVP secretion may lead to undesirable clinical effects such as AVP V1 receptor-mediated vasoconstriction and AVP V2 receptor-mediated water reabsorption. However, although an increase in the AVP level was observed, no increase in blood pressure was seen, suggesting that the elevated AVP did not induce adverse effects via the AVP V1 receptor because of antagonism by tolvaptan. Furthermore, a consistent increase in urine volume was observed throughout the study, suggesting no enhancement in AVP V2 receptor activity.
Potential effects of tolvaptan on the renin–angiotensin system or on renal function, which are affected by conventional diuretics, were not evaluated in this study. However, it has been reported that tolvaptan does not upregulate the renin–angiotensin system or the sympathetic nervous system in a study of conscious canines [15]. Furthermore, tolvaptan did not affect markers of renal function, glomerular filtration rate or renal plasma flow in a clinical study [16]. Furthermore, no increases in serum creatinine or blood urea nitrogen were reported in the EVEREST trial, which evaluated long-term tolvaptan use [17].
Safety
Tolvaptan was not associated with any major clinical concerns and was clinically tolerable at all doses tested. Higher doses of tolvaptan led to more episodes of thirst and dehydration. If excessive diuresis occurs, water intake is recommended.
Limitations of the study
This study aimed to evaluate the efficacy of tolvaptan when used in combination with furosemide, and was not designed to evaluate the efficacy of tolvaptan given as a single agent. Such evaluation is needed in patients with decreased electrolyte levels in whom the use of conventional diuretics may not be effective. In addition, the efficacy of tolvaptan was evaluated over the short-term (7 days) rather than the long-term. A previous study showed that, in patients with decompensated HF, tolvaptan did not worsen long-term prognosis [17], although this was not evaluated in the current study. Future studies are needed to evaluate the efficacy of tolvaptan in maintaining the observed improvements in volume overload-associated signs.
Conclusions
In this study of Japanese HF patients with volume overload despite the use of furosemide (≥40 mg), tolvaptan given at doses of 15, 30 or 45 mg once-daily for 7 days significantly reduced body weight and improved the signs of volume overload. Tolvaptan was efficacious as add-on treatment to conventional diuretic therapy without decreasing serum electrolyte levels. Tolvaptan exhibited dose-dependent aquaretic effects leading to a dose-dependent increase in the frequency of adverse reactions such as thirst and dehydration. Therefore, 15 mg tolvaptan is recommended as a therapeutic dose for Japanese patients with HF to improve volume overload. The use of tolvaptan at a dose lower than 15 mg may be considered in patients at high risk for acute or severe aquaresis. Phase III placebo-controlled studies are now needed to confirm the efficacy of tolvaptan, particularly 15 mg tolvaptan, in improving volume overload-associated sequelae in Japanese patients.
References
Nohria A, Tsang SW, Fang JC, et al. Clinical assessment identified hemodynamic profiles that predict outcomes in patients admitted with heart failure. J Am Coll Cardiol. 2003;41:1797–804.
Gupta S, Neyses L. Diuretic usage in heart failure: a continuing conundrum in 2005. Eur Heart J. 2005;26:644–9.
Francis GS, Siegel RM, Goldsmith SR, Olivari MT, Levine TB, Cohn JN. Acute vasoconstrictor response to intravenous furosemide in patients with chronic congestive heart failure. Activation of the neurohormonal axis. Ann Intern Med. 1985;103:1–6.
Gheorghiade M, Niazi I, Ouyang J, et al. Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial. Circulation. 2003;107:2690–6.
Japanese Circulation Society. Guidelines for the Treatment of Chronic Heart Failure, 2010. Available at: http://www.j-circ.or.jp/guideline/pdf/JCS2010_matsuzaki_h.pdf. Accessed 13 April 2011.
Hunt SA, Abraham WT, Chin MH, et al. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation. 2005;112:e154–235.
Mancia G, De Backer G, Dominiczak A, et al. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. 2007;25:1105–87.
Schrier R. The patient with hyponatremia or hypernatremia. In: Schrier R, editor. Manual of nephrology. 4th ed. Boston: Little, Brown & Co; 1995. p. 20–36.
Zeidel M. Special diuretics. In: Seldin D, Giebisch G, editors. Diuretic agents: clinical physiology and pharmacology. San Diego: Academic Press Inc; 1997. p. 113–34.
Gheorghiade M, Konstam MA, Burnett JC Jr, et al. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials. JAMA. 2007;297:1332–43.
Ghali JK, Hamad B, Yasothan U, Kirkpatrick P. Tolvaptan. Nat Rev Drug Discov. 2009;8:611–12.
Hunt SA, Abraham WT, Chin MH, et al. 2009 focused updated incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration with the International Society for Heart and Lung Transplantation. J Am Coll Cardiol. 2009;53:e1–e90.
Muller K, Gamba G, Jaquet F, Hess B. Torasemide vs. furosemide in primary care patients with chronic heart failure NYHA II to IV—efficacy and quality of life. Eur J Heart Fail. 2003;5:793–801.
Chen HH, Schrier RW. Pathophysiology of volume overload in acute heart failure syndromes. Am J Med. 2006;119:S11–6.
Miyazaki T, Fujiki H, Yamamura Y, Nakamura S, Mori T. Tolvaptan, an orally active vasopressin V(2)-receptor antagonist—pharmacology and clinical trials. Cardiovasc Drug Rev. 2007;25:1–13.
Costello-Boerringter LC, Smith WB, Boerrigter G, et al. Vasopressin-2-receptor antagonism augments water excretion without changes in renal hemodynamics or sodium and potassium excretion in human heart failure. Am J Physiol Renal Physiol. 2006;290:F273–8.
Konstam MA, Gheorghiade M, Burnett JC Jr, et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297:1319–31.
Disclosures
None of the authors have any conflicts of interest associated with this study.
Author information
Authors and Affiliations
Consortia
Corresponding author
Appendix: Participating investigators and institutions
Appendix: Participating investigators and institutions
The following investigators and institutions participated in this trial: Akihiro Azuma, University Hospital, Kyoto Prefectural University of Medicine, Kyoto; Yoshinori Doi, Kochi Medical School Hospital, Nankoku, Kochi; Kenji Fujii, Sakurabashi Watanabe Hospital, Osaka; Kazuteru Fujimoto, National Hospital Organization Kumamoto Medical Center, Kumamoto; Jun Fuse, National Hospital Organization Tokyo Medical Center, Meguro, Tokyo; Hiroyuki Hanada, Hirosaki University School of Medicine & Hospital, Hirosaki, Aomori; Michiaki Hiroe, National Center for Global Health and Medicine, Shinjuku, Tokyo; Hitoshi Hishida, Fujita Health University, Toyoake, Aichi; Hiroaki Hosokawa, National Hospital Organization Toyohashi Medical Center, Toyohashi, Aichi; Takayuki Inomata, Kitasato University Hospital, Sagamihara, Kanagawa; Ryoji Ishiki, Toyota Memorial Hospital, Toyota, Aichi; Hideo Izawa, Nagoya University Hospital, Nagoya, Aichi; Toshio Kasai, National Hospital Organization Nagano National Hospital, Nagano; Shu Kasama, Cardiovascular Hospital of Central Japan, Seta-gun, Gunma; Shun-ichi Kobayashi, Kanagawa Cardiovascular and Respiratory Center, Yokohama, Kanagawa; Masahiko Koda, Gifu Prefectural General Medical Center, Gifu; Sunao Kojima, Kumamoto University Hospital, Kumamoto; Kengo Matsumoto, National Hospital Organization Kure Medical Center, Kure, Hiroshima; Takahiro Matsumoto, National Hospital Organization Kyushu Medical Center, Fukuoka; Tatsuru Matsuoka, National Hospital Organization Kagoshima Medical Center, Kagoshima; Shinji Miki, Mitsubishi Kyoto Hospital, Kyoto; Kazuaki Mitsudo, Kurashiki Central Hospital, Kurashiki, Okayama; Toshiro Miura, Yamaguchi University Hospital, Ube, Yamaguchi; Shigeyuki Nishimura, Saitama Medical University Hospital, Iruma-gun, Saitama; Ryuji Nohara, Kitano Hospital, Osaka; Akira Nozaki, Kanto Central Hospital of the Mutual Aid Association of Public School Teachers, Setagaya, Tokyo; Hiroshi Ogawa, Tokuyama Central Hospital, Shunan, Yamaguchi; Yoshihiko Sakai, Dokkyo Medical University Koshigaya Hospital, Koshigoe, Saitama; Mamoru Sato, Iwate Medical University Hospital, Morioka, Iwate; Ikuo Segawa, Iwate Medical University Hospital, Morioka, Iwate; Yoshihiko Seino, Nippon Medical School Hospital, Bunkyo, Tokyo; Junya Shite, Kobe University Hospital, Kobe, Hyogo; Jun-ichi Suzuki, Tokyo Medical and Dental University, Bunkyo, Tokyo; Masahiro Suzuki, National Hospital Organization Saitama National Hospital, Wako, Saitama; Shuichi Taguchi, National Hospital Organization Mito Medical Center, Ibaraki; Yoshifumi Takada, Tokyo Medical University Hospital, Shinjuku, Tokyo; Keiji Tanaka, Nippon Medical School Hospital, Bunkyo, Tokyo; Masaru Tanaka, Osaka Red Cross Hospital, Osaka; Jun Tanouchi, Osaka Rosai Hospital, Sakai, Osaka; Chuwa Tei, Kagoshima University Medical And Dental Hospital, Kagoshima; Hitoshi Toda, Kagoshima City Hospital, Kagoshima; Kazufumi Tsuchihashi, Sapporo Medical University Hospital, Sapporo, Hokkaido; Hitoshi Yasumoto, Kokura Memorial Hospital, Kitakyushu, Fukuoka; Yoshio Yasumura, National Hospital Organization Osaka National Hospital, Osaka; Hiroyuki Yokoyama, National Hospital Organization Shizuoka Medical Center, Suntou gun, Shizuoka.
Rights and permissions
About this article
Cite this article
Matsuzaki, M., Hori, M., Izumi, T. et al. Effects of Tolvaptan on Volume Overload in Japanese Patients with Heart Failure: Results of a Phase II, Multicenter, Randomized, Double-blind, Placebo-controlled, Parallel-group Study. Cardiovasc Drugs Ther 25 (Suppl 1), 19–31 (2011). https://doi.org/10.1007/s10557-011-6303-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-011-6303-y