
Abstract
The spread of primary tumor cells to distant organs, termed metastasis, is the principal cause of cancer mortality and is a critical therapeutic target in oncology. Thus, a better understanding of metastatic progression is critical for improved therapeutic approaches requiring insight into the timing of tumor cell dissemination and seeding of distant organs, which can lead to the formation of occult lesions. However, due to limitations in imaging techniques, primary tumors can only be detected when they reach a relatively large size (e.g., > 1 cm3), which, based on our understanding of tumor evolution, is 10 to 20 years (30 doubling times) following tumor initiation. Recent insights into the timing of metastasis are based on the genomic profiling of paired primary tumors and metastases, suggesting that tumor cell seeding of secondary sites occurs early during tumor progression and years prior to diagnosis. Following seeding, tumor cells may remain in a dormant state as single cells or micrometastases before emerging as overt lesions. This timeline and the role of metastatic dormancy are regulated by interactions between the tumor, its microenvironment, and tumor-specific T cell responses. An improved understanding of the mechanisms and interactions responsible for immune evasion and tumor cell release from dormancy would support the development of novel targeted therapeutics. We posit herein that the immunosuppressive mechanisms mediated by myeloid-derived suppressor cells (MDSCs) are a major contributor to tumor progression, and that these mechanisms promote tumor cell escape from dormancy. Thus, while extensive studies have demonstrated a role for MDSCs in the escape from adoptive and innate immune responses (T-, natural killer (NK)-, and B cell responses), facilitating tumor progression and metastasis, few studies have considered their role in dormancy. In this review, we discuss the role of MDSC expansion, driven by tumor burden, and its role in escape from dormancy, resulting in occult metastases, and the potential for MDSC inhibition as an approach to prolong the survival of patients with advanced malignancies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
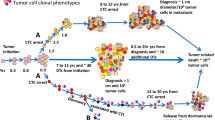
In tumor progression, the invasion and metastasis of tumor cells to other organs result in the onset of overt metastases and, eventually, patient mortality. It is established that the clinical detection of metastases is associated with poor patient outcomes [1]. However, many patients with no detectable metastases at diagnosis develop overt metastases or primary tumor relapse months, years, or even decades following “curative surgery” [2]. This latency period, during which cancer cells do not grow and remain in a quiescent or equilibrium state, is known as “cancer dormancy” [3]. In these patients, disseminated tumor cells (DTCs) can remain in a latent state as micrometastases, designated as “dormant metastases.” Furthermore, it is widely reported that the dormancy of primary tumor and metastatic cells occurs during tumor progression, with a duration that varies among tumors and cancer phenotypes [4] Fig. 1.

Overview of clonal selection and tumor progression and dormancy within the life history of a tumor. During tumor initiation, progression, and metastasis, ~ 30 doubling times (DTs) or an estimated 7 to 10 years (yrs) is required to achieve the 1-cm tumor (109 tumor cells) that can allow a clinical diagnosis. During this time, cancer-associated genetic instability results in metastatic variants such that circulating tumor cells (CTCs) circulate and arrest with the potential to form a disseminated tumor cell (DTC) early during tumor growth. Furthermore, host immunity via tumor-specific cytotoxic T-lymphocytes (CTL) can control the DTC growth, resulting in a dormant lesion that can be held in stasis for years or decades until released from dormancy in association with an increase in myeloid-derived suppressor cells (MDSCs) reversing host T cell responses. Regardless of the timing associated with the process metastases, in the absence of intervention, a lethal tumor volume (~ 1012 tumor cells) occurs within 10 doubling times and 0.5 to 20 years post-diagnosis. Differences in tumor cell colors represent different clonal phenotypes
2 MDSCs and metastasis
Cancer patients with a high tumor burden and/or advanced clinical stage or histologic grade often have an increased frequency of myelopoietic progenitor cells (MPCs) in their peripheral blood (PB) [5, 6] and an increased number of circulating neutrophils, termed neutrophilia [7, 8]. Cancer-associated neutrophilia and the associated circulating MPCs are suggested to be due to tumor secretion [7] of hematopoietic growth factors, including colony-stimulating factors (CSF)s. These CSFs stimulate the proliferation of myeloid cells and mobilize hematopoietic progenitor cells from the bone marrow (BM) into the circulation [9, 10]. Mobilized MPCs may establish themselves in the spleen or liver where they proliferate, forming sites of extramedullary myelopoiesis (EMM), further increasing the frequency of circulating myeloid cells.
A subset of circulating and tumor-infiltrating MPCs is identified as MDSCs due to their innate immune suppressive activity [11, 12]. Currently, there are three recognized types of MDSCs [13,14,15]: granulocytic (G) or polymorphonuclear (PMN), monocytic (M), and immature or early (i or e). Their human phenotype remains somewhat controversial; however, at present, most investigators agree that human MDSCs are lineage-negative (Lin−), i.e., lacking T-(CD3), B-(CD19), and NK- (CD56) cell markers, and are CD11b+CD33+HLA-DR- [16]. The three known MDSC subsets are differentiated based on their expression of the granulocyte markers CD15 or CD66b and the monocyte marker CD14. The third MDSC subset (i-MDSC) has been identified as lacking markers for granulocytes and monocytes but expresses CD33.
MDSCs are functionally associated with immune evasion by inducing T cell dysfunction through the production of reactive oxygen species (ROS) [17, 18], arginase-1 (ARG1) [19], and nitric oxide synthase (NOS2) [20]. ARG1 hydrolyzes L-arginine [21], which is critical for T cell proliferation [19], cytokine production [19], and expression of the T cell receptor zeta chain (TCR- ζ) [19], into urea and ornithine [21]. Thus, depletion of extracellular L-arginine by MDSC-derived ARG1 thereby inhibits T cell proliferation, function, and anti-tumor activity [22,23,24,25], and potentially inducing cell cycle arrest [26]. The downregulation of TCR- ζ chain expression on T cells is associated with reduced stability of the TCR-ζ mRNA [27], which may also be responsible for inhibiting T cell proliferation [28]. Furthermore, L-arginine is also a substrate for inducible (i)-NOS [21] that results in the production of nitric oxide (NO): a potent signaling molecule with lymphotoxic potential [29]. ROS produced by MDSCs reacts with NO to form peroxynitrite, which can modify the T cell receptor by nitrosylating a tyrosine residue within the major histocompatibility complex (MHC)-TCR complex [30], inhibiting antigen recognition and T cell function [30]. In addition, MDSCs can secrete immunosuppressive cytokines, most notably IL-10 [31], contributing to their immunosuppressive activity.
In patients with solid tumors, the primary cause of mortality is metastasis, which is the growth of tumor cells at sites discontinuous from the primary tumor. MDSCs are thought to have a role in the development of tumor metastases by suppressing the immune system, contributing to vasculogenesis, and possibly by aiding in the formation of a pre-metastatic niche. A pre-metastatic niche provides a microenvironment consisting of infiltrating inflammatory cells and extracellular matrix proteins that can aid in metastatic cell colonization; the nature of this microenvironment is suggested to contribute to the arrest and survival of DTC [32]. The concept of a pre-metastatic niche originated in a paper published by Steven Paget in 1889, stating that tumor cells exhibit site-specific preferences, with specific tumor types having the tendency to “seed” specific organs [33]. Paget posited in his “seed-and-soil” hypothesis that the spread of tumor cells was not random, but rather governed by regulated processes [33]. This hypothesis was elaborated upon by Kaplan et al., who observed that the formation of a pre-metastatic niche was supported by vascular endothelial growth factor receptor-1 positive (VEGFR1+) BM-derived hematopoietic progenitor cells [34], a cell population now known as MDSCs.
MDSCs are associated with myelopoiesis, a process commonly dysregulated in cancer patients and associated with the failure of immature myeloid cell differentiation. This creates a source of immature myeloid cells in the BM that can then mobilize into the circulation and arrest in common metastatic organ sites. Hematopoietic progenitor cell mobilization occurs through tumor secretion of growth factors, such as G-CSF, and is mediated in part by the CXC chemokine receptor 4 (CXCR4) [35] which is expressed on CD34+ hematopoietic progenitor cells and its ligand, stromal cell-derived factor 1 (SDF-1/CXCL12) [35]. G-CSF induces progenitor cell mobilization and secretion of proteases that can cleave CXCR4 [36], making it unable to bind to its ligand (CXCL12) and thereby increasing progenitor cell mobilization [36]. In cancer patients, this clinically presents as neutrophilia and monocytosis. Furthermore, the administration of growth factors in both cancer patients and normal stem cell donors [37] has been shown to increase the serum levels of the stromal factor matrix metalloproteinase-9 (MMP-9). MMP-9 aids in MDSC migration through the bone marrow [38, 39], as well as tumor progression [40,41,42]. Circulating MDSCs can then arrest in secondary lymphoid tissues, such as the spleen or liver, and proliferate, i.e., EMM.
Indeed, increased frequencies of myeloid (CD33+) and progenitor (CD34+) cells have been found in the splenic tissue of cancer patients with solid tumors as compared to patients without neoplastic disease [43, 44]. Furthermore, the frequencies of myeloid and progenitor cells in the spleens of cancer patients negatively correlate with overall survival (OS) [44]. Though rare, patients with solid tumors can develop an enlarged spleen [45], termed splenomegaly. This is caused by increased EMM [46], which can also occur in the liver [47, 48], following dysregulation of the BM microenvironment [49]. Perturbation of the BM microenvironment by tumor progression, metastasis, and/or chemotherapy can also result in myelofibrosis (MF) resulting from an inability of the BM to undergo hematopoiesis and extensive EMM in organs such as the spleen or liver. As observed in a case report by Kiely et al. [46], 6 out of 8 patients with metastatic carcinoma experienced bone pain and marrow fibrosis [46]. Similarly, BM metastases secondary to MF has also been documented in a breast cancer patient [50]. Studies such as these provide insight into how tumor growth can cause immune dysregulation in the marrow, leading to increased myelopoiesis and MDSC frequencies that can further neoplastic progression.
3 Timing and processes of tumor initiation, metastasis, and progression
Metastasis is initiated when tumor cells leave the primary tumor and enter the hematologic or lymphatic circulation as circulating tumor cells (CTCs). However, a clinically relevant metastasis requires that CTCs survive innate immune cell interactions in the circulation, as well as vascular turbulence, arrest, and extravasation in a vascular bed. The arrest of CTCs is a mechanical process, whereas the survival and growth of the arrested cells are in part, capillary bed-selective, as specific tumor cell phenotypes preferentially establish themselves in distinct secondary organs [51]. Thus, the circulatory anatomy has a role in the dissemination of tumor cells; however, this is not the complete mechanism regulating site-specific distribution of CTCs. As such, the arrest of tumor cells in a given organ is a necessary, but insufficient process for the initiation of a metastasis [52]. For example, breast adenocarcinoma metastases are first observed in axillary lymph nodes, then eventually, if not successfully excised, the liver, lungs, bones, and lastly, brain, supporting a process of lymphatic followed by hematogenous spread [33, 53]. Similarly, lung cancers initially metastasize to the regional lymph nodes, then to the brain [53, 54]. Thus, when CTCs survive arrest, they may begin to proliferate and become vascularized, developing into DTCs [55]. However, due to selective pressures, the presence of CTCs and DTCs are, again, not predictive of overt metastasis [56], as most tumor cells that enter the circulation are rapidly eliminated [57]. Paget suggested that specific organ microenvironments nourish the formation of secondary tumor foci by providing congenial “soils” integral to metastasis formation, contributing to site-specificity and establishing appropriate microenvironmental support for tumor seeding [33]. Supporting Paget’s observation, a later study observed that BM metastases of breast cancer cells had genetic alterations and BM DTCs had different phenotypes relative to the primary tumor [58]. Thus, the selection of metastatic sites is not due to chance, but rather due to the affinity of tumor cells for the milieu of specific organs. Despite metastatic inefficiency [54], i.e., the low frequency of CTCs that survives to form DTCs, it is DTCs and their growth into micrometastatic foci that represent the primary challenges to adjuvant therapy following primary tumor resection. Therefore, successful adjuvant therapy [59] for metastatic disease requires an understanding of the factors contributing to the survival of DTCs and the intra- and interlesional heterogeneity of micro- and overt metastases [60].
This lesional heterogeneity is associated, in part, with the independent progression of metastases arising from early DTCs rather than from the primary tumor cells: a process known as parallel progression [61,62,63]. Early dissemination of cancer cells is supported by the finding that DTCs can be detected in the BM of patients with early-stage breast cancer [64], including patients with pre-invasive ductal carcinoma in situ (DCIS) [65, 66]. Several studies comparing the genotypes of DTCs with those of primary tumor cells have shown that DTCs are less evolved than the primary tumor [67], further supporting parallel evolution given that they likely arose earlier than the bulk of primary tumor cells [61, 67,68,69]. For example, in breast cancer patients, BM DTCs have been shown to display fewer chromosomal aberrations, sub-chromosomal allelic losses, and gene amplification events than cells from primary tumors, as assayed by comparative genomic hybridization [70]. Indeed, only half of BM DTCs display abnormal karyograms, as opposed to 100% of cells from the primary tumor [58, 70]. This divergent genetic/mutational progression between primary tumors and DTCs suggests that a prolonged time period occurs between CTC arrest, formation of an overt metastasis, and when a diagnosis can be made, supporting the initiation of metastases when the primary tumor is at a less-progressed genomic state. Furthermore, the occurrence of parallel evolution is supported by microarray studies of primary tumors and the identification of expression profiles that predict the risk of developing a metastasis [58, 71, 72]. These microarray studies, in addition to timeline studies that have focused on cell doubling times, support the occurrence of DTCs and micrometastases years before diagnosis, such that gross metastases develop late in the life history of a tumor, providing time for cellular heterogeneity of metastatic cells and selection to occur [67].
Studies of parallel evolution in breast cancer patients have been confirmed and extended by studies in colorectal carcinoma (CRC) patients, further improving our understanding of the genetic modifications undergone by primary tumor cells that lead to the development of cells with metastatic potential. In the CRC studies, tumor cells in early-stage patients were shown to have a subset of primary tumor cells with invasive potential [73]. Genetic mapping of clonal and subclonal cancer progression, using whole-exome sequencing data from 23 metastatic CRC patients, showed, in agreement with prior studies [74], that despite their phenotypic and functional differences, the majority of single-nucleotide variants were shared between the primary tumor and its metastases. A separate study employing phylogenetic analysis of CRC progression extended these observations and documented that the divergence of metastatic lineages occurs early in cancer progression [75], supporting the origin of metastases from a single founder tumor cell. These studies suggest that, in a majority of patients, a metastatic lesion is derived from a single primary tumor cell before it is clinically detectable. Furthermore, it has been concluded that the primary tumors of ~ 90% of patients with metastatic CRC exhibit a subclonal selection of cells with a selective growth advantage. Together, these studies demonstrate that parallel evolution of primary and secondary tumors occurs in association with selection pressures [76] and that metastasis occurs early in tumor progression [71, 77].
In this review, we emphasize the need for a translation and clinical focus on therapy, rather than the prevention of metastasis, based on our understanding of the metastatic process [55] and its timeline [78]. As discussed herein, CTCs frequently seed at secondary sites years prior to the primary tumor diagnosis, at a time when the primary tumor is only a few millimeters in diameter [33, 73]. Current diagnostic methods can identify primary tumors or metastases only when they grow to ~ 1 cm3 volume, which contains about 109 cells [79], depending on the tumor phenotype. However, a tumor does not reach this size until ~ 7 to 10 years after initiation, and, by then, CTCs will have seeded secondary sites. During the exponential growth phase of a solid mammary tumor [80], the cellular doubling time is between 44 and > 1800 days, and with this information, a calculation of the timeline from tumor induction to a diagnosable tumor volume is possible [78]. For example, primary breast cancers have an average doubling time of 130 days during their exponential growth phase [63, 81]. This suggests that, following tumor initiation, a primary tumor will undergo about 30 cell divisions over approximately 10 years to achieve an overt tumor mass (~ 1 cm in diameter). Furthermore, a tumor burden of approximately 1000 cm3 is lethal [82], which would occur in about 10 doubling times from a 1-cm3 diagnosable primary tumor. Thus, with a 130-day doubling time, a tumor would become lethal in approximately 10 doubling times [83] from when the tumor can be diagnosed, versus 30 doubling times to a diagnostic window. However, the timing from diagnosis to mortality differs between tumors due to heterogeneity in doubling time, therapeutic intervention, and the impact of growth retardation associated with an increasing primary and secondary tumor mass, a variable phenomenon known as tumor dormancy [79].
4 Metastatic tumor cell dormancy
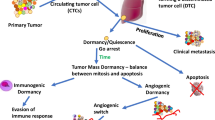
Consistent with the chronology of the metastatic process and parallel evolution is the observation that DTCs can enter into a dormant state in which they survive, but do not increase in size, and thus can remain undiagnosed for years or even decades [84]. Only when DTCs are released from dormancy do they proliferate and form an overt metastatic focus. There are two overall mechanisms for the establishment of dormancy: quiescence and tumor mass dormancy. Quiescence is a state of dormancy in which cells do not proliferate, and is induced by exogenous cues such as changes in the extracellular matrix, the formation of metastatic niches, hypoxia, and/or endoplasmic reticulum stress [85]. The occurrence of quiescent dormancy is supported by a meta-analysis of over 60,000 early-stage breast cancer patients treated with endocrine therapy, which revealed that metastases can be detected 5 to 20 years following primary tumor diagnosis, surgical resection, and adjuvant chemotherapy [86]. However, the relative contribution of quiescent DTCs or micrometastases to cancer relapse and survival, relative to chemo-resistance or other treatment challenges, remains unclear.
The second mechanism of dormancy, termed tumor mass dormancy [85, 87], is suggested to occur when a balance is achieved between cancer cell proliferation and cancer cell death, generating a “steady state” in which the DTC size remains constant [25]. This balance is achieved by angiogenic or immune-mediated regulation. The growth of cancer cells requires significant energy and resources, which is provided by an increase in vascularity [88]. Angiogenesis, the process by which additional blood supply (vascularity) is generated, is regulated by pro-angiogenic and anti-angiogenic factors [89]. By regulating the tumor vascular supply, these factors affect the transport of oxygen and nutrients to the tumor site, supporting tumor growth and reducing the incidence of apoptosis [90].
Immune-mediated tumor mass dormancy is critical to controlling tumor development and growth. Thus, the survival and continued growth of tumors requires that they evade the host immune response [91, 92]. This mechanism of dormancy is evoked when the rate at which immune-mediated cancer cell cytotoxicity matches the rate of cancer cell proliferation, resulting in a malignant mass with a stable size [93]. The cancer cell cytotoxicity that is thought to invoke immune-mediated dormancy is suggested to be the result of tumor-specific cytostatic and cytolytic CD8+ T-effector cell activity [94]. However, T cell responses that can cause dormancy can also lead to the secretion of cytokines that inhibit angiogenesis [95, 96], which would then lead to angiogenic dormancy. This response suggests the potential for cross-talk between these two dormancy mechanisms.
5 MDSCs, pre-metastatic niches, and release from metastatic dormancy
Metastatic site microenvironments contribute to the establishment of DTCs by providing support for tumor cell survival and outgrowth [55] and are identified as metastatic niches [97]. In addition, organ microenvironments contribute to metastatic site-specificity, due in part to primary tumor-induced conditioning associated with the systemic expansion of, and infiltration by, tumor-associated macrophages (TAM), neutrophils, T-regulatory (T-regs) cells, and MDSCs [98]. Tumor-infiltrating MDSCs and T-regs are derived from the BM, where they are found in high numbers [99], which is in part why bone is a common site for DTCs to escape from cancer-directed immunity. Prior to or following CTC arrest, a metastatic niche incorporates a number of additional functional elements critical to tumor cell survival and proliferation, including vascularization, modification of the extracellular matrix (ECM), recruitment and infiltration of MPCs, establishment of hypoxia, and organ-associated epithelial cell secretion of hormones and growth factors [100]. Indeed, breast cancer metastasis to bone involves cross-talk between tumor and bone such that tumor-secreted factors stimulate bone marrow cells to release growth factors and cytokines that support the growth and survival of DTCs in bone [101]. Additionally, tumor-associated MDSCs and fibroblasts can support the infiltration and expansion of non-transformed cells, including endothelial cells and their precursors, contributing to the vascularization of a micrometastatic site [102]. Together, these processes support the survival of DTCs and the formation of micrometastatic foci by inhibiting tumor-specific cytotoxic T cell infiltration and facilitating the survival and growth of tumor cells. Furthermore, T-regs and MDSCs can inhibit adaptive immune responses [103] that would otherwise limit the growth of micrometastatic lesions [87], supporting their release from dormancy.
Although primary tumors can modulate the microenvironment of distant organs and metastases, they may not have a direct role in tumor cell arrest via the formation of a metastatic niche. While primary tumors frequently result in neutrophilia and circulating and organ-infiltrating MDSCs, due to growth factor secretion, this is associated with the presence of a bulky tumor burden. Thus, this may not be relevant to the arrest of CTCs, their extravasation, and the establishment of DTCs, as these metastatic processes occur prior to the primary tumor growth into a sizable tumor mass. However, given the role of MDSCs in the suppression of adaptive immunity, we posit that their role in tumor progression may include the release of DTC from dormancy, associated with adaptive host immunity and the subsequent development of overt metastatic foci. This would occur following MDSC infiltration of organ capillary beds when the primary tumor has achieved a substantial size. Given that MDSC frequency is directly correlated with tumor burden [104], it is unlikely that they affect CTC arrest at secondary sites, as there is minimal tumor burden when DTCs first begin to form and CTCs arrest. Given their potential function in the release of micrometastatic foci from dormancy, we posit that MDSCs are involved in the metastatic process late in cancer progression, at the time of or after cancer diagnosis. Furthermore, it is suggested that MDSCs have an important role in sustaining the survival and growth of DTCs and micrometastatic foci by conferring resistance to host immunosurveillance and immunotherapy.
In addition to releasing metastases from dormancy, MDSCs may also contribute to metastatic organ specificity, as their presence can support tumor cell escape from host adoptive immunity [16]. As mentioned previously, the organ specificity of CTCs is supported by organ-specific growth factors that regulate the survival and growth of DTCs and micrometastatic foci. Thus, while lesion-infiltrating lymphocytes may contribute to tumor cell dormancy, MDSCs may release DTCs from dormancy, allowing outgrowth of micrometastatic foci. The spleen, arguably a rare site for metastasis formation [105], is one example of organ-specific controlled tumor cell growth [106] due in part to it being a relatively hostile environment for tumor cells due to high number of T cells [107]. Thus, T cells can control tumor cell growth, while metastatic site-infiltration by MDSCs, in association with their immunosuppressive activity, may facilitate the survival and growth of DTCs and micrometastatic foci.
The role of MDSCs in CTC arrest, DTC formation, release of DTCs from dormancy, and in tumor progression remains controversial. Sophisticated models and techniques need to be developed to answer questions regarding pre-metastatic niche development and maintenance, and how we can use these tools to explore therapies to inhibit neoplastic progression. Although dormancy has been recognized for several decades, many unknowns remain concerning its regulation: for example, what changes to the microenvironment initiate the release of DTCs from dormancy, and what external events trigger these changes? To answer these questions, a better understanding of the microenvironmental changes to DTC niches is required. Specifically, what are the key processes in the evolution of a DTC to a micrometastatic focus? How does the form and function of niches change as they mature? Similarly, do endogenous niches differ between solid organs, and what role do the various environmental constituents (myeloid, lymphoid, fibroblast, ECM, and vascular) contribute to the evolving niche, and what is their relevance to tumor progression? Answering these questions will not only enrich our understanding of the metastatic process but will also help identify new strategies for the effective treatment and management of metastatic cancer. Thus, it may be that our clinical goal should be to slow/prevent the release of DTCs from dormancy as a less toxic and practical therapeutic approach to limit development of metastatic clinical disease.
6 Therapeutic regulation of MDSCs in neoplasia
Emerging evidence regarding the role of MDSCs in cancer progression and their association with poor clinical outcomes [108] has stimulated an exploitation of MDSCs as targets for cancer treatment. In addition to studying novel therapies specifically targeting MDSCs and their effector functions, many of the current clinical trials employ off-label use of FDA-approved drugs to assess their efficacy against MDSCs as monotherapeutics or in combination with other immune-modulating therapeutic strategies. Currently, MDSC-targeting modalities aim to deplete MDSCs [109], inhibit MDSC-mediated immunosuppressive mechanisms [109], induce MDSC differentiation [110], and block MDSC proliferation and recruitment to tumor sites [109, 111]. The following discussion is focused on clinical strategies that have been suggested in clinical trials to target MDSCs and their bioactivities. Although a number of drugs with potential activity against MDSCs, both off target and novel, have been studied in preclinical models, in this review, we emphasize therapeutics that have been examined clinically and which have shown provisional activity against immune surrogates (Table 1).
7 Inhibition of MDSC proliferation
In addition to tumor cytotoxicity, off-target immune regulation has been documented for some chemotherapeutic agents. Capecitabine, a 5-fluorouracil (5-FU) prodrug, can significantly decrease the frequency of circulating MDSCs and increase cytotoxic T cell infiltration, as shown with glioblastoma multiforme (GBM) tumors, theoretically enhancing anti-tumor activity [108]. Similarly, the antimetabolite, gemcitabine, can significantly reduce the frequency of circulating G-MDSCs, but not M-MDSCs, as shown in patients with pancreatic adenocarcinoma (PDAC) [112]. However, the study documenting this effect measured MDSC frequency using cryopreserved PB mononuclear cells (PBMC), despite reports that MDSCs are sensitive to freeze-thaw lysis [152, 153]. In a separate study, administration of gemcitabine with capecitabine (GemCap) in PDAC patients resulted in a decreased frequency of total circulating MDSCs (Lin−HLA-DR−CD11b+) in 8 out of 19 patients [113]. In another arm of this study, 21 patients were treated with GemCap, an anti-cancer vaccine, and granulocyte–macrophage (GM)-CSF to reduce chemotherapy-associated myelosuppression, with a decreased frequency of circulating MDSCs observed in 18 of the 21 patients [113]. The decreased frequencies of total MDSCs in both groups were associated with reduced serum levels of tumor-secreted cytokines and inversely correlated with changes in tumor size [113]. However, this study also measured MDSC frequencies from previously cryopreserved PBMCs [113] limiting conclusions regarding cytotoxic agent reduction in the MDSC frequency. In patients with metastatic CRC, treatment with the FOLFOX (folic acid, 5-FU, and oxaliplatin) regimen in combination with the tyrosine kinase inhibitor (TKI), bevacizumab, was reported to significantly reduce the frequency of circulating G-MDSCs, but not M-MDSCs [115] in 15 out of 25 patients, and was associated with a better progression-free survival (PFS) [115]. Taken together, these data show that the frequency of MDSCs, particularly G-MDSCs, may be lowered with some cytotoxic therapies. However, some chemotherapeutic protocols may elevate the frequency of MDSCs, negatively affecting the tumor microenvironment. This difference in response was found in a study of 23 metastatic CRC patients treated with either FOLFOX or FOLFIRI (folic acid, 5-FU, and irinotecan) chemotherapeutic regimens. The FOLFOX-treated patients had a decrease in the frequency of MDSCs (CD33+HLA-DR−CD11b+), whereas the FOLFIRI-treated patients had an increase in the frequency of MDSCs [114]. The increased frequency of MDSCs and its chemotherapy-dependence was also documented in a clinical study of breast cancer patients given 4 cycles of dose-dense doxorubicin plus cyclophosphamide (AC) every 2 weeks followed by 4 cycles of dose-dense paclitaxel [5]. In all cycles of chemotherapy, peg-filgrastim (PEGylated G-CSF) was administered on protocol day 2 to reduce therapy-induced neutropenia [154]. Both the AC and paclitaxel regimens resulted in a different impact on the percentage and absolute number of circulating MDSCs as compared to baseline [5]. AC was associated with a significantly greater frequency and absolute number of circulating MDSCs (11.72% and 1157 cells/μL) as compared to paclitaxel (3.65% and 257 cells/μL) [5]. The increase in MDSCs frequency was suggested to be due to the use of peg-filgrastim and repeated cycles of chemotherapy [5]. Furthermore, the dose-dense AC therapy was associated with significantly higher frequency and absolute number of MDSCs relative to paclitaxel therapy cycles [5]. Furthermore, peg-filgrastim has been shown to elevate the frequency of MDSCs in numerous cancer studies [155], as have repeated cycles of chemotherapy that may be positively associated with increased suppressor cell activity [156]. It is noted that this dose-dense protocol of 4 cycles of AC followed by 4 cycles of taxane therapy has been shown to improve clinical outcomes, as opposed to a 21-day dosing regimen [157]. As demonstrated in the aforementioned studies, given that cancer therapy is frequently a poly-therapeutic approach, more research is needed to assess how anti-neoplastics in combination with other agents can affect MDSCs and, thus, the cancer-associated immunosuppressive milieu.
8 Inhibition of MDSC immunosuppression
PDE-5 inhibitors, such as tadalafil, promote the accumulation of intracellular cyclic guanosine monophosphate (cGMP), triggering inhibitory downstream effects on MDSC function [117, 158]. In a patient with refractory multiple myeloma (MM), treatment with tadalafil was found to reduce serum monoclonal immunoglobulin (M-protein) levels [116]. Later studies in MM patients [116] demonstrated that administration of tadalafil, to their concurrent treatment regimens, significantly reduced the frequency of BM M-MDSCs (HLA-DR−CD14+) at 6 months. However, following 6–11 months of treatment, although the frequency of BM M-MDSCs increased, a significant decrease in the expression of iNOS and ARG1 was observed [116]. Concomitant with a decrease in iNOS and ARG1 expression on M-MDSCs in the BM, a significant decrease in tyrosine nitrosylation was observed at 11+ months post-treatment [116] in association with an increase in the frequency and proliferation of stimulated BM CD8+IFN-γ+ and TCR-ζ+ cytotoxic T cells [116]. As mentioned previously, iNOS, ROS, and ARG1 promote MDSC-mediated T cell suppression by contributing to the nitrosylation of the T cell receptor [159, 160], depletion of L-arginine, downregulation of TCR- ζ chain expression [161], and upregulation of toxic mediators, including activated O2 radicals and NO [22,23,24]. Thus, it has been suggested that by significantly decreasing iNOS and ARG1 expression and increasing CD8+ T cell activity, tadalafil can successfully target MDSCs in MM patients [116].
The response of MDSCs to PDE-5 inhibition has also been reported in other cancers. In metastatic melanoma patients with stable disease (SD), tadalafil treatment did not significantly reduce the frequency of circulating M-MDSCs; however, it did reduce NO production by M-MDSCs in the metastatic lesions of 2 out of 3 patients who developed SD and significantly increased cytotoxic T cell recruitment to these secondary lesions [118]. In head and neck squamous cell carcinoma (HNSCC) patients, PDE-5 inhibition significantly decreased the expression of the immunosuppressive mediators ARG1 and iNOS and the frequency of circulating MDSCs from baseline [117]. Additionally, T cell proliferation and activation were significantly increased in tadalafil-treated patients [117]. The above evidence suggests that PDE-5 inhibitors can be useful in mitigating T cell inhibition and thus potentially improving tumor-specific T cell responses by reducing MDSC frequency and inhibiting iNOS and ARG1 levels [117, 162].
ARG1 expression is frequently increased in cancers, including breast cancer [163, 164] and CRC [165]; however, it is unclear whether a relationship exists between ARG1 expression and cancer patient survival [166]. In a cohort of hospitalized critically ill patients, the frequency of G-MDSCs was negatively correlated with plasma L-arginine concentration, and patient mortality was associated with the frequency of G-MDSCs (CD15+CD14−HLA-DR− and Lin−CD33+HLA-DR−) on day 1 of hospitalization [167]. Based on these observations, it was suggested that L-arginine depletion by G-MDSCs may contribute to an elevated risk of mortality in patients with higher levels of G-MDSCs. A number of investigational new drugs (INDs) targeting ARG1 have been studied in preclinical models and are currently in clinical trials. For example, the arginase inhibitor, CB-1158, has been shown to increase T cell proliferation in medium conditioned by G-MDSCs isolated from lung cancer patients [119] and, as of 2019, has shown minimal toxicities in a cohort of metastatic CRC patients in an ongoing clinical trial [120] (NCT02903914).
iNOS also contributes to MDSC immunosuppression by increasing signal transducer and activator of transcription 3 (STAT3) activation, elevating ROS levels via the production of NO [30, 168, 169], contributing to the formation of peroxynitrites via an interaction between NO and ROS [30, 170] and results in nitrosylation of T cell receptors [110, 159, 160, 171]. Investigational iNOS inhibitors such as N(G)-monomethyl L-arginine (L-NMMA) have been shown to decrease MDSC-suppressive activity in murine tumor models [172] and are currently being investigated in clinical cancer trials (NCT04095689, NCT02834403, NCT03236935). Preliminary results from an ongoing phase Ib/II clinical trial in triple-negative breast cancer patients showed a 22.2% overall response rate (ORR) to higher-dose L-NMMA (17.5 mg/kg) and docetaxel (100 mg/m2) [121] (NCT02834403).
NCX-4016, an NO-releasing aspirin derivative, does not reduce NO, but rather increases plasma NO levels nearly twofold [173], thereby inhibiting iNOS activity [174]. Furthermore, NCX-4016 has been reported to inhibit iNOS and ARG1 activity by CD11b+ cells [123], increase anti-tumor immune activity [123], elevate CD8+ T cells [123], and increase IFN-γ release [123]. Administration of NCX-4016 in healthy volunteers was shown to inhibit monocyte activation, lower IL-6 levels, and reduce CD11b expression on monocytes, indicating that it may inhibit MDSC in humans, as well [122]. Despite the evidence regarding iNOS inhibition, preclinical studies have indicated that NO and peroxynitrites produced by macrophages can induce cytotoxicity of tumor cells [172], potentially overcoming the effects of iNOS- and NO-induced inhibition of T cell cytotoxicity [30, 170]. Therefore, it remains unclear whether iNOS inhibition or NO supplementation may be worthwhile in the treatment of cancer given the diverse bioactivities of NO, despite their potential to curb tumor immune evasion.
Inhibition of myeloid cell growth factors has been suggested to reduce MDSC proliferation, as well as tumor infiltration by other immunosuppressive myeloid populations such as TAMs [175]. We note that the literature for circulating and tissue-infiltrating myeloid cells is controversial, as the phenotypes of TAMs and MDSCs are not clearly defined. Therefore, we applied the nomenclature used within each referenced manuscript, with the understanding that the cellular phenotype and function may be independent of terminology. The most extensively studied drugs have targeted colony-stimulating factor-1 receptors (CSF1R), which are also known as c-fms proto-oncogene and CD115. Several CSF1R inhibitors are currently in clinical trials, with some showing promising results [176], and in a few instances, their impact on monocytes and MDSCs have been studied in addition to patient outcomes. In AML patients, administration of the CSF1R inhibitor, GW-2580, has shown to target CSF1Rhi monocytes, and patients with CSF1Rhi monocytes that co-expressed HLA-DR and CD33 were particularly sensitive to GW-2580 [124]. Conversely, CSF1Rhi monocytes that co-expressed CD16 and CD66b were more resistant to intervention. While all MDSCs express the common myeloid linage marker, CD33, and G-MDSCs express CD66b, M-MDSCs are known to be HLA-DRlow/− and CD16− [177]. Thus, it is unknown whether the CSF1Rhi monocyte subsets include of MDSCs. However, in vitro studies of GW-2580 on blood samples from chronic lymphocytic leukemia (CLL) patients showed that GW-2580 treatment decreased the total number of nurse-like cells (NLCs), a monocyte subset in CLL patients that interacts with TAMs and is associated with CLL progression [178, 179].
Pexidartinib (formerly PLX-3397) is a small molecule inhibitor of tyrosine kinases (TK) involved in hematopoiesis [180] such as CSF1R, c-KIT (stem cell factor receptor), Flt-3 (Fms-related tyrosine kinase 3), and VEGFR which has been studied clinically. In patients with advanced solid tumors, administration of pexidartinib in combination with paclitaxel showed clinical benefit (complete and partial response and stable disease) in 19 of 38 patients and an overall response rate (ORR) of 16% [181]. Furthermore, this was associated with a decrease (between 57 and 100%) in the frequency of circulating CD14dimCD16+ non-classical monocytes, supporting a relationship between clinical benefit and CSF1R inhibition, as non-classical monocytes highly express CSF1R as compared to other monocyte subsets [181, 182]. Similarly, pexidartinib administration to patients with GBM reduced the frequency of circulating CD14dimCD16+ non-classical monocytes by nearly 50% and significantly decreased the expression of intratumoral Iba1+ microglia, a resident macrophage of neural tissue and a form of TAM in neurologic malignancies [126]. However, pexidartinib did not improve PFS in this subset of GBM patients [126]. Another TKI, imatinib, with an inhibitory profile of activity similar to pexidartinib, is currently used in the treatment of BCR-ABL+ chronic myeloid leukemia (CML) and also targets CSF1R [183]. Imatinib has been shown to significantly reduce the frequency of circulating G-MDSCs (CD11b+CD33+CD14−HLA-DR−) [127] in patients with CML.
The bisphosphonate derivative, zoledronic acid (ZA), inhibits bone-resorbing osteoclasts, residential tissue macrophages in bone that are derived from the myeloid linage, and is thus used to treat osteopenia [184] and prolong disease-free survival (DFS) in post-menopausal breast cancer patients [185]. Treatment with ZA in murine studies has documented a decrease in circulating MDSCs and increased tumor infiltration by T cells [186, 187]. To date, one clinical study of 15 PDAC patients with non-metastatic disease examined the activity of ZA on MDSCs [147]. These patients received ZA 2 weeks prior to surgery and twice, 4 weeks apart, following surgery [147]. Samples were obtained prior to ZA injection and 3 months after surgery for flow cytometry analysis following cell isolation by Ficoll-density centrifugation and cryopreservation, to identify G-MDSCs (CD45+CD33+CD11b+CD15+) [147]. In these studies, no difference was observed in the frequency of G-MDSCs in the PB or BM following ZA treatment [147]. The authors suggested that the lack of MDSC response to ZA administration could be due to an insufficient dose or duration of ZA administration [147]. However, as mentioned previously, MDSCs and granulocytes are sensitive to freeze/thaw lysis [152, 153], potentially obscuring flow cytometric assessment.
Immune checkpoint inhibitors (ICIs) are a class of drugs that block checkpoint proteins on tumor cells, such as programmed death-ligand 1 (PD-L1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4), from binding to their ligands, PD-1, and CD80/86, respectively. These receptor-ligand interactions contribute to tumor evasion of immunosurveillance by CD8+ T cells [188]. The CTLA-4 inhibitor, ipilimumab, approved by the FDA for metastatic melanoma [189], improved OS in patients with lower frequencies of MDSCs [128, 129]. Furthermore, administration of ipilimumab reduced the frequency of M-MDSCs, NO production by M-MDSCs, and the frequency of PD-1+ G-MDSCs [129]. Melanoma patients given ipilimumab over the course of 9 weeks had a significant reduction in frequencies of G-MDSCs and ARG1+ myeloid cells in their PB as compared to baseline levels [130, 190]. In a separate study in melanoma patients, treatment with ipilimumab significantly decreased the frequency of G-MDSCs and iNOS and ARG1 expression relative to baseline [131], suggesting that ICIs may be effective in reducing the frequency of G-MDSCs and cells that express the ICI-targeted checkpoint marker, as well as a possible use for MDSCs as biomarkers to predict and/or monitor patient outcomes. Recent studies indicate that a low frequency of MDSCs may be a predictive response marker for melanoma patient benefit from ipilimumab treatment [191]. Interestingly, patients treated with multiple doses of the PD-L1 inhibitors, atezolizumab, or avelumab have been reported to have a significantly decreased percentage of PD-L1+ M-MDSCs in their PB [132], while multiple doses of the PD-1 inhibitor, pembrolizumab, significantly decreased the PB frequency of PD-1+ M- and i-MDSCs [132]. Consistent with these observations, several studies have documented a relationship between MDSC infiltration and PD1 blockade resistance, and that selective depletion of MDSCs could restore anti-PD1 therapy efficacy [192, 193].
9 Inhibition of MDSC recruitment
Ibrutinib, a Bruton’s tyrosine kinase (BTK) and IL-2-inducible kinase (ITK) inhibitor, has off-target effects on MDSC mobilization, lending to its exploration as an immunomodulatory therapy against a number of solid tumors [194]. Human and murine MDSCs express BTK [134], and treatment with ibrutinib inhibits BTK phosphorylation of MDSCs generated in vitro from patients with metastatic melanoma [134]. In vitro studies using human donor PB samples observed that administration of ibrutinib inhibits MDSC migration, generation, and nitrate production [134]. Furthermore, a clinical study in CLL patients treated with ipilimumab observed an increase in the absolute numbers of CD4+ and CD8+ T cells and a decrease in circulating G-MDSCs [135]. Despite the rationale for its efficacy, current clinical studies report no survival benefit with ibrutinib alone or in combination with other therapies for patients with pancreatic [195], breast [196, 197], or neuroendocrine cancers [198]. However, preliminary results of an ongoing study in solid tumors treated with ibrutinib observed a decrease in plasma levels of chemokines involved in MDSC trafficking [136], stimulating a number of clinical trials with ibrutinib against solid tumors [196, 199].
Antigen presenting cells and tumor cells can secrete chemokines that promote recruitment of inflammatory cells, including MDSCs, into the tumor microenvironment. Inhibition of chemokine function [200] has been used to block MDSC recruitment [201] and MDSC-associated regulation of tumor progression. One such inhibitor, HuMax-IL8 (formerly BMS-986253) is a monoclonal antibody that binds IL-8: a chemotactic factor that is associated with cancer progression and MDSC accumulation at tumor sites [202]. In a 3 × 3 dose escalation study of patients with metastatic or unresectable solid tumors, HuMax-IL8 decreased serum IL-8 levels in all doses studied, with a significant reduction in IL-8 occurring on day 3 compared to baseline. In addition, stable disease was observed in 73% of patients with a median treatment duration of 24 weeks [137]. While preclinical studies showed that HuMax-IL8 significantly inhibited MDSC recruitment in combination with docetaxel and improved T cell-mediated cytotoxicity [203], the clinical study by Bilusic et al. revealed no significant changes in circulating the frequency of MDSCs. This study incorporated a mix of solid tumor patients with an N of 3 during dose escalation and an N of 6 in the dose-expansion phase [137]. In contrast to the Hu Max- IL8 studies, combination therapy with the CXCR4 antagonist BL-8040 and the PD-1 inhibitor pembrolizumab and chemotherapy in a larger cohort of patients with metastatic PDAC was found to elevate tumor-infiltrating CD8+ effector T cells and to reduce infiltration by MDSCs in biopsies of tumor metastases [138]. Additionally, a significant increase in circulating CD4+ and CD8+ T cells and a significant decrease in T-regs were observed. In the first cohort, administration of BL-8040 and pembrolizumab resulted in a disease control rate (DCR) of 34.5%, while the second cohort (BL-8040, pembrolizumab, and chemotherapy) had a DCR of 77% and an ORR of 32%. These results suggest that there may be potential for combination chemokine and checkpoint inhibitor therapy in mitigating MDSC recruitment and immunosuppression at tumor sites.
10 Induction of MDSC differentiation
All-trans retinoic acid (ATRA) specifically targets immature myeloid cells, including MDSCs, by inducing their differentiation through the upregulation of glutathione synthase and glutathione production [148, 204]. In one study, administration of ATRA significantly reduced the frequency of Lin−HLA-DR−CD33+ myeloid cells in the PB of metastatic kidney cancer patients 7 and 14 days post-treatment [148]. In a different study of advanced stage melanoma patients, administration of ATRA with ipilimumab significantly reduced the frequency of circulating MDSCs and their expression of immunosuppressive genes [149].
Similar to ATRA, vitamin D3 has long been of interest in cancer therapy due to its anti-inflammatory and immunomodulatory activity [205]. Furthermore, vitamin D3 has been shown to inhibit cancer progression and recurrence and improve DFS [150]. A recent study in mice reported that MDSCs, especially M-MDSCs, express the vitamin D receptor (VDR) and respond to vitamin D3 binding with reduced immunosuppressive activity [206]. A clinical study in HNSCC patients given escalating doses of vitamin D3 observed a significant decrease in the frequency of CD34+ suppressor cells, likely MDSCs in the higher-dose groups, and a significant decrease in serum GM-CSF levels at week two in the highest dose group [151]. Unfortunately, the frequency of CD34+ suppressor cells and serum cytokine levels increased following 6 weeks of treatment [151]. The initial reduction followed by the increase in the frequency of circulating suppressor cells may be due to the lower serum GM-CSF levels in the beginning of the study as compared to week six [151]. Additional studies are needed to determine which cancers and associated treatment regimens, as well as patient characteristics, would most benefit from vitamin D3 supplementation.
11 Tyrosine kinase and JAK inhibitors
The TKI, sunitinib, has been shown to inhibit the VEGFR-1/2/3, PDGFR-α/β, c-Kit receptor, FLT-3, and receptor encoded by the ret proto-oncogene (RET) [207], most of which stimulate the proliferation of MDSCs [208] and are a frontline therapy for renal carcinoma (RCC) [209]. A study in RCC patients reported that a significant decrease in the frequency of MDSCs was observed when patients’ myeloid cells were cultured in vitro with varying concentrations of sunitinib for 48 h, with the concentrations of sunitinib-inducing MDSC apoptosis in a dose-dependent manner [139]. Furthermore, a subset of patients treated with two cycles of sunitinib had decreased frequencies of MDSCs from baseline, with this MDSC reduction significantly correlated with an increase in IFN-γ production [139]. In a separate study, cancer patients with oligometastatic disease were treated with sunitinib prior to stereotactic body radiotherapy [140]. Post-7 days of sunitinib therapy, patients had a significantly lower frequency of circulating MDSCs (CD33+HLA-DR−/lo) [140]. Of note in these studies, is that M-MDSCs were defined as CD33+CD14+CD16+, differing from most studies, which include HLA-DR− cells and/or CD11b+ cells in the M-MDSC phenotype.
Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathways involve various TKs and downstream effectors that are important for MDSC recruitment, expansion, and suppression of T cell function via binding of tumor-derived signaling molecules such as VEGF, G-CSF, and GM-CSF. Therefore, JAK/STAT pathway inhibition poses a promising MDSC-targeting strategy [210]. Ruxolitinib (Rux), an inhibitor of JAK1/2, has been shown to significantly decrease the frequency of circulating G-MDSCs, numbers of T-regs [141], and levels of pSTAT5 in patients with myeloproliferative disease [141]. However, treatment-induced STAT5 activation and reduction in the numbers of T cells and pro-inflammatory cytokines can contribute to loss of T cell function [141]. Rux treatment was not found to be as beneficial for patients with solid tumors such as non-small cell lung cancer (NSCLC) [211] and metastatic pancreatic cancer [212].
12 Improving checkpoint inhibitor therapy by targeting MDSCs
While ICIs are effective in reducing MDSCs, as demonstrated in patients with metastatic melanoma [128, 131, 189], their efficacy may be improved in combination with drugs targeting MDSCs. MDSCs inhibit T cell activation and immune responses to tumor antigens [156, 213], despite the use of PD-1 and PD-L1 checkpoint inhibitors [109, 214]. Therefore, the development of therapeutic strategies promoting anti-tumor immune responses, i.e., ICIs, and those that deplete or inhibit MDSCs, has been suggested to potentially improve patient outcomes and prevent the development of MDSC-associated ICI resistance [215]. A meta-analysis of anti-PD-1 therapy studies in NSCLC patients demonstrated that the addition of PD-1 inhibitor, pembrolizumab, to chemotherapy resulted in a significantly improved OS [133]. Similarly, combining the anti-angiogenic drug, bevacizumab, with the antimetabolite capecitabine, reduced circulating MDSCs following tumor resection [108]. Furthermore, in these studies, mass cytometry time of flight (CyTOF) analysis of tumor samples determined that adding capecitabine to the treatment regimen aided in tumor-directed immune activation, with a significant reduction in the expression of CTLA-4 and PD-1 on macrophages and CTLA-4 on lymphocytes [108]. Preliminary data in a phase I clinical trial in patients with metastatic tumors using the BTK inhibitor, ibrutinib, in combination with the PD-1 inhibitor, nivolumab, indicates that the two inhibitors significantly decrease plasma chemokine levels (IL-12, CCL2, CCL3, and CCL4) and circulating MDSCs in the first cycle of therapy [216]. These studies provide early evidence of the efficacy of combining ICIs with chemotherapeutics or immunomodulatory agents and reinforce the theory that targeting MDSCs is critical to the sustained efficacy of ICI therapy.
13 Combination immunotherapy with cancer vaccines
The aim of cancer vaccines is to sensitize and activate T cells to kill tumor cells. Despite decades of study, only one vaccine, sipuleucel-T (Provenge®), has received FDA approval, though it only extends the OS of metastatic prostate cancer patients by 4 months [217]. Therefore, similar to ICI combination therapy, strategies that simultaneously target MDSCs, through their inactivation or depletion, may improve cancer vaccine efficacy. An investigational nelipepimut-S vaccine (NeuVax™) for breast cancer uses GM-CSF as an adjuvant [218]. The use of GM-CSF as an adjuvant increases tumor antigen presentation and, thus, potential anti-tumor responses [218]; however, it also expands MDSCs at high doses [218, 219]. This demonstrates one of the challenges in maximizing antigen presentation, while minimizing MDSC expansion, as documented in a study in relapsed prostate cancer patients given daily high doses of GM-CSF that experienced an increase in the absolute number of MDSCs and T-regs, whereas patients given lower, intermittent doses had fewer MDSCs and T-regs [220]. Additionally, tumor-secreted GM-CSF in mesothelioma patients was shown to bolster the immunosuppressive activities of CD15+CD33−HLA-DRlow/-CD11b+CD66b+CD16+ granulocytic cells [221]. In a phase I/II trial of the nelipepimut-S vaccine, cytotoxic T cell responses were increased and the frequency of T-regs was decreased following the course of treatment and indicated favorable responses in patients; however, circulating MDSCs were not assessed [222].
Aside from selecting an appropriate adjuvant to maximize antigen presentation and anti-tumor immunity, chemotherapeutics, as with ICIs, may also minimize the negative impact of inhibitory immune factors and cell populations, such as MDSCs, on vaccine efficacy. In a clinical trial, patients with advanced pancreatic cancer were given personalized cancer peptide vaccines plus gemcitabine. In this study, 11 of the 13 patients showed clinical responses defined as a reduction in tumor size [143]. In another study in small cell lung cancer (SCLC) patients, co-administration of INGN-225 (a vaccine comprised of autologous dendritic cells transduced with a modified p53 adenovirus) and ATRA resulted in the development of a tumor-specific T cell response in 43.3% of vaccine and ATRA-treated patients versus 20% of patients treated with the vaccine alone [223]. In a similar study, administration of INGN-225 to SCLC patients who had been receiving cytotoxic chemotherapy resulted in a significant p53-specific T cell response in 52% of patients, along with persistently elevated IFN-γ production 2–3 weeks after the last vaccine cycle; however, a significant elevation in immature myeloid cells (Lin−HLA-DR−CD33+) was also observed, which included MDSCs [224]. Of the patients who received second-line chemotherapy following vaccine administration, 61.9% achieved a complete or partial response to therapy [224]. In another study, patients with platinum-resistant ovarian cancer were given a non-dendritic cell p53 vaccine (p53MVA; day 15) with gemcitabine (days 1 and 8) over 3 cycles of chemotherapy. Approximately, half of the patients had p53-reactive CD4+ and CD8+ T cells, despite no change in the frequency of circulating MDSCs or T-regs after the first treatment cycle [225]. Although no association was observed between MDSC or T-reg levels and treatment response, an inverse trend in patient PFS and MDSC and T-reg frequency was observed, but was not statistically significant [225]. Cancer vaccines can augment tumor-specific immunity, but their use would arguably be most effective in patients with minimal MDSC frequencies [143, 217,218,219, 222,223,224,225]. The mixed responses to vaccine-based immunotherapy describes a need for therapeutic strategies involving vaccine preparation and combination therapy with other immune-modulating agents to abrogate the immunosuppressive effects of MDSCs, to ensure that patients are able to mount sufficient T cell anti-tumor responses.
In summary, the goal of adjuvant therapy is to reduce the risk of recurrence and outgrowth of metastases after primary tumor treatment. The process of tumor dormancy contributes to the time between initial diagnosis and metastatic relapse, which is the primary cause of cancer mortality. Thus, improving our understanding of why DTCs enter dormancy, later reawaken, and proliferate into an occult metastasis is vital to developing effective therapeutic strategies against neoplastic disease. Dormant DTCs are rarely detectable with current diagnostic technologies, making it critical to better understand reawakening and preventing the outgrowth of gross metastasis. Adaptive anti-tumor immune responses can maintain tumor cell dormancy, whereas chronic inflammation, i.e., circulating MDSCs, can reactivate DTC from immune-mediated dormancy. Thus, we suggest that limiting the expansion and function of MDSCs may maintain DTC dormancy and inhibit outgrowth of micrometastases, resulting in prolonged survival. In conclusion, disrupting tumor immune microenvironmental interactions provides an attractive therapeutic option, bringing us closer to the goal of preventing tumor relapse and improving patient quality of life, while posing as a pragmatic strategy to preventing relapse.
References
DeSantis, C. E., Lin, C. C., Mariotto, A. B., Siegel, R. L., Stein, K. D., Kramer, J. L., Alteri, R., Robbins, A. S., & Jemal, A. (2014). Cancer treatment and survivorship statistics, 2014. CA: a Cancer Journal for Clinicians, 64, 252–271.
Jahanban-Esfahlan, R., et al. (2019). Tumor cell dormancy: threat or opportunity in the fight against cancer. Cancers (Basel), 11, 1207.
Yuhas, J. M., & Tarleton, A. E. (1978). Dormancy and spontaneous recurrence of human breast cancer in vitro. Cancer Research, 38, 3584–3589.
Romero, I., Garrido, F., & Garcia-Lora, A. M. (2014). Metastases in immune-mediated dormancy: a new opportunity for targeting cancer. Cancer Research, 74, 6750–6757.
Diaz-Montero, C. M., Salem, M. L., Nishimura, M. I., Garrett-Mayer, E., Cole, D. J., & Montero, A. J. (2009). Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunology, Immunotherapy, 58, 49–59.
Wang, L., Chang, E. W. Y., Wong, S. C., Ong, S. M., Chong, D. Q. Y., & Ling, K. L. (2013). Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. Journal of Immunology, 190, 794–804.
Hocking, W., Goodman, J., & Golde, D. (1983). Granulocytosis associated with tumor cell production of colony-stimulating activity. Blood, 61, 600–603.
Fredeau, L., Bohelay, G., Shourick, J., Piver, D., Guyot, A., Schlageter, M. H., Caux, F., & Maubec, E. (2020). Paraneoplastic neutrophilic leukaemoid reaction in a patient with melanoma: association between tumour volume and leucocytosis. The British Journal of Dermatology, 183, 579–580.
Sieff, C. A. (1987). Hematopoietic growth factors. The Journal of Clinical Investigation, 79, 1549–1557.
Lane, T. A., Ho, A. D., Bashey, A., Peterson, S., Young, D., & Law, P. (1999). Mobilization of blood-derived stem and progenitor cells in normal subjects by granulocyte-macrophage- and granulocyte-colony-stimulating factors. Transfusion, 39, 39–47.
Slavin, S., & Strober, S. (1979). Induction of allograft tolerance after total lymphoid irradiation (TLI): development of suppressor cells of the mixed leukocyte reaction (MLR). Journal of Immunology, 123, 942–946.
Pak, A. S., Wright, M. A., Matthews, J. P., Collins, S. L., Petruzzelli, G. J., & Young, M. R. (1995). Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34(+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clinical Cancer Research, 1, 95–103.
Dumitru, C. A., Moses, K., Trellakis, S., Lang, S., & Brandau, S. (2012). Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunology, Immunotherapy, 61, 1155–1167.
Poschke, I., Mougiakakos, D., Hansson, J., Masucci, G. V., & Kiessling, R. (2010). Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Research, 70, 4335–4345.
Rodriguez, P. C., Ernstoff, M. S., Hernandez, C., Atkins, M., Zabaleta, J., Sierra, R., & Ochoa, A. C. (2009). Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Research, 69, 1553–1560.
Talmadge, J. E., & Gabrilovich, D. I. (2013). History of myeloid-derived suppressor cells. Nature Reviews Cancer, 13, 739–752.
Schmielau, J., & Finn, O. J. (2001). Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Research, 61, 4756–4760.
Corzo, C. A., Cotter, M. J., Cheng, P., Cheng, F., Kusmartsev, S., Sotomayor, E., Padhya, T., McCaffrey, T. V., McCaffrey, J. C., & Gabrilovich, D. I. (2009). Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. Journal of Immunology, 182, 5693–5701.
Zea, A. H., Rodriguez, P. C., Atkins, M. B., Hernandez, C., Signoretti, S., Zabaleta, J., McDermott, D., Quiceno, D., Youmans, A., O’Neill, A., Mier, J., & Ochoa, A. C. (2005). Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Research, 65, 3044–3048.
Khadge, S., Sharp, J. G., McGuire, T. R., Thiele, G. M., Black, P., DiRusso, C., Cook, L., Klassen, L. W., & Talmadge, J. E. (2018). Immune regulation and anti-cancer activity by lipid inflammatory mediators. International Immunopharmacology, 65, 580–592.
Durante, W., Johnson, F. K., & Johnson, R. A. (2007). Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clinical and Experimental Pharmacology & Physiology, 34, 906–911.
Czystowska-Kuzmicz, M., Sosnowska, A., Nowis, D., Ramji, K., Szajnik, M., Chlebowska-Tuz, J., Wolinska, E., Gaj, P., Grazul, M., Pilch, Z., Zerrouqi, A., Graczyk-Jarzynka, A., Soroczynska, K., Cierniak, S., Koktysz, R., Elishaev, E., Gruca, S., Stefanowicz, A., Blaszczyk, R., Borek, B., Gzik, A., Whiteside, T., & Golab, J. (2019). Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nature Communications, 10, 3000.
Geiger, R., et al. (2016). L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell, 167, 829–842.e813.
Raber, P., Ochoa, A. C., & Rodríguez, P. C. (2012). Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunological Investigations, 41, 614–634.
Rodriguez, P. C., Quiceno, D. G., Zabaleta, J., Ortiz, B., Zea, A. H., Piazuelo, M. B., Delgado, A., Correa, P., Brayer, J., Sotomayor, E. M., Antonia, S., Ochoa, J. B., & Ochoa, A. C. (2004). Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Research, 64, 5839–5849.
Rodriguez, P. C., Quiceno, D. G., & Ochoa, A. C. (2006). l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood, 109, 1568–1573.
Rodriguez, P. C., Zea, A. H., Culotta, K. S., Zabaleta, J., Ochoa, J. B., & Ochoa, A. C. (2002). Regulation of T cell receptor CD3zeta chain expression by L-arginine. The Journal of Biological Chemistry, 277, 21123–21129.
Bronte, V., Serafini, P., Mazzoni, A., Segal, D. M., & Zanovello, P. (2003). L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends in Immunology, 24, 302–306.
Eiserich, J. P. (2003). Nitric oxide: a simple free radical with complex chemistry and biology (pp. 1–19). Dordrecht: Springer Netherlands.
Nagaraj, S., Gupta, K., Pisarev, V., Kinarsky, L., Sherman, S., Kang, L., Herber, D. L., Schneck, J., & Gabrilovich, D. I. (2007). Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nature Medicine, 13, 828–835.
Yaseen, M. M., Abuharfeil, N. M., Darmani, H., & Daoud, A. (2020). Mechanisms of immune suppression by myeloid-derived suppressor cells: the role of interleukin-10 as a key immunoregulatory cytokine. Open Biology, 10, 200111.
Nguyen, D. X., Bos, P. D., & Massague, J. (2009). Metastasis: from dissemination to organ-specific colonization. Nature Reviews. Cancer, 9, 274–284.
Paget, S. (1989). The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Reviews, 8, 98–101.
Kaplan, R. N., Riba, R. D., Zacharoulis, S., Bramley, A. H., Vincent, L., Costa, C., MacDonald, D. D., Jin, D. K., Shido, K., Kerns, S. A., Zhu, Z., Hicklin, D., Wu, Y., Port, J. L., Altorki, N., Port, E. R., Ruggero, D., Shmelkov, S. V., Jensen, K. K., Rafii, S., & Lyden, D. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature, 438, 820–827.
Hoggatt, J., & Pelus, L. M. (2011). Many mechanisms mediating mobilization: an alliterative review. Current Opinion in Hematology, 18, 231–238.
Levesque, J. P., Hendy, J., Takamatsu, Y., Simmons, P. J., & Bendall, L. J. (2003). Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. The Journal of Clinical Investigation, 111, 187–196.
Saito, T., Usui, N., Asai, O., Dobashi, N., Yano, S., Osawa, H., Takei, Y., Takahara, S., Ogasawara, Y., Otsubo, H., Yamaguchi, Y., Minami, J., Aiba, K., Otsubo, H., Hoshi, Y., & Kataoka, M. (2007). Elevated serum levels of human matrix metalloproteinase-9 (MMP-9) during the induction of peripheral blood stem cell mobilization by granulocyte colony-stimulating factor (G-CSF). Journal of Infection and Chemotherapy, 13, 426–428.
Xu, M., Bruno, E., Chao, J., Huang, S., Finazzi, G., Fruchtman, S. M., Popat, U., Prchal, J. T., Barosi, G., Hoffman, R., & for the MPD Research Consortium. (2005). Constitutive mobilization of CD34+ cells into the peripheral blood in idiopathic myelofibrosis may be due to the action of a number of proteases. Blood, 105, 4508–4515.
Heissig, B., Hattori, K., Dias, S., Friedrich, M., Ferris, B., Hackett, N. R., Crystal, R. G., Besmer, P., Lyden, D., Moore, M. A. S., Werb, Z., & Rafii, S. (2002). Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell, 109, 625–637.
Zeng, Z. S., Cohen, A. M., & Guillem, J. G. (1999). Loss of basement membrane type IV collagen is associated with increased expression of metalloproteinases 2 and 9 (MMP-2 and MMP-9) during human colorectal tumorigenesis. Carcinogenesis, 20, 749–755.
Davies, B., Waxman, J., Wasan, H., Abel, P., Williams, G., Krausz, T., Neal, D., Thomas, D., Hanby, A., & Balkwill, F. (1993). Levels of matrix metalloproteases in bladder cancer correlate with tumor grade and invasion. Cancer Research, 53, 5365–5369.
Hamdy, F. C., Fadlon, E. J., Cottam, D., Lawry, J., Thurrell, W., Silcocks, P. B., Anderson, J. B., Williams, J. L., & Rees, R. C. (1994). Matrix metalloproteinase 9 expression in primary human prostatic adenocarcinoma and benign prostatic hyperplasia. British Journal of Cancer, 69, 177–182.
Cortez-Retamozo, V., Etzrodt, M., Newton, A., Rauch, P. J., Chudnovskiy, A., Berger, C., Ryan, R. J. H., Iwamoto, Y., Marinelli, B., Gorbatov, R., Forghani, R., Novobrantseva, T. I., Koteliansky, V., Figueiredo, J. L., Chen, J. W., Anderson, D. G., Nahrendorf, M., Swirski, F. K., Weissleder, R., & Pittet, M. J. (2012). Origins of tumor-associated macrophages and neutrophils. Proceedings of the National Academy of Sciences of the United States of America, 109, 2491–2496.
Wu, C., Ning, H., Liu, M., Lin, J., Luo, S., Zhu, W., Xu, J., Wu, W. C., Liang, J., Shao, C. K., Ren, J., Wei, B., Cui, J., Chen, M. S., & Zheng, L. (2018). Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. The Journal of Clinical Investigation, 128, 3425–3438.
Klein, B., Stein, M., Kuten, A., Steiner, M., Barshalom, D., Robinson, E., & Gal, D. (1987). Splenomegaly and solitary spleen metastasis in solid tumors. Cancer, 60, 100–102.
Kiely, J. M., & Silverstein, M. N. (1969). Metastatic carcinoma simulating agnogenic myeloid metaplasia and myelofibrosis. Cancer, 24, 1041–1044.
Schlitt, H. J., Schäfers, S., Deiwick, A., Eckardt, K. U., Pietsch, T., Ebell, W., Nashan, B., Ringe, B., Wonigeit, K., & Pichlmayr, R. (1995). Extramedullary erythropoiesis in human liver grafts. Hepatology, 21, 689–696.
Craig, C. E. H., Quaglia, A., & Dhillon, A. P. (2004). Extramedullary haematopoiesis in massive hepatic necrosis. Histopathology, 45, 518–525.
Mohyuddin, G. R., & Yacoub, A. (2016). Primary myelofibrosis presenting as extramedullary hematopoiesis in a transplanted liver graft: case report and review of the literature. Case Rep Hematol, 2016, 9515404.
Yablonski-Peretz, T., Sulkes, A., Polliack, A., Weshler, Z., Okon, E., & Catane, R. (1985). Secondary myelofibrosis with metastatic breast cancer simulating agnogenic myeloid metaplasia: Report of a case and review of the literature. Medical and Pediatric Oncology, 13, 92–96.
Rusciano, D., & Burger, M. M. (1992). Why do cancer cells metastasize into particular organs? Bioessays, 14, 185–194.
Nicolson, G. L. (1988). Cancer metastasis: tumor cell and host organ properties important in metastasis to specific secondary sites. Biochimica et Biophysica Acta, 948, 175–224.
Graf, A. H., Buchberger, W., Langmayr, H., & Schmid, K. W. (1988). Site preference of metastatic tumours of the brain. Virchows Archiv. A, Pathological Anatomy and Histopathology, 412, 493–498.
Riihimäki, M., Hemminki, A., Fallah, M., Thomsen, H., Sundquist, K., Sundquist, J., & Hemminki, K. (2014). Metastatic sites and survival in lung cancer. Lung Cancer, 86, 78–84.
Talmadge, J. E., Donkor, M., & Scholar, E. (2007). Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metastasis Reviews, 26, 373–400.
Tarin, D., Price, J. E., Kettlewell, M. G., Souter, R. G., Vass, A. C., & Crossley, B. (1984). Mechanisms of human tumor metastasis studied in patients with peritoneovenous shunts. Cancer Research, 44, 3584–3592.
Fidler, I. J. (1970). Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125 I-5-iodo-2′-deoxyuridine. Journal of the National Cancer Institute, 45, 773–782.
Schmidt-Kittler, O., Ragg, T., Daskalakis, A., Granzow, M., Ahr, A., Blankenstein, T. J. F., Kaufmann, M., Diebold, J., Arnholdt, H., Muller, P., Bischoff, J., Harich, D., Schlimok, G., Riethmuller, G., Eils, R., & Klein, C. A. (2003). From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proceedings of the National Academy of Sciences of the United States of America, 100, 7737–7742.
Talmadge, J. E., & Fidler, I. J. (2010). AACR centennial series: the biology of cancer metastasis: Historical perspective. Cancer Research, 70, 5649–5669.
Bartlett, E. K., Fetsch, P. A., Filie, A. C., Abati, A., Steinberg, S. M., Wunderlich, J. R., White, D. E., Stephens, D. J., Marincola, F. M., Rosenberg, S. A., & Kammula, U. S. (2014). Human melanoma metastases demonstrate nonstochastic site-specific antigen heterogeneity that correlates with T-cell infiltration. Clinical Cancer Research, 20, 2607–2616.
Klein, C. A. (2009). Parallel progression of primary tumours and metastases. Nature Reviews. Cancer, 9, 302–312.
Weiss, L. (1983). Random and nonrandom processes in metastasis, and metastatic inefficiency. Invasion & Metastasis, 3, 193–207.
Arnerlöv, C., et al. (1992). Breast carcinoma growth rate described by mammographic doubling time and S-phase fraction. Correlations to clinical and histopathologic factors in a screened population. Cancer, 70, 1928–1934.
Klein, C. A., Blankenstein, T. J. F., Schmidt-Kittler, O., Petronio, M., Polzer, B., Stoecklein, N. H., & Riethmüller, G. (2002). Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet, 360, 683–689.
Gruber, I. V., Hartkopf, A. D., Hahn, M., Taran, F. A., Staebler, A., Wallwiener, D., Brucker, S. Y., Hanke, J., & Fehm, T. (2016). Relationship between hematogenous tumor cell dissemination and cellular immunity in DCIS patients. Anticancer Research, 36, 2345–2351.
Sänger, N., Effenberger, K. E., Riethdorf, S., van Haasteren, V., Gauwerky, J., Wiegratz, I., Strebhardt, K., Kaufmann, M., & Pantel, K. (2011). Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. International Journal of Cancer, 129, 2522–2526.
Hu, Z., & Curtis, C. (2020). Looking backward in time to define the chronology of metastasis. Nature Communications, 11, 3213.
Alix-Panabieres, C., & Pantel, K. (2014). Challenges in circulating tumour cell research. Nature Reviews. Cancer, 14, 623–631.
Dasgupta, A., Lim, A. R., & Ghajar, C. M. (2017). Circulating and disseminated tumor cells: harbingers or initiators of metastasis? Molecular Oncology, 11, 40–61.
Schardt, J. A., Meyer, M., Hartmann, C. H., Schubert, F., Schmidt-Kittler, O., Fuhrmann, C., Polzer, B., Petronio, M., Eils, R., & Klein, C. A. (2005). Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell, 8, 227–239.
Gray, J. W. (2003). Evidence emerges for early metastasis and parallel evolution of primary and metastatic tumors. Cancer Cell, 4, 4–6.
Weigelt, B. (2003). Gene expression profiles of primary breast tumors maintained in distant metastases. Proceedings of the National Academy of Sciences, 100, 15901–15905.
Magrì, A., & Bardelli, A. (2019). Does early metastatic seeding occur in colorectal cancer? Nature Reviews. Gastroenterology & Hepatology, 16, 651–653.
Jones, S., Chen, W. D., Parmigiani, G., Diehl, F., Beerenwinkel, N., Antal, T., Traulsen, A., Nowak, M. A., Siegel, C., Velculescu, V. E., Kinzler, K. W., Vogelstein, B., Willis, J., & Markowitz, S. D. (2008). Comparative lesion sequencing provides insights into tumor evolution. Proceedings of the National Academy of Sciences of the United States of America, 105, 4283–4288.
Hu, Z., Ding, J., Ma, Z., Sun, R., Seoane, J. A., Scott Shaffer, J., Suarez, C. J., Berghoff, A. S., Cremolini, C., Falcone, A., Loupakis, F., Birner, P., Preusser, M., Lenz, H. J., & Curtis, C. (2019). Quantitative evidence for early metastatic seeding in colorectal cancer. Nature Genetics, 51, 1113–1122.
Stoecklein, N. H., Hosch, S. B., Bezler, M., Stern, F., Hartmann, C. H., Vay, C., Siegmund, A., Scheunemann, P., Schurr, P., Knoefel, W. T., Verde, P. E., Reichelt, U., Erbersdobler, A., Grau, R., Ullrich, A., Izbicki, J. R., & Klein, C. A. (2008). Direct genetic analysis of single disseminated cancer cells for prediction of outcome and therapy selection in esophageal cancer. Cancer Cell, 13, 441–453.
Birkbak, N. J., & McGranahan, N. (2020). Cancer genome evolutionary trajectories in metastasis. Cancer Cell, 37, 8–19.
Talmadge, J. E. (2007). Clonal selection of metastasis within the life history of a tumor. Cancer Research, 67, 11471–11475.
Friberg, S., & Mattson, S. (1997). On the growth rates of human malignant tumors: implications for medical decision making. Journal of Surgical Oncology, 65, 284–297.
von Fournier, D., et al. (1980). Growth rate of 147 mammary carcinomas. Cancer, 45, 2198–2207.
Shackney, S. E., McCormack, G. W., & Cuchural Jr., G. J. (1978). Growth rate patterns of solid tumors and their relation to responsiveness to therapy: an analytical review. Annals of Internal Medicine, 89, 107–121.
Fidler, I. J., & Balch, C. M. (1987). The biology of cancer metastasis and implications for therapy. Current Problems in Surgery, 24, 129–209.
Ulmschneider, M. B., & Searson, P. C. (2015). Mathematical models of the steps involved in the systemic delivery of a chemotherapeutic to a solid tumor: from circulation to survival. Journal of Controlled Release, 212, 78–84.
Willis, R. The spread of tumours in the human body. By Rupert A. Willis. M.D., B.S., D.Sc. (Melbourne), 1934 London: J. & A. Churchill. 25s. net. BJS (British Journal of Surgery) 22, 196–196 (1934).
Endo, H., & Inoue, M. (2019). Dormancy in cancer. Cancer Science, 110, 474–480.
Pan, H., Gray, R., Braybrooke, J., Davies, C., Taylor, C., McGale, P., Peto, R., Pritchard, K. I., Bergh, J., Dowsett, M., Hayes, D. F., & EBCTCG. (2017). 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. The New England Journal of Medicine, 377, 1836–1846.
Goddard, E. T., Bozic, I., Riddell, S. R., & Ghajar, C. M. (2018). Dormant tumour cells, their niches and the influence of immunity. Nature Cell Biology, 20, 1240–1249.
Folkman, J. (1971). Tumor angiogenesis: therapeutic implications. The New England Journal of Medicine, 285, 1182–1186.
Hanahan, D., & Folkman, J. (1996). Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell, 86, 353–364.
Siemann, D. W., & Horsman, M. R. (2015). Modulation of the tumor vasculature and oxygenation to improve therapy. Pharmacology & Therapeutics, 153, 107–124.
Farrar, J. D., et al. (1999). Cancer dormancy. VII. A regulatory role for CD8+ T cells and IFN-gamma in establishing and maintaining the tumor-dormant state. Journal of Immunology, 162, 2842–2849.
Dunn, G. P., Old, L. J., & Schreiber, R. D. (2004). The immunobiology of cancer immunosurveillance and immunoediting. Immunity, 21, 137–148.
Wheelock, E. F., Weinhold, K. J., & Levich, J. (1981). The tumor dormant state. Advances in Cancer Research, 34, 107–140.
Koebel, C. M., Vermi, W., Swann, J. B., Zerafa, N., Rodig, S. J., Old, L. J., Smyth, M. J., & Schreiber, R. D. (2007). Adaptive immunity maintains occult cancer in an equilibrium state. Nature, 450, 903–907.
Sidky, Y. A., & Borden, E. C. (1987). Inhibition of angiogenesis by interferons: effects on tumor- and lymphocyte-induced vascular responses. Cancer Research, 47, 5155–5161.
Voest, E. E., Kenyon, B. M., O’Reilly, M. S., Truitt, G., D’Amato, R. J., & Folkman, J. (1995). Inhibition of angiogenesis in vivo by interleukin 12. Journal of the National Cancer Institute, 87, 581–586.
Sleeman, J. P. (2012). The metastatic niche and stromal progression. Cancer Metastasis Reviews, 31, 429–440.
Fane, M., & Weeraratna, A. T. (2020). How the ageing microenvironment influences tumour progression. Nature Reviews. Cancer, 20, 89–106.
Baschuk, N., Rautela, J., & Parker, B. S. (2015). Bone specific immunity and its impact on metastasis. Bonekey Reports, 4, 665.
Nakajima, M., Morikawa, K., Fabra, A., Bucana, C. D., & Fidler, I. J. (1990). Influence of organ environment on extracellular matrix degradative activity and metastasis of human colon carcinoma cells. Journal of the National Cancer Institute, 82, 1890–1898.
Casimiro, S., Guise, T. A., & Chirgwin, J. (2009). The critical role of the bone microenvironment in cancer metastases. Molecular and Cellular Endocrinology, 310, 71–81.
Sceneay, J., Smyth, M. J., & Möller, A. (2013). The pre-metastatic niche: finding common ground. Cancer Metastasis Reviews, 32, 449–464.
Lindau, D., Gielen, P., Kroesen, M., Wesseling, P., & Adema, G. J. (2013). The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology, 138, 105–115.
Mitchell, K. G., et al. (2020). Neutrophil expansion defines an immunoinhibitory peripheral and intratumoral inflammatory milieu in resected non-small cell lung cancer: a descriptive analysis of a prospectively immunoprofiled cohort. Journal for Immunotherapy of Cancer, 8, e000405.
Marymont Jr., J. H., & Gross, S. (1963). Patterns of metastatic cancer in the spleen. American Journal of Clinical Pathology, 40, 58–66.
Sauer, J., Sobolewski, K., & Dommisch, K. (2009). Splenic metastases--not a frequent problem, but an underestimate location of metastases: epidemiology and course. Journal of Cancer Research and Clinical Oncology, 135, 667–671.
Watson, G. F., Diller, I. C., & Ludwick, N. V. (1947). Spleen Extract and Tumor Growth. Science, 106, 348.
Peereboom, D. M., et al. (2019). Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight, 4, e130748.
Weber, R., Fleming, V., Hu, X., Nagibin, V., Groth, C., Altevogt, P., Utikal, J., & Umansky, V. (2018). Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Frontiers in Immunology, 9, 1310.
Wesolowski, R., Markowitz, J., & Carson, W. E. (2013). Myeloid derived suppressor cells – a new therapeutic target in the treatment of cancer. Journal for Immunotherapy of Cancer, 1, 10.
Sun, L., et al. (2019). Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight, 4, e126853.
Eriksson, E., Wenthe, J., Irenaeus, S., Loskog, A., & Ullenhag, G. (2016). Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. Journal of Translational Medicine, 14, 282.
Annels, N. E., Shaw, V. E., Gabitass, R. F., Billingham, L., Corrie, P., Eatock, M., Valle, J., Smith, D., Wadsley, J., Cunningham, D., Pandha, H., Neoptolemos, J. P., & Middleton, G. (2014). The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunology, Immunotherapy, 63, 175–183.
Kanterman, J., Sade-Feldman, M., Biton, M., Ish-Shalom, E., Lasry, A., Goldshtein, A., Hubert, A., & Baniyash, M. (2014). Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Research, 74, 6022–6035.
Limagne, E., Euvrard, R., Thibaudin, M., Rébé, C., Derangère, V., Chevriaux, A., Boidot, R., Végran, F., Bonnefoy, N., Vincent, J., Bengrine-Lefevre, L., Ladoire, S., Delmas, D., Apetoh, L., & Ghiringhelli, F. (2016). Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX–bevacizumab drug treatment regimen. Cancer Research, 76, 5241–5252.
Noonan, K. A., Ghosh, N., Rudraraju, L., Bui, M., & Borrello, I. (2014). Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer Immunology Research, 2, 725–731.
Califano, J. A., Khan, Z., Noonan, K. A., Rudraraju, L., Zhang, Z., Wang, H., Goodman, S., Gourin, C. G., Ha, P. K., Fakhry, C., Saunders, J., Levine, M., Tang, M., Neuner, G., Richmon, J. D., Blanco, R., Agrawal, N., Koch, W. M., Marur, S., Weed, D. T., Serafini, P., & Borrello, I. (2015). Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clinical Cancer Research, 21, 30–38.
Hassel, J. C., et al. (2017). Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology, 6, e1326440.
Steggerda, S. M., Bennett, M. K., Chen, J., Emberley, E., Huang, T., Janes, J. R., Li, W., MacKinnon, A. L., Makkouk, A., Marguier, G., Murray, P. J., Neou, S., Pan, A., Parlati, F., Rodriguez, M. L. M., van de Velde, L. A., Wang, T., Works, M., Zhang, J., Zhang, W., & Gross, M. I. (2017). Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. Journal for Immunotherapy of Cancer, 5, 101.
Naing, A., Bauer, T., Papadopoulos, K. P., Rahma, O., Tsai, F., Garralda, E., Naidoo, J., Pai, S., Gibson, M. K., Rybkin, I., Wang, D., McDermott, D. F., Fasolo, A., de Miguel, M., Shaheen, M., Jenkins, Y., Kallender, H., Gogov, S., Kuriakose, E., & Pishvaian, M. (2019). Phase I study of the arginase inhibitor INCB001158 (1158) alone and in combination with pembrolizumab (PEM) in patients (Pts) with advanced/metastatic (adv/met) solid tumours. Annals of Oncology, 30, v160.
Chung, A., et al. (2019). Abstract OT3-08-01: A phase Ib/II clinical trial investigating the efficacy of nitric oxide deprivation and docetaxel in triple negative breast cancer. Cancer Research, 79, OT3–08-01.
Fiorucci, S., Mencarelli, A., Meneguzzi, A., Lechi, A., Renga, B., del Soldato, P., Morelli, A., & Minuz, P. (2004). Co-administration of nitric oxide-aspirin (NCX-4016) and aspirin prevents platelet and monocyte activation and protects against gastric damage induced by aspirin in humans. Journal of the American College of Cardiology, 44, 635–641.
De Santo, C., et al. (2005). Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proceedings of the National Academy of Sciences of the United States of America, 102, 4185–4190.
Edwards, D. K. T., et al. (2019). CSF1R inhibitors exhibit antitumor activity in acute myeloid leukemia by blocking paracrine signals from support cells. Blood, 133, 588–599.
Wesolowski, R., et al. (2019). Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther Adv Med Oncol, 11, 1758835919854238.
Butowski, N., Colman, H., de Groot, J. F., Omuro, A. M., Nayak, L., Wen, P. Y., Cloughesy, T. F., Marimuthu, A., Haidar, S., Perry, A., Huse, J., Phillips, J., West, B. L., Nolop, K. B., Hsu, H. H., Ligon, K. L., Molinaro, A. M., & Prados, M. (2016). Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology, 18, 557–564.
Giallongo, C., Parrinello, N. L., la Cava, P., Camiolo, G., Romano, A., Scalia, M., Stagno, F., Palumbo, G. A., Avola, R., Li Volti, G., Tibullo, D., & di Raimondo, F. (2018). Monocytic myeloid-derived suppressor cells as prognostic factor in chronic myeloid leukaemia patients treated with dasatinib. Journal of Cellular and Molecular Medicine, 22, 1070–1080.
Martens, A., Wistuba-Hamprecht, K., Foppen, M. G., Yuan, J., Postow, M. A., Wong, P., Romano, E., Khammari, A., Dreno, B., Capone, M., Ascierto, P. A., di Giacomo, A. M., Maio, M., Schilling, B., Sucker, A., Schadendorf, D., Hassel, J. C., Eigentler, T. K., Martus, P., Wolchok, J. D., Blank, C., Pawelec, G., Garbe, C., & Weide, B. (2016). Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clinical Cancer Research, 22, 2908–2918.
Gebhardt, C., Sevko, A., Jiang, H., Lichtenberger, R., Reith, M., Tarnanidis, K., Holland-Letz, T., Umansky, L., Beckhove, P., Sucker, A., Schadendorf, D., Utikal, J., & Umansky, V. (2015). Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clinical Cancer Research, 21, 5453–5459.
de Coaña, Y. P., et al. (2013). Ipilimumab treatment results in an early decrease in the frequency of circulating granulocytic myeloid-derived suppressor cells as well as their Arginase1 production. Cancer Immunology Research, 1, 158–162.
Pico de Coana, Y., et al. (2017). Ipilimumab treatment decreases monocytic MDSCs and increases CD8 effector memory T cells in long-term survivors with advanced melanoma. Oncotarget, 8, 21539–21553.
Tzeng, A., Diaz-Montero, C. M., Rayman, P. A., Kim, J. S., Pavicic Jr., P. G., Finke, J. H., Barata, P. C., Lamenza, M., Devonshire, S., Schach, K., Emamekhoo, H., Ernstoff, M. S., Hoimes, C. J., Rini, B. I., Garcia, J. A., Gilligan, T. D., Ornstein, M. C., & Grivas, P. (2018). Immunological correlates of response to immune checkpoint inhibitors in metastatic urothelial carcinoma. Targeted Oncology, 13, 599–609.
Parker, N., Al-Obaidi, A., Truong, Q. V., & Badgett, R. (2019). Pembrolizumab versus the standard of care for cancer therapy: a meta-analysis of 12 KEYNOTE trials comparing overall survival. Journal of Clinical Oncology, 37, e14159–e14159.
Stiff, A., Trikha, P., Wesolowski, R., Kendra, K., Hsu, V., Uppati, S., McMichael, E., Duggan, M., Campbell, A., Keller, K., Landi, I., Zhong, Y., Dubovsky, J., Howard, J. H., Yu, L., Harrington, B., Old, M., Reiff, S., Mace, T., Tridandapani, S., Muthusamy, N., Caligiuri, M. A., Byrd, J. C., & Carson III, W. E. (2016). Myeloid-derived suppressor cells express Bruton’s tyrosine kinase and can be depleted in tumor-bearing hosts by Ibrutinib treatment. Cancer Research, 76, 2125–2136.
Ferrer, G., Jung, B., Rukhsana, A., Chiu, P. Y., Mazzarello, A. N., Palacios, F., Chen, S. S., Yan, X. J., Barrientos, J. C., Burger, J. A., Kolitz, J. E., Allen, S. L., Rai, K. R., Sherry, B., & Chiorazzi, N. (2018). Ibrutinib treatment reduces myeloid derived suppressor cell numbers and function in chronic lymphocytic leukemia. Blood, 132, 239–239.
Benner, B., Quiroga, D. M., Good, L., Sun, S., Savardekar, H., Duggan, M. C., Konda, B., Verschraegen, C. F., Kendra, K. L., Shah, M. H., Rupert, R., Monk, P., Shah, H. A., Noonan, A. M., Bixel, K. L., Hays, J. L., Behbehani, G., Pietrzak, M., Carson, W. E., Wesolowski, R., & ASCO Authors Group. (2020). A pilot study of Bruton’s tyrosine kinase inhibitor ibrutinib alone and in combination with PD-1 inhibitor nivolumab in patients with metastatic solid tumors. Journal of Clinical Oncology, 38, 3111–3111.
Bilusic, M., Heery, C. R., Collins, J. M., Donahue, R. N., Palena, C., Madan, R. A., Karzai, F., Marté, J. L., Strauss, J., Gatti-Mays, M. E., Schlom, J., & Gulley, J. L. (2019). Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. Journal for Immunotherapy of Cancer, 7, 240.
Bockorny, B., Semenisty, V., Macarulla, T., Borazanci, E., Wolpin, B. M., Stemmer, S. M., Golan, T., Geva, R., Borad, M. J., Pedersen, K. S., Park, J. O., Ramirez, R. A., Abad, D. G., Feliu, J., Muñoz, A., Ponz-Sarvise, M., Peled, A., Lustig, T. M., Bohana-Kashtan, O., Shaw, S. M., Sorani, E., Chaney, M., Kadosh, S., Vainstein Haras, A., von Hoff, D. D., & Hidalgo, M. (2020). BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nature Medicine, 26, 878–885.
Ko, J. S., Zea, A. H., Rini, B. I., Ireland, J. L., Elson, P., Cohen, P., Golshayan, A., Rayman, P. A., Wood, L., Garcia, J., Dreicer, R., Bukowski, R., & Finke, J. H. (2009). Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clinical Cancer Research, 15, 2148–2157.
Chen, H. M., Ma, G., Gildener-Leapman, N., Eisenstein, S., Coakley, B. A., Ozao, J., Mandeli, J., Divino, C., Schwartz, M., Sung, M., Ferris, R., Kao, J., Wang, L. H., Pan, P. Y., Ko, E. C., & Chen, S. H. (2015). Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clinical Cancer Research, 21, 4073–4085.
Parampalli Yajnanarayana, S., Stübig, T., Cornez, I., Alchalby, H., Schönberg, K., Rudolph, J., Triviai, I., Wolschke, C., Heine, A., Brossart, P., Kröger, N., & Wolf, D. (2015). JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. British Journal of Haematology, 169, 824–833.
Schneble, E. J., Berry, J. S., Trappey, F. A., Clifton, G. T., Ponniah, S., Mittendorf, E., & Peoples, G. E. (2014). The HER2 peptide nelipepimut-S (E75) vaccine (NeuVax) in breast cancer patients at risk for recurrence: correlation of immunologic data with clinical response. Immunotherapy, 6, 519–531.
Yanagimoto, H., Mine, T., Yamamoto, K., Satoi, S., Terakawa, N., Takahashi, K., Nakahara, K., Honma, S., Tanaka, M., Mizoguchi, J., Yamada, A., Oka, M., Kamiyama, Y., Itoh, K., & Takai, S. (2007). Immunological evaluation of personalized peptide vaccination with gemcitabine for pancreatic cancer. Cancer Science, 98, 605–611.
Chiappori, A. A., Soliman, H., Janssen, W. E., Antonia, S. J., & Gabrilovich, D. I. (2010). INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert Opinion on Biological Therapy, 10, 983–991.
Antonia, S. J., et al. (2006). Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clinical Cancer Research, 12, 878–887.
Hardwick, N. R., Frankel, P., Ruel, C., Kilpatrick, J., Tsai, W., Kos, F., Kaltcheva, T., Leong, L., Morgan, R., Chung, V., Tinsley, R., Eng, M., Wilczynski, S., Ellenhorn, J. D. I., Diamond, D. J., & Cristea, M. (2018). p53-reactive T cells are associated with clinical benefit in patients with platinum-resistant epithelial ovarian cancer after treatment with a p53 vaccine and gemcitabine chemotherapy. Clinical Cancer Research, 24, 1315–1325.
Sanford, D. E., Porembka, M. R., Panni, R. Z., Mitchem, J. B., Belt, B. A., Plambeck-Suess, S. M., Lin, G., Denardo, D. G., Fields, R. C., Hawkins, W. G., Strasberg, S. M., Lockhart, A. C., Wang-Gillam, A., Goedegebuure, S. P., & Linehan, D. C. (2013). A study of zoledronic acid as neo-adjuvant, perioperative therapy in patients with resectable pancreatic ductal adenocarcinoma. Journal of Cancer Therapy, 4, 797–803.
Mirza, N., Fishman, M., Fricke, I., Dunn, M., Neuger, A. M., Frost, T. J., Lush, R. M., Antonia, S., & Gabrilovich, D. I. (2006). <em>all-trans</em>-retinoic acid improves differentiation of myeloid cells and immune response in Cancer patients. Cancer Research, 66, 9299–9307.
Tobin, R. P., Jordan, K. R., Robinson, W. A., Davis, D., Borges, V. F., Gonzalez, R., Lewis, K. D., & McCarter, M. D. (2018). Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. International Immunopharmacology, 63, 282–291.
Walsh, J. E., Clark, A.-M., Day, T. A., Gillespie, M. B., & Young, M. R. I. (2010). Use of α,25-Dihydroxyvitamin D3 treatment to stimulate immune infiltration into head and neck squamous cell carcinoma. Human Immunology, 71, 659–665.
Lathers, D. M. R., Clark, J. I., Achille, N. J., & Young, M. R. I. (2004). Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D3. Cancer Immunology, Immunotherapy, 53, 422–430.
Kotsakis, A., Harasymczuk, M., Schilling, B., Georgoulias, V., Argiris, A., & Whiteside, T. L. (2012). Myeloid-derived suppressor cell measurements in fresh and cryopreserved blood samples. Journal of Immunological Methods, 381, 14–22.
Trellakis, S., Bruderek, K., Hütte, J., Elian, M., Hoffmann, T. K., Lang, S., & Brandau, S. (2013). Granulocytic myeloid-derived suppressor cells are cryosensitive and their frequency does not correlate with serum concentrations of colony-stimulating factors in head and neck cancer. Innate Immunity, 19, 328–336.
Crawford, J., Dale, D. C., & Lyman, G. H. (2004). Chemotherapy-induced neutropenia. Cancer, 100, 228–237.
Wesolowski, R., Duggan, M. C., Stiff, A., Markowitz, J., Trikha, P., Levine, K. M., Schoenfield, L., Abdel-Rasoul, M., Layman, R., Ramaswamy, B., Macrae, E. R., Lustberg, M. B., Reinbolt, R. E., Mrozek, E., Byrd, J. C., Caligiuri, M. A., Mace, T. A., & Carson III, W. E. (2017). Circulating myeloid-derived suppressor cells increase in patients undergoing neo-adjuvant chemotherapy for breast cancer. Cancer Immunology, Immunotherapy, 66, 1437–1447.
Talmadge, J. E., Reed, E. C., Kessinger, A., Kuszynski, C. A., Perry, G. A., Gordy, C. L., Mills, K. C., Thomas, M. L., Pirruccello, S. J., Letheby, B. A., Arneson, M. A., & Jackson, J. D. (1996). Immunologic attributes of cytokine mobilized peripheral blood stem cells and recovery following transplantation. Bone Marrow Transplantation, 17, 101–109.
Citron, M. L., Berry, D. A., Cirrincione, C., Hudis, C., Winer, E. P., Gradishar, W. J., Davidson, N. E., Martino, S., Livingston, R., Ingle, J. N., Perez, E. A., Carpenter, J., Hurd, D., Holland, J. F., Smith, B. L., Sartor, C. I., Leung, E. H., Abrams, J., Schilsky, R. L., Muss, H. B., & Norton, L. (2003). Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. Journal of Clinical Oncology, 21, 1431–1439.
Serafini, P., Meckel, K., Kelso, M., Noonan, K., Califano, J., Koch, W., Dolcetti, L., Bronte, V., & Borrello, I. (2006). Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. Journal of Experimental Medicine, 203, 2691–2702.
Markowitz, J., Wang, J., Vangundy, Z., You, J., Yildiz, V., Yu, L., Foote, I. P., Branson, O. E., Stiff, A. R., Brooks, T. R., Biesiadecki, B., Olencki, T., Tridandapani, S., Freitas, M. A., Papenfuss, T., Phelps, M. A., & Carson, W. E. (2017). Nitric oxide mediated inhibition of antigen presentation from DCs to CD4+ T cells in cancer and measurement of STAT1 nitration. Scientific Reports, 7, 15424.
Nagaraj, S., Schrum, A. G., Cho, H.-I., Celis, E., & Gabrilovich, D. I. (2010). Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. The Journal of Immunology, 184, 3106–3116.
Ezernitchi, A. V., Vaknin, I., Cohen-Daniel, L., Levy, O., Manaster, E., Halabi, A., Pikarsky, E., Shapira, L., & Baniyash, M. (2006). TCR zeta down-regulation under chronic inflammation is mediated by myeloid suppressor cells differentially distributed between various lymphatic organs. Journal of Immunology, 177, 4763–4772.
Weed, D. T., Vella, J. L., Reis, I. M., de la fuente, A. C., Gomez, C., Sargi, Z., Nazarian, R., Califano, J., Borrello, I., & Serafini, P. (2015). Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clinical Cancer Research, 21, 39–48.
Poschke, I., De Boniface, J., Mao, Y., & Kiessling, R. (2012). Tumor-induced changes in the phenotype of blood-derived and tumor-associated T cells of early stage breast cancer patients. International Journal of Cancer, 131, 1611–1620.
Avtandilyan, N., Javrushyan, H., Petrosyan, G., & Trchounian, A. (2018). The involvement of arginase and nitric oxide synthase in breast cancer development: arginase and NO synthase as therapeutic targets in cancer. BioMed Research International, 2018, 8696923.
Ma, Z., Lian, J., Yang, M., Wuyang, J., Zhao, C., Chen, W., Liu, C., Zhao, Q., Lou, C., Han, J., & Zhang, Y. (2019). Overexpression of Arginase-1 is an indicator of poor prognosis in patients with colorectal cancer. Pathology, Research and Practice, 215, 152383.
Grzywa, T. M., et al. (2020). Myeloid cell-derived arginase in cancer immune response. Frontiers in Immunology, 11, 938.
Gey, A., Tadie, J. M., Caumont-Prim, A., Hauw-Berlemont, C., Cynober, L., Fagon, J. Y., Terme, M., Diehl, J. L., Delclaux, C., & Tartour, E. (2015). Granulocytic myeloid-derived suppressor cells inversely correlate with plasma arginine and overall survival in critically ill patients. Clinical and Experimental Immunology, 180, 280–288.
Jayaraman, P., Parikh, F., Lopez-Rivera, E., Hailemichael, Y., Clark, A., Ma, G., Cannan, D., Ramacher, M., Kato, M., Overwijk, W. W., Chen, S. H., Umansky, V. Y., & Sikora, A. G. (2012). Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. Journal of Immunology, 188, 5365–5376.
Mazzoni, A., Bronte, V., Visintin, A., Spitzer, J. H., Apolloni, E., Serafini, P., Zanovello, P., & Segal, D. M. (2002). Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. Journal of Immunology, 168, 689–695.
Kusmartsev, S., Nefedova, Y., Yoder, D., & Gabrilovich, D. I. (2004). Antigen-specific inhibition of CD8<sup>+</sup> T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. The Journal of Immunology, 172, 989–999.
Feng, S., Cheng, X., Zhang, L., Lu, X., Chaudhary, S., Teng, R., Frederickson, C., Champion, M. M., Zhao, R., Cheng, L., Gong, Y., Deng, H., & Lu, X. (2018). Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proceedings of the National Academy of Sciences of the United States of America, 115, 10094–10099.
Fauskanger, M., Haabeth, O. A. W., Skjeldal, F. M., Bogen, B., & Tveita, A. A. (2018). Tumor killing by CD4(+) T cells is mediated via induction of inducible nitric oxide synthase-dependent macrophage cytotoxicity. Frontiers in Immunology, 9, 1684.
Carini, M., Aldini, G., Orioli, M., Piccoli, A., Tocchetti, P., & Maffei Facino, R. (2004). Chemiluminescence and LC–MS/MS analyses for the study of nitric oxide release and distribution following oral administration of nitroaspirin (NCX 4016) in healthy volunteers. Journal of Pharmaceutical and Biomedical Analysis, 35, 277–287.
Chang, K., Lee, S. J., Cheong, I., Billiar, T. R., Chung, H. T., Han, J. A., Kwon, Y. G., Ha, K. S., & Kim, Y. M. (2004). Nitric oxide suppresses inducible nitric oxide synthase expression by inhibiting post-translational modification of IkappaB. Experimental & Molecular Medicine, 36, 311–324.
Li, W., Zhang, X., Chen, Y., Xie, Y., Liu, J., Feng, Q., Wang, Y., Yuan, W., & Ma, J. (2016). G-CSF is a key modulator of MDSC and could be a potential therapeutic target in colitis-associated colorectal cancers. Protein & Cell, 7, 130–140.
Cannarile, M. A., et al. (2017). Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. Journal for Immunotherapy of Cancer, 5, 53.
Apodaca, M. C., Wright, A. E., Riggins, A. M., Harris, W. P., Yeung, R. S., Yu, L., & Morishima, C. (2019). Characterization of a whole blood assay for quantifying myeloid-derived suppressor cells. Journal for Immunotherapy of Cancer, 7, 230.
Tsukada, N., Burger, J. A., Zvaifler, N. J., & Kipps, T. J. (2002). Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood, 99, 1030–1037.
Edwards, V. D., et al. (2018). Targeting of colony-stimulating factor 1 receptor (CSF1R) in the CLL microenvironment yields antineoplastic activity in primary patient samples. Oncotarget, 9, 24576–24589.
Curfs, J. H., Meis, J. F., & Hoogkamp-Korstanje, J. A. (1997). A primer on cytokines: sources, receptors, effects, and inducers. Clinical Microbiology Reviews, 10, 742–780.
Wesolowski, R., et al. (2019). Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Therapeutic Advances in Medical Oncology, 11, 1758835919854238.
Olingy, C. E., Dinh, H. Q., & Hedrick, C. C. (2019). Monocyte heterogeneity and functions in cancer. Journal of Leukocyte Biology, 106, 309–322.
Dewar, A. L., Cambareri, A. C., Zannettino, A. C. W., Miller, B. L., Doherty, K. V., Hughes, T. P., & Lyons, A. B. (2005). Macrophage colony-stimulating factor receptor c-fms is a novel target of imatinib. Blood, 105, 3127–3132.
Hines, S. L., Sloan, J. A., Atherton, P. J., Perez, E. A., Dakhil, S. R., Johnson, D. B., Reddy, P. S., Dalton, R. J., Mattar, B. I., & Loprinzi, C. L. (2010). Zoledronic acid for treatment of osteopenia and osteoporosis in women with primary breast cancer undergoing adjuvant aromatase inhibitor therapy. Breast, 19, 92–96.
Coleman, R., Cameron, D., Dodwell, D., Bell, R., Wilson, C., Rathbone, E., Keane, M., Gil, M., Burkinshaw, R., Grieve, R., Barrett-Lee, P., Ritchie, D., Liversedge, V., Hinsley, S., Marshall, H., & AZURE investigators. (2014). Adjuvant zoledronic acid in patients with early breast cancer: final efficacy analysis of the AZURE (BIG 01/04) randomised open-label phase 3 trial. The Lancet Oncology, 15, 997–1006.
Melani, C., Sangaletti, S., Barazzetta, F. M., Werb, Z., & Colombo, M. P. (2007). Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Research, 67, 11438–11446.
Porembka, M. R., Mitchem, J. B., Belt, B. A., Hsieh, C. S., Lee, H. M., Herndon, J., Gillanders, W. E., Linehan, D. C., & Goedegebuure, P. (2012). Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunology, Immunotherapy, 61, 1373–1385.
Granier, C., de Guillebon, E., Blanc, C., Roussel, H., Badoual, C., Colin, E., Saldmann, A., Gey, A., Oudard, S., & Tartour, E. (2017). Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open, 2, e000213.
Robert, C., Thomas, L., Bondarenko, I., O’Day, S., Weber, J., Garbe, C., Lebbe, C., Baurain, J. F., Testori, A., Grob, J. J., Davidson, N., Richards, J., Maio, M., Hauschild, A., Miller Jr., W. H., Gascon, P., Lotem, M., Harmankaya, K., Ibrahim, R., Francis, S., Chen, T. T., Humphrey, R., Hoos, A., & Wolchok, J. D. (2011). Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. The New England Journal of Medicine, 364, 2517–2526.
Pico de Coaña, Y., Masucci, G., Hansson, J., & Kiessling, R. (2014). Myeloid-derived suppressor cells and their role in CTLA-4 blockade therapy. Cancer Immunology, Immunotherapy, 63, 977–983.
Meyer, C., Cagnon, L., Costa-Nunes, C. M., Baumgaertner, P., Montandon, N., Leyvraz, L., Michielin, O., Romano, E., & Speiser, D. E. (2014). Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunology, Immunotherapy, 63, 247–257.
Highfill, S. L., et al. (2014). Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Science Translational Medicine, 6, 237ra267.
De Henau, O., et al. (2016). Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature, 539, 443–447.
Molina-Cerrillo, J., Alonso-Gordoa, T., Gajate, P., & Grande, E. (2017). Bruton’s tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treatment Reviews, 58, 41–50.
Tempero, M., et al. (2019). Ibrutinib in combination with nab-paclitaxel and gemcitabine as first-line treatment for patients with metastatic pancreatic adenocarcinoma: results from the phase 3 RESOLVE study. Annals of Oncology, 30, iv126.
Metzler, J. M., Burla, L., Fink, D., & Imesch, P. (2020). Ibrutinib in gynecological malignancies and breast cancer: a systematic review. International Journal of Molecular Sciences, 21, 4154.
Hong, D., Rasco, D., Veeder, M., Luke, J. J., Chandler, J., Balmanoukian, A., George, T. J., Munster, P., Berlin, J. D., Gutierrez, M., Mita, A., Wakelee, H., Samakoglu, S., Guan, S., Dimery, I., Graef, T., & Borazanci, E. (2019). A phase 1b/2 study of the Bruton tyrosine kinase inhibitor Ibrutinib and the PD-L1 inhibitor durvalumab in patients with pretreated solid tumors. Oncology, 97, 102–111.
Al-Toubah, T., et al. (2020). A phase II study of Ibrutinib in advanced neuroendocrine neoplasms. Neuroendocrinology, 110, 377–383.
Massó-Vallés, D., Jauset, T., & Soucek, L. (2016). Ibrutinib repurposing: From B-cell malignancies to solid tumors. Oncoscience, 3, 147–148.
Yokochi, S., Hashimoto, H., Ishiwata, Y., Shimokawa, H., Haino, M., Terashima, Y., & Matsushima, K. (2001). An anti-inflammatory drug, propagermanium, may target GPI-anchored proteins associated with an MCP-1 receptor, CCR2. Journal of Interferon & Cytokine Research, 21, 389–398.
Masuda, T., Noda, M., Kogawa, T., Kitagawa, D., Hayashi, N., Jomori, T., Nakanishi, Y., Nakayama, K. I., Ohno, S., & Mimori, K. (2020). Phase I dose-escalation trial to repurpose propagermanium, an oral CCL2 inhibitor, in patients with breast cancer. Cancer Science, 111, 924–931.
Tobin, R. P., Jordan, K. R., Kapoor, P., Spongberg, E., Davis, D., Vorwald, V. M., Couts, K. L., Gao, D., Smith, D. E., Borgers, J. S. W., Robinson, S., Amato, C., Gonzalez, R., Lewis, K. D., Robinson, W. A., Borges, V. F., & McCarter, M. D. (2019). IL-6 and IL-8 are linked with myeloid-derived suppressor cell accumulation and correlate with poor clinical outcomes in melanoma patients. Frontiers in Oncology, 9, 1223.
Dominguez, C., McCampbell, K. K., David, J. M., & Palena, C. (2017). Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight, 2, e94296.
Nefedova, Y., Fishman, M., Sherman, S., Wang, X., Beg, A. A., & Gabrilovich, D. I. (2007). Mechanism of all-<em>trans</em> retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Research, 67, 11021–11028.
Liu, W., et al. (2018). The anti-inflammatory effects of vitamin D in tumorigenesis. International Journal of Molecular Sciences, 19, 2736.
Fleet, J. C., Burcham, G. N., Calvert, R., & Ratliff, T. L. (2019). Abstract 2364: 1α, 25 dihydroxyvitamin D (1,25(OH)<sub>2</sub>D) inhibits the T cell suppressive function of myeloid derived suppressor sells (MDSC). Cancer Research, 79, 2364–2364.
Motzer, R. J., Escudier, B., Gannon, A., & Figlin, R. A. (2017). Sunitinib: ten years of successful clinical use and study in advanced renal cell carcinoma. Oncologist, 22, 41–52.
Finke, J., Ko, J., Rini, B., Rayman, P., Ireland, J., & Cohen, P. (2011). MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. International Immunopharmacology, 11, 856–861.
Jonasch, E. (2019). NCCN guidelines updates: management of metastatic kidney cancer. Journal of the National Comprehensive Cancer Network, 17, 587–589.
de Haas, N., de Koning, C., Spilgies, L., de Vries, I. J. M., & Hato, S. V. (2016). Improving cancer immunotherapy by targeting the STATe of MDSCs. OncoImmunology, 5, e1196312.
Giaccone, G., Sanborn, R. E., Waqar, S. N., Martinez-Marti, A., Ponce, S., Zhen, H., Kennealey, G., Erickson-Viitanen, S., & Schaefer, E. (2018). A placebo-controlled phase II study of ruxolitinib in combination with pemetrexed and cisplatin for first-line treatment of patients with advanced nonsquamous non–small-cell lung cancer and systemic inflammation. Clinical Lung Cancer, 19, e567–e574.
Hurwitz, H. I., Uppal, N., Wagner, S. A., Bendell, J. C., Beck, J. T., Wade 3rd, S. M., Nemunaitis, J. J., Stella, P. J., Pipas, J. M., Wainberg, Z. A., Manges, R., Garrett, W. M., Hunter, D. S., Clark, J., Leopold, L., Sandor, V., & Levy, R. S. (2015). Randomized, double-blind, phase II study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. Journal of Clinical Oncology, 33, 4039–4047.
Talmadge, J. E. (2007). Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clinical Cancer Research, 13, 5243–5248.
Fleming, V., et al. (2018). Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Frontiers in Immunology, 9, 398.
Hou, A., Hou, K., Huang, Q., Lei, Y., & Chen, W. (2020). Targeting myeloid-derived suppressor cell, a promising strategy to overcome resistance to immune checkpoint inhibitors. Frontiers in Immunology, 11, 783.
Benner, B., Quiroga, D. M., Good, L., Sun, S., Savardekar, H., Duggan, M. C., Konda, B., Verschraegen, C. F., Kendra, K. L., Shah, M. H., Rupert, R., Monk, P., Shah, H. A., Noonan, A. M., Bixel, K. L., Hays, J. L., Behbehani, G., Pietrzak, M., Carson, W. E., Wesolowski, R., & ASCO Authors Group. (2020). A pilot study of Bruton’s tyrosine kinase inhibitor ibrutinib alone and in combination with PD-1 inhibitor nivolumab in patients with metastatic solid tumors. Journal of Clinical Oncology, 38, 3111–3111.
Fernández, A., Oliver, L., Alvarez, R., Fernández, L. E., Lee, K. P., & Mesa, C. (2014). Adjuvants and myeloid-derived suppressor cells: enemies or allies in therapeutic cancer vaccination. Human Vaccines & Immunotherapeutics, 10, 3251–3260.
Dranoff, G. (2003). GM-CSF-secreting melanoma vaccines. Oncogene, 22, 3188–3192.
Serafini, P., Carbley, R., Noonan, K. A., Tan, G., Bronte, V., & Borrello, I. (2004). High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Research, 64, 6337–6343.
Triozzi, P. L., Achberger, S., Aldrich, W., Elson, P., Garcia, J., & Dreicer, R. (2012). Differential immunologic and microRNA effects of 2 dosing regimens of recombinant human granulocyte/macrophage colony stimulating factor. Journal of Immunotherapy, 35, 587–594.
Khanna, S., Graef, S., Mussai, F., Thomas, A., Wali, N., Yenidunya, B. G., Yuan, C., Morrow, B., Zhang, J., Korangy, F., Greten, T. F., Steinberg, S. M., Stetler-Stevenson, M., Middleton, G., de Santo, C., & Hassan, R. (2018). Tumor-derived GM-CSF promotes granulocyte immunosuppression in mesothelioma patients. Clinical Cancer Research, 24, 2859–2872.
Schneble, E. J., Berry, J. S., Trappey, F. A., Clifton, G. T., Ponniah, S., Mittendorf, E., & Peoples, G. E. (2014). The HER2 peptide nelipepimut-S (E75) vaccine (NeuVax™) in breast cancer patients at risk for recurrence: correlation of immunologic data with clinical response. Immunotherapy, 6, 519–531.
Chiappori, A. A., Soliman, H., Janssen, W. E., Antonia, S. J., & Gabrilovich, D. I. (2010). INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert Opinion on Biological Therapy, 10, 983–991.
Antonia, S. J., et al. (2006). Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clinical Cancer Research, 12, 878–887.
Hardwick, N. R., Frankel, P., Ruel, C., Kilpatrick, J., Tsai, W., Kos, F., Kaltcheva, T., Leong, L., Morgan, R., Chung, V., Tinsley, R., Eng, M., Wilczynski, S., Ellenhorn, J. D. I., Diamond, D. J., & Cristea, M. (2018). p53-reactive T cells are associated with clinical benefit in patients with platinum-resistant epithelial ovarian cancer after treatment with a p53 vaccine and gemcitabine chemotherapy. Clinical Cancer Research, 24, 1315–1325.
Funding
This work was supported, in whole or in part, by National Institutes of Health Grants for Specialized Programs of Research Excellence, Grant P50 CA127297, and by the Fred & Pamela Buffett Cancer Center Support Grant P30 CA036727.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
All authors agree with submission to Cancer and Metastasis Reviews.
Code availability
Not applicable
Informed consent
Not applicable
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cole, K., Pravoverov, K. & Talmadge, J.E. Role of myeloid-derived suppressor cells in metastasis. Cancer Metastasis Rev 40, 391–411 (2021). https://doi.org/10.1007/s10555-020-09947-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-020-09947-x