
Abstract
Objective
The protective properties of heme oxygenase 1 (HO-1) give reason to study this mechanism as a potential therapeutic target for inflammatory and cardiovascular diseases. Recent evidence suggests a possible interaction between the HO-1/CO- and the protein kinase Akt/NO-pathway. This study was designed to examine the effects of continuous HO-1 overexpression in endothelial cells.
Methods
Oncoretroviral vectors were constructed to achieve constitutive overexpression of HO-1, Akt, and green fluorescence protein in human umbilical vein endothelial cells. [3H]thymidine-incorporation and lipid-peroxidation were measured following exposure to heme and H2O2. Expression of HO-1, Akt and its downstream-target endothelial NO-synthase were quantified by Western blot analysis. NO-synthase-activity was measured using the citrulline-conversion-assay.
Results
HO-1-overexpression reduced proliferative rates and DNA-synthesis of HUVEC, but provided potent protection from oxidative stress induced by heme and H2O2. Phosphorylated-Akt and eNOS was downregulated in HO-1-HUVEC. eNOS-activity was reduced in HO-1-HUVEC. Co-infection with the Akt-retrovirus restored proliferative rates and eNOS-expression and -activity.
Conclusion
Continuously elevated HO-1-activity protects EC from oxidative stress but inhibits Akt-mediated proliferation and eNOS-expression. This inhibitory feedback mechanism could be a limitation of HO-1 as a target for the treatment of vascular disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Heme oxygenases (HOs) are the rate limiting enzymes in heme degradation. They catalyze the oxidative cleavage of the heme ring to form biliverdin IXα, releasing ferrous iron and carbon monoxide (CO). The transcriptional activity of the hmox1 gene is upregulated by various chemical, physical and other noxious stimuli, providing the attacked cell prompt assistance by the expression of the 32 kDa heat shock protein heme oxygenase-1 (HO-1). The HO-1 pathway has been the subject of extensive studies and all HO reaction products were shown to be integrated into the anti-oxidant and anti-inflammatory defense cascades [1–3] in a complex manner. On a cellular level, HO-1/CO appears to exhibit dose-dependent effects with respect to cell survival [1, 4–6] and apoptotic cell death [7]. Several experimental studies provide evidence that HO-1 is a potential target for the treatment of various diseases of the cardiovascular system [8–12], organ transplantation [13–15], inflammatory bowel disease [16], autoimmune neuroinflammation [17] and TNFα-induced liver failure [18].
HO-1 expression and activity are regulated on a transcriptional and posttranslational level, involving the Jak/STAT pathway [19], the p38β mitogen activated protein kinase (MAPK) signaling pathway [6, 20, 21] and the extracellular signal regulated protein kinase ERK1/2 [22]. HO-1 was shown to be localized in plasma membrane caveolae, with caveolin-1 partly modulating its enzymatic activity [23, 24].
Recent evidence suggests a possible interaction between the HO-1/CO- and the protein kinase Akt-pathway. It was reported that the incubation of rat pheochromocytoma cells with nerve growth factor or the phenol carnesol reduced reactive oxygen species by an upregulation of HO-1 in a phosphatidylinositol 3-kinase PI3K/Akt-dependent manner [25, 26]. Brunt et al. reported that the protection of rat vascular smooth muscle cells (VSMCs) in vitro from H2O2 induced oxidative stress by HO-1 depends on the coactivation of the PI3K/Akt-pathway [4]. Akt appears to phosphorylate HO-1 at Ser188 [27].
It is well established that the Akt-pathway plays a key role in the vascular homeostasis. Especially in EC, numerous important pathways including cell cycle progression, telomerase activity, handling of lipid particles, susceptibility to inflammatory stimuli, anti-thrombotic mechanisms etc. are regulated by Akt and its main effector eNOS. Prolonged imbalance of the Akt/NO-pathway is involved in the pathophysiology of endothelial dysfunction, impaired angiogenesis, hypertension, thrombosis, atherosclerosis and plaque stability [28–34].
The aim of this study was to examine the impact of continuous HO-1 overexpression in EC with respect to Akt dependent effects.
2 Materials and methods
2.1 Recombinant retroviruses
Bicistronic pC.MMP-vector constructs were generated as previously described [35, 36]. Briefly, the human HO-1 cDNA was PCR-amplified to introduce a 6-histidine-tag at the 5′-end and flanking 5′-NcoI- and 3′-BamHI-restriction sites and TA-cloned into pGEM-T (Promega, Mannheim, Germany). Ligations were performed with the Rapid DNA-Ligation kit (Roche Applied Science, Mannheim, Germany) in a 5 min-reaction at room temperature. The fragment was then inserted together with an IRES-GFP-element into the murine retroviral cassette pC.MMP. The HA-tagged constitutively active, myristoylated, murine Akt1-cDNA [31] was kindly provided by Dr. R. Braun-Dulläus, Dresden, Germany, and cloned into pC.MMP. We constructed this vector to achieve nuclear red-fluorescence in the infected cell via an IRES-DsRed2.Nuc-element derived from pIRES2-DsRed2 (BD Biosciences Clontech, Heidelberg, Germany) with two PCR-added copies of the nuclear-localization-signal of the SV40-large T-antigen at the 3′-end of the DsRed2-reading frame. Cloning sites and reading frames were confirmed by automated sequencing. Replication-defective MMP-retroviruses were generated in 293T-cells by co-transfection of the vector constructs (empty vector for the mock-infection negative controls) with the helper plasmids pMD.M-MLV.gag/pol and pMD.M-VSV.G.env. Retroviral supernatant was collected 48 h later and titered on NIH/3T3-cells. The target cells were infected by an 8-h-exposure with the retroviral particles at 37°C in the presence of polybrene (8 μg/ml; Sigma-Aldrich, Germany). The cells were used at day 5 after infection.
2.2 Cell culture
2nd-passage human umbilical vein endothelial cells (HUVEC) were purchased from PromoCell, Heidelberg, Germany and grown in the recommended EGM-media supplemented with 5% FCS, 0.4% ECGS/H, 10 ng/ml EGF, and antibiotics (media and all supplements were purchased from PromoCell). All the experiments were performed with HUVEC before passage 6. 293T-cells were cultivated in DMEM (Invitrogen, Karlsruhe Germany) with 10% fetal calf serum, and antibiotics. NIH/3T3-cells were maintained in DMEM with 10% calf serum, and antibiotics.
2.3 Measurement of HO-activity and immunolocalization
Total HO-activity was measured in cell-lysates in the absence or presence of the HO-1 inhibitor zinc-protoporphyrin IX (ZnPP, 25 μM; Frontier Scientific Porphyrin Products, UT, USA) by a spectrophotometric quantification of bilirubin production as previously described [10]. Briefly, subconfluent cultures were lysed in 500 μl (per 100 mm dish) ice-cold SDS sample buffer (Tris pH 6.8, 62.5 mM, SDS 2%, glycerol 10%, DTT 50 mM). 160 μg of sample protein were then incubated for 40 min at 37°C in the dark in a final volume of 700 μl 0.1 M phosphate buffer (pH 7.4) containing 5 mM MgCl2, 1 mM NADPH, 2 mM Glucose-6-P, 1 U Glucose-6-P-dehydrogenase, 25 μM hemin chloride (reagents from Sigma-Aldrich), and 2 mg of biliverdin reductase (prepared from rat liver homogenates by ultracentrifugation with 100,000×g, for 1 h at 4°C in a SW41Ti rotor). Reaction was stopped by transferring the mixtures on ice for 5 min. Absorption was measured at 464 nm (for bilirubin, extinction coefficient 40 mM−1 cm−1) and 530 nm (basal absorption). HO-activity could then be quantified in nmol of bilirubin production per mg of sample protein per hour. For transgene immunolocalization, infected HUVEC were plated in 4-well chamber slides and fixed with 10% buffered formalin. Blocking with 5% BSA was followed by an overnight incubation at 4°C with antibodies recognizing the 6His-tag (Qiagen, Hilden, Germany). Rhodamine-conjugated secondary antibody was purchased from Jackson ImmunoResearch (distributed by Dianova, Hamburg, Germany). The antibodies were used at recommended dilutions. After washing, the cells were covered with Vectashield containing DAPI (Vector Laboratories, Burlingame, CA, USA) for nuclear staining.
2.4 Oxidative stress and [3H]thymidine incorporation assay
1.0 × 104/well infected HUVEC were plated into 24-well plates, and allowed to attach overnight. Oxidative stress was induced by H2O2 (200 μM; Sigma-Aldrich, Germany), or heme (50 μM; Sigma-Aldrich, Germany) for 48 h. DNA-synthesis was quantified by [3H]thymidine-incorporation (1 μCi/ml; Amersham Pharmacia Biotech, Freiburg, Germany) during the following 24 h with complete growth medium. Control cells were cultered in parallel without the noxious stimuli. Radioactivity was measured after fixation and cell lysis with NaOH in a standard liquid scintillation counter. Rates of DNA-synthesis were analyzed in relation to the values obtained from the control HUVEC.
2.5 Lipid peroxidation assay
The magnitude of oxidative stress was assessed by measuring total lipid peroxides (malondialdehyde and 4-hydroxynonenal) using a commercially available spectrophotometric kit (Calbiochem-Novabiochem, Bad Soden, Germany).
2.6 Antibodies and western blotting
For Western blot analysis, HUVEC were plated at 2.0 × 105 in 60 mm dishes and allowed to attach overnight. A 12-h-serum deprivation was followed by a 12-h period under regular culture conditions. The cells were lysed in 100 μl SDS sample buffer (Tris pH 6.8, 62.5 mM, SDS 2%, glycerol 10%, DTT 50 mM) on ice. The extracts were transferred to microcentrifuge tubes and heated to 95°C for denaturation. For immunoprecipitation, a commercially available kit (Protein G; Roche Applied Science, Mannheim, Germany) was used. Protein content was photometrically determined with a BCA assay using a BSA standard curve. 30 μg of protein were separated under denaturing conditions by SDS-PAGE (4–15% gradient gels; Bio-Rad Laboratories, Munich, Germany), transferred to nitrocellulose membranes (Whatman, Dassel, Germany) and probed with the following antibodies at recommended dilutions: human HO-1 (Research Diagnostics, Germany), Akt, p-Akt (Ser473), eNOS, p-eNOS(Ser1177) (Cell Signaling Technology, MA, USA), GFP and β-actin (Santa Cruz Biotechnology, CA, USA). Specific signal intensities were quantified using a Bio-Rad GelDoc 2000-System.
2.7 Citrulline assay for eNOS activity
eNOS activity was determined by measuring the conversion of [3H]l-arginine (1 μCi/ml; Amersham Pharmacia Biotech, Freiburg, Germany) to [3H]l-citrulline in the presence or absence of l-NAME (1 mM) using an NOS assay kit (Calbiochem-Novabiochem, Bad Soden, Germany) according to the manufacturers instructions.
2.8 Statistical analysis
Results are expressed as mean±SEM. The experimental data were analyzed by ANOVA with Bonferroni’s correction or unpaired two-tailed t-test using SPSS software. The probability values P < 0.05 were considered to be significant.
3 Results
3.1 Retroviral vectors allow continuous and moderate elevation of HO-1 activity in HUVEC
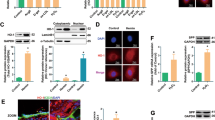
In order to evaluate the cytoprotective properties of HO-1 in endothelial cells, we cloned a series of replication defective retroviral vectors. The construct for gene transfer of HO-1 was bicistronic and resulted in cytoplasmic coexpression of the green fluorescence protein (GFP) in the infected cell. GFP-virus- and mock-infected-cells served as negative controls. Figure 1 summarizes the biochemical and functional characterization of MMP-mediated transduction of HO-1 in HUVEC. Robust expression of the 6His-tagged HO-1-transgene was found 5 days after the infection in an MOI (multiplicity of infection)-dependent fashion (Fig. 1a). We used an MOI of 4 for all future experiments resulting very reproducably in transduction rates of >95% (data not shown). Immunoprecipitation verified the tagged polypeptide as HO-1 (Fig. 1b). Fluorescence immunohistochemistry demonstrated that the transferred gene product was—like the endogenous HO-1 enzyme—localized in the microsomal compartment (Fig. 1c). Actual HO-activity was quantified photometrically by measuring bilirubin-production, and showed that our retroviral transduction protocol resulted in a moderate and stable elevation of the enzyme activity in HUVEC (5.4-fold vs GFP) (Fig. 1d).

Retrovirus-mediated gene transfer of HO-1 results in moderately elevated enzyme activity levels in HUVEC. Western blot analysis for expression of 6HisHO-1 (32 kDa) and GFP (27 kDa) in HUVEC 5 days after infection with GFP- or 6HisHO-1-IRES-GFP retroviruses using indicated multiplicities of infection (MOI). Bicistronic vector design results in equimolar expression of the marker protein. Note linear increase of gene expression levels with increasing MOIs (a). Immunoprecipitation verifies 6His-tagged protein as human HO-1. Five days after the infection HO-1-HUVEC were lysed, and protein homogenates were incubated with the anti-6His-antibody and protein G for separation. Whole cell lysates (left lane) and the immunoprecipitated fraction (right lane) were separated. Membranes were probed with anti-human HO-1 antibody (b). 6HisHO-1 transgene (Rhodamine Red) shows microsomal localization in 6HisHO-1-IRES-GFP infected HUVEC (GFP green, nucleus DAPI-blue) (c). HO-activity is elevated 5.4-fold in HUVEC 5 days following retroviral infection at an MOI of 4. Photometric quantification of bilirubin-production in the absence or presence of the HO-1-inhibitor ZnPP (Data are mean±SEM, n = 5 for each group, *P < 0.05 vs GFP) (d)
3.2 HO-1 is antiproliferative, and still provides protection from oxidative stress induced by H2O2 and heme
We noted that 5 days after retroviral infection, HO-1-HUVEC had 30% to 50% lower cell counts compared to mock- or GFP-infected negative-control cells. Nevertheless, when simultaneously replated at identical cell numbers and exposed to oxidative stress mediated by heme and H2O2 for a period of 48 h, more of the HO-1-overexpressing cells survived, so that the rate of DNA-synthesis in the following 24 h was still 76 ± 10% (heme) and 83 ± 7% (H2O2) compared to the HO-1-HUVEC that were not exposed to the noxius chemicals. DNA-synthesis of mock- and GFP-HUVEC dropped to 12–39%, respectively (Fig. 2a). We directly assessed the magnitude of oxidative stress under these conditions, and found that lipid peroxidation was markedly reduced in HO-1-HUVEC (Fig. 2b), indicating superior handling of reactive oxygen species in HO-1-HUVEC to prevent oxidative damage and functional destruction of membrane lipids.

Retrovirus-mediated gene transfer of HO-1 provides protection from oxidative stress to HUVEC. Reduction of lipid peroxidation. (a) HUVEC were infected with indicated retroviral vectors at an MOI of 4 and 5 days later plated at 1.0 × 104/well in 24-well plates, six repeats per group. Cells were allowed to attach overnight. One half was exposed to heme (50 μM) or H2O2 (200 μM) in culture media for 48 h. The other half remained in the culture media only. After washing fresh media was added, and DNA-synthesis was measured by [3H]thymidine incorporation during the next 24 h. Data are shown as relative counts. (b) To directly quantify the magnitude of oxidative stress the lipid peroxides malondialdehyde (MDA) and 4-hydroxy-alkene (4-HNE) were spectrophotometrically quantified in above cells following 48 h of incubation with heme (50 μM) or H2O2 (200 μM). Control cells remained in regular culture media. Data are mean±SEM of six independent experiments (*P < 0.05 vs control. **P < 0.05 vs GFP)
3.3 Antiproliferative effect of HO-1 can be overcome by Akt gene transfer but not by exogenous stimulation with Insulin
We were interested in the modification of the protein kinase Akt in our studies and constructed a retroviral cassette to overexpress a constitutively active form of Akt. We inserted in this vector an IRES-DsRed2.Nuc element resulting in predominantly nuclear red fluorescence of the infected cell. This allowed double infection experiments with the GFP-(GFP/Akt) and 6HisHO-1-IRES-GFP-viruses (HO-1/Akt) (Fig. 3a–c). As mentioned above, continuously elevated levels of HO-1 markedly reduced the proliferative rates of HUVEC compared to mock- or GFP-infected cells. For instance, as shown in Fig. 3d, when cells were replated at 2.0 × 105 5 days after the initial retroviral infection, and grown in complete growth medium, only 0.67 ± 0.40 viable HO-1-HUVEC were counted by trypan-blue exclusion 4 days later. Co-infection with the Akt virus completely overcame the antiproliferative effect of HO-1 and restored proliferation to levels of GFP/Akt-HUVEC (Fig. 3d). This was confirmed by quantification of DNA-synthesis during a 24-h pulsing experiment in the presence of radioactively-labeled thymidine. The counts obtained from HO-1 cells were significantly reduced (Fig. 3e). Myr.Akt-overexpression resulted in higher rates of DNA-synthesis with comparable levels in GFP/Akt- and HO-1/Akt-cells. In this experiment, HUVEC were additionally stimulated with insulin to activate the endogenous Akt pathway. This resulted in significantly higher counts in mock- and GFP-infected HUVEC, but not in the HO-1 cells (Fig. 3e).

Co-infection with the Akt-retrovirus but not exogenous insulin restores proliferative rates in HO-1-HUVEC. HUVEC (phase contrast, a) were co-infected with retroviral vectors for HO-1-IRES-GFP and myr.Akt-IRES-DsRed2.Nuc. Cells could easily be identified by fluorescence microscopy showing cytoplasmic green GFP- (b), and predominantly nuclear DsRed2-red fluorescence (c). Five days after infection HUVEC were plated at 2.0 × 105 in 60 mm-dishes. Cells were trypsinized and counted with trypan blue 4 days later. Note significant anti-proliferative effect of HO-1 in HUVEC (*P < 0.05 vs GFP). Akt-overexpressing cells grew significantly faster (**P < 0.05 vs HO-1) (d). Five days after retroviral infection HUVEC were plated at 1.0 × 104 in 24 well-plates, and were allowed to attach overnight. [3H]thymidine-incorporation was measured during the following 24 h-period in regular culture media in the absence or presence of human insulin (10 μM) (e). Data are mean±SEM of four independent experiments (*P < 0.05 vs GFP without insulin, **P < 0.05 vs HO-1 without insulin, ***P < 0.05 vs no insulin)
3.4 Continuous overexpression of HO-1 suppresses Akt and the Akt-target eNOS
Quantitative Western blot analysis for the expression of Akt and its effector eNOS revealed that 5 days after retroviral infection with the HO-1-virus, HUVEC had significantly lower levels of phosphorylated-Akt (−46% compared to mock, P < 0.05) (Fig. 4a). Total eNOS (−32% compared to mock, P < 0.05) and phosphorylated-eNOS (−66%, P < 0.05) were also significantly lower compared to negative control cells (Fig. 4b). Co-infection of HUVEC with the myr.Akt vector resulted in the marked upregulation of the Akt-effector eNOS. There was no upregulation of endogenous HO-1 in the GFP/Akt cells 5 days after transduction (Fig. 4c).

The active form of Akt and its downstream target eNOS is downregulated in HO-1-HUVEC. This is reversed by co-infection with the Akt-retrovirus. HUVEC were single- or co-infected (MOI of 4) using indicated retroviruses, and cultured for additional 5 days. (a) The expression of total Akt and phosphorylated-Akt (Ser473), and (b) total eNOS and phosphorylated-eNOS (Ser1177) was quantified by Western blotting. Data are mean±SEM of four independent experiments (*P < 0.05 vs GFP). (c) Shows a representative panel of immunoblots for HO-1, Akt, phospho-Akt, eNOS, phospho-eNOS. GFP and β-actin lanes document equal loading
3.5 Continuously elevated HO-1 suppresses eNOS-activity in vitro
NOS-activity was assessed via measurement of the conversion of radioactively labeled arginine to citrulline, and was found to be significantly lower in HO-1-HUVEC compared to mock- or GFP-infected cells. Co-infection with the myr.Akt construct could completely restore eNOS activity to the levels of GFP/Akt cells (Fig. 5).

eNOS-activity is reduced in HO-1-HUVEC. HUVEC were infected with indicated vectors (MOI 4). Five days later 5.0 × 105 cells were lysed in 50 μl sample buffer. Reaction was incubated for 30 min at room temperature. Converted [3H]l-citrulline was quantified as c.p.m. and expressed relative to the values obtained from mock-infected cells (dashed line). Data are mean±SEM of four independent experiments. eNOS-activity was significantly lower in HO-1-HUVEC (*P < 0.05 vs GFP). Co-infection with the Akt-retrovirus elevated eNOS-activity (*P < 0.05 vs GFP, **P < 0.05 vs HO-1)
4 Discussion
The heat shock protein HO-1 has demonstrated potent therapeutic efficacy in several animal models of inflammatory, proliferative and cardiovascular disease [2, 3]. The detailed mechanisms underlying this effect are not completely understood. All byproducts of the catalyzed heme degradation appear to be involved in the HO associated defense cascade with the gaseous CO likely to play a key effector role [6, 13, 18, 20, 37, 38]. The bile pigments with their strong antioxidant properties together with the promoted ferritin synthesis have a beneficial impact on the cellular redox homeostasis [1]. HO-1 is therefore proposed as an attractive candidate for the development of specific drug interventions aiming at future clinical applications. Most vascular biology studies of HO-1/CO were focused on the effects on VSMCs and could clearly document the anti-oxidant properties. Interestingly, unlike other cellular defense mechanisms, HO-1/CO interferes for unknown reasons with cell cycle progression. The anti-proliferative effect can in high doses even result in apoptosis [7]. This is likely to explain most of the improved remodeling seen after arterial injury in animal models of vasculoproliferative disease [8, 12]. The impact of HO-1/CO on endothelial function has not been thoroughly investigated so far. However, especially with reference to the recent evidence indicating functional interactions between the HO-1 and Akt pathways, we wanted to address this relationship. In order to mimick a pharmacological approach, we used a retroviral vector in our study to achieve stable overexpression of HO-1 in HUVEC, resulting in an 5.4-fold increased activity level. We could confirm for HUVEC that elevated HO-1-levels confer a survival advantage under conditions of non-receptor mediated oxidative stress induced in our experiments by heme and H2O2. The lipid peroxidation-assay uncovered that HO-1-HUVEC displayed advantageous handling of reactive oxygen species to prevent functional destruction of membrane lipids. It was recently reported that in VSMCs HO-1-mediated protection from H2O2-induced oxidative stress depends on the coactivation of the Akt pathway [4]. Pretreatment with a PI3K-inhibitor diminished the anti-oxidant properties in VSMCs under high H2O2 concentrations presumably by delaying the upregulation of HO-1. The authors could demonstrate that in the first few hours of H2O2 treatment, Akt is activated by phosphorylation and accelerates HO-1-upregulation through the transcription factor Nrf2. However, our data indicate that for prolonged episodes of oxidative damage, HO-1 is still capable of providing its protective effect to HUVEC even in the absence of simultaneous and continuous Akt-upregulation. The reported induction of HO-1 shortly after Akt-phosphorylation does not seem to be permanent as we did not observe any endogenous HO-1 expression in HUVEC 5 days after infection with the myr.Akt virus.
The moderate elevation of HO-1 achieved by our retroviral vector was sufficient to dramatically interfere with cell cycle progression of HUVEC. The anti-proliferative properties of HO-1/CO have been described in various other cell types including VSMCs. At supraphysiological doses, HO-1/CO even induces apoptotic cell death. Our data provide evidence that the growth arrest mediated by HO-1 in HUVEC seems to involve the Akt-pathway. Akt-activity was found to be significantly lower in HUVEC 5 days after retrovirus-mediated continuous overexpression of HO-1.
The suppression of Akt-activity in HO-1 overexpressing HUVEC also influenced the expression of downstream-targets of Akt. We found both eNOS-level and -activity markedly reduced in HO-1 overexpressing HUVEC. After co-infection with the myr.Akt virus, HO-1-HUVEC regained their proliferation rate and eNOS activity to the same level that was observed in GFP/Akt cells. Interestingly, treatment of HO-1-HUVEC with insulin, known to be a strong activator of the endogenous PI3K/Akt pathway in EC [32], failed to overcome the HO-1-associated cell cycle retardation.
An in vivo-relationship between chronically elevated HO-1/CO in the vessel wall and marked endothelial dysfunction had previously been observed in salt induced hypertensive Dahl rats. These animals have high levels of HO-1-expression in the vessel wall. Only treatment with the HO-1-inhibitor chromium mesoporphyrin could restore some of the endothelium dependent vasoreactivity and lower blood pressure values [38–40]. Our data suggesting a negative feedback loop of the HO-1/CO- and Akt/NO-pathways in endothelial cells offer a possible explanation for this phenomenon.
The limitations of our study have to be mentioned: all in vitro experiments were performed in a single EC type. The antiproliferative effect of HO-1 has been described for several other cell types, but we cannot rule out that other EC and non-EC display different handling of Akt upon retroviral gene transfer of HO-1. We concentrated on a single time; here, kinetic experiments might provide additional information.
In summary, both the Akt as well as the HO-1/CO pathway play important roles in cellular defense strategies and seem to interfere with each other in a time- and dose-dependent manner. In the initial phase of oxidative stress, both pathways are rapidly and almost simultaneously activated to achieve optimal cytoprotection for the attacked cell. Here, the transcription factor Nrf2 seems to play a linking role [4, 22, 25]. Our data would indicate that there is no codependence of the two systems for prolonged periods; they even appear to have inhibitory feedback mechanisms on each other. Continuous overexpression of a constitutively active Akt does not result in stable upregulation of HO-1 and on the other hand, continuously elevated HO-1 levels result in the suppression of Akt and its effectors. One could speculate that HO-1/CO is not necessarily the proposed “good player” for maintaining cellular and tissue homeostasis especially in chronic proliferative and/or inflammatory vascular disease like atherosclerosis, hypertension, neointima formation and plaque stability. Initiation and progression of these diseases are known to crucially depend on a properly functioning Akt/NO-system. A possible negative interaction on the Akt/NO pathway in endothelial cells has also to be taken into consideration when proposing continuous stimulation of HO-1/CO as a potential therapeutical strategy in clinical applications. It could also be that prolonged administration of non-cell specific activators of HO-1/CO would rather worsen vascular disease. Further studies are needed to clarify this interaction.
References
Ferris CD, Jaffrey SR, Sawa A, Takahashi M, Brady SD, Barrow RK, et al. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat Cell Biol 1999;1:152–7.
Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 2006;86:583–650.
Ryter SW, Morse D, Choi AM. Carbon monoxide and bilirubin: potential therapies for pulmonary/vascular injury and disease. Am J Respir Cell Mol Biol 2007;36:175–82.
Brunt KR, Fenrich KK, Kiani G, Tse MY, Pang SC, Ward CA, et al. Protection of human vascular smooth muscle cells from H2O2-induced apoptosis through functional codependence between HO-1 and AKT. Arterioscler Thromb Vasc Biol 2006;26:2027–34.
Lee PJ, Alam J, Wiegand GW, Choi AM. Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc Natl Acad Sci U S A 1996;93:10393–8.
Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, et al. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med 2000;192:1015–26.
Liu XM, Chapman GB, Wang H, Durante W. Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation 2002;105:79–84.
Duckers HJ, Boehm M, True AL, Yet SF, San H, Park JL, et al. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat Med 2001;7:693–8.
Liu X, Pachori AS, Ward CA, Davis JP, Gnecchi M, Kong D, et al. Heme oxygenase-1 (HO-1) inhibits postmyocardial infarct remodeling and restores ventricular function. FASEB J 2006;20:207–16.
Melo LG, Agrawal R, Zhang L, Rezvani M, Mangi AA, Ehsan A, et al. Gene therapy strategy for long-term myocardial protection using adeno-associated virus-mediated delivery of heme oxygenase gene. Circulation 2002;105:602–7.
Pachori AS, Melo LG, Zhang L, Solomon SD, Dzau VJ. Chronic recurrent myocardial ischemic injury is significantly attenuated by pre-emptive adeno-associated virus heme oxygenase-1 gene delivery. J Am Coll Cardiol 2006;47:635–43.
Tulis DA, Durante W, Liu X, Evans AJ, Peyton KJ, Schafer AI. Adenovirus-mediated heme oxygenase-1 gene delivery inhibits injury-induced vascular neointima formation. Circulation 2001;104:2710–5.
Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med 2003;9:183–90.
Gerbitz A, Ewing P, Wilke A, Schubert T, Eissner G, Dietl B, et al. Induction of heme oxygenase-1 before conditioning results in improved survival and reduced graft-versus-host disease after experimental allogeneic bone marrow transplantation. Biol Blood Marrow Transplant 2004;10:461–72.
Nakao A, Neto JS, Kanno S, Stolz DB, Kimizuka K, Liu F, et al. Protection against ischemia/reperfusion injury in cardiac and renal transplantation with carbon monoxide, biliverdin and both. Am J Transplant 2005;5:282–91.
Paul G, Bataille F, Obermeier F, Bock J, Klebl F, Strauch U, et al. Analysis of intestinal haem-oxygenase-1 (HO-1) in clinical and experimental colitis. Clin Exp Immunol 2005;140:547–55.
Chora AA, Fontoura P, Cunha A, Pais TF, Cardoso S, Ho PP, et al. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest 2007;117:438–47.
Zuckerbraun BS, Billiar TR, Otterbein SL, Kim PK, Liu F, Choi AM, et al. Carbon monoxide protects against liver failure through nitric oxide-induced heme oxygenase 1. J Exp Med 2003;198:1707–16.
Tron K, Samoylenko A, Musikowski G, Kobe F, Immenschuh S, Schaper F, et al. Regulation of rat heme oxygenase-1 expression by interleukin-6 via the Jak/STAT pathway in hepatocytes. J Hepatol 2006;45:72–80.
Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 2000;6:422–8.
Wijayanti N, Kietzmann T, Immenschuh S. Heme oxygenase-1 gene activation by the NAD(P)H oxidase inhibitor 4-(2-aminoethyl) benzenesulfonyl fluoride via a protein kinase B, p38-dependent signaling pathway in monocytes. J Biol Chem 2005;280:21820–9.
Wu CC, Hsu MC, Hsieh CW, Lin JB, Lai PH, Wung BS. Upregulation of heme oxygenase-1 by Epigallocatechin-3-gallate via the phosphatidylinositol 3-kinase/Akt and ERK pathways. Life Sci 2006;78:2889–97.
Kim HP, Wang X, Nakao A, Kim SI, Murase N, Choi ME, et al. Caveolin-1 expression by means of p38beta mitogen-activated protein kinase mediates the antiproliferative effect of carbon monoxide. Proc Natl Acad Sci U S A 2005;102:11319–24.
Kim HP, Wang X, Galbiati F, Ryter SW, Choi AM. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. Faseb J 2004;18:1080–9.
Salinas M, Diaz R, Abraham NG, Ruiz de Galarreta CM, Cuadrado A. Nerve growth factor protects against 6-hydroxydopamine-induced oxidative stress by increasing expression of heme oxygenase-1 in a phosphatidylinositol 3-kinase-dependent manner. J Biol Chem 2003;278:13898–904.
Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, et al. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem 2004;279:8919–29.
Salinas M, Wang J, Rosa de Sagarra M, Martin D, Rojo AI, Martin-Perez J, et al. Protein kinase Akt/PKB phosphorylates heme oxygenase-1 in vitro and in vivo. FEBS Lett 2004;578:90–4.
Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol 2004;15:1983–92.
Breitschopf K, Zeiher AM, Dimmeler S. Pro-atherogenic factors induce telomerase inactivation in endothelial cells through an Akt-dependent mechanism. FEBS Lett 2001;493:21–5.
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999;399:601–5.
Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999;399:597–601.
Hermann C, Assmus B, Urbich C, Zeiher AM, Dimmeler S. Insulin-mediated stimulation of protein kinase Akt: a potent survival signaling cascade for endothelial cells. Arterioscler Thromb Vasc Biol 2000;20:402–9.
Hoffmann J, Haendeler J, Aicher A, Rossig L, Vasa M, Zeiher AM, et al. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ Res 2001;89:709–15.
Dimmeler S, Zeiher AM. Exercise and cardiovascular health: get active to “AKTivate” your endothelial nitric oxide synthase. Circulation 2003;107:3118–20.
Griese DP, Ehsan A, Melo LG, Kong D, Zhang L, Mann MJ, et al. Isolation and transplantation of autologous circulating endothelial cells into denuded vessels and prosthetic grafts: implications for cell-based vascular therapy. Circulation 2003;108:2710–5.
Griese DP, Achatz S, Batzlsperger CA, Strauch UG, Grumbeck B, Weil J, et al. Vascular gene delivery of anticoagulants by transplantation of retrovirally-transduced endothelial progenitor cells. Cardiovasc Res 2003;58:469–77.
Peyton KJ, Reyna SV, Chapman GB, Ensenat D, Liu XM, Wang H, et al. Heme oxygenase-1-derived carbon monoxide is an autocrine inhibitor of vascular smooth muscle cell growth. Blood 2002;99:4443–8.
Teran FJ, Johnson RA, Stevenson BK, Peyton KJ, Jackson KE, Appleton SD, et al. Heme oxygenase-derived carbon monoxide promotes arteriolar endothelial dysfunction and contributes to salt-induced hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 2005;288:R615–22.
Gaine SP, Booth G, Otterbein L, Flavahan NA, Choi AM, Wiener CM. Induction of heme oxygenase-1 with hemoglobin depresses vasoreactivity in rat aorta. J Vasc Res 1999;36:114–9.
Johnson FK, Durante W, Peyton KJ, Johnson RA. Heme oxygenase inhibitor restores arteriolar nitric oxide function in dahl rats. Hypertension 2003;41:149–55.
Author information
Authors and Affiliations
Corresponding author
Additional information
Christian A. Batzlsperger and Stefan Achatz contributed equally to this work.
Rights and permissions
About this article
Cite this article
Batzlsperger, C.A., Achatz, S., Spreng, J. et al. Evidence for a Possible Inhibitory Interaction between the HO-1/CO- and Akt/NO-Pathways in Human Endothelial Cells. Cardiovasc Drugs Ther 21, 347–355 (2007). https://doi.org/10.1007/s10557-007-6051-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-007-6051-1