
Abstract
Several pieces of evidence support the role of activated platelets in the development of the chronic inflammation-related diseases, such as atherothrombosis and cancer, mainly via the release of soluble factors and microparticles (MPs). Platelets and MPs contain a repertoire of proteins and genetic material (i.e., mRNAs and microRNAs) which may be influenced by the clinical condition of the individuals. In fact, platelets are capable of up-taking proteins and genetic material during their lifespan. Moreover, the content of platelet-derived MPs can be delivered to other cells, including stromal, immune, epithelial, and cancer cells, to change their phenotype and functions, thus contributing to cancer promotion and its metastasization. Platelets and MPs can play an indirect role in the metastatic process by helping malignant cells to escape from immunological surveillance. Furthermore, platelets and their derived MPs represent a potential source for blood biomarker development in oncology. This review provides an updated overview of the roles played by platelets and MPs in cancer and metastasis formation. The possible analysis of platelet and MP molecular signatures for the detection of cancer and monitoring of anticancer treatments is discussed. Finally, the potential use of MPs as vectors for drug delivery systems to cancer cells is put forward.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Numerous pieces of evidence convincingly support the contribution of platelets in cancer development and progression. The first clue comes from the observation that the use of the antiplatelet agent low-dose aspirin is associated with reduced incidence and mortality for colorectal cancer (CRC) and other types of cancer [1, 2]. Importantly, aspirin was found to prevent the risk of distant metastasis [3,4,5]. These findings open the way to perform a large number of studies to clarify the possible mechanisms of aspirin action. Based on the preferential action on the platelet by aspirin when administered at low doses, once daily, it was hypothesized that the antiplatelet effect of the drug might play a central role in the protection against cancer [6,7,8,9]. This idea was the driving force to study in detail how platelets may contribute to tumorigenesis and metastasis. The results of these studies uncovered novel biological roles of platelets beyond hemostasis and thrombosis. Interestingly, it was also discovered that the assessment of platelet content (including RNAs and proteins) describes specific signatures that give information on cancer diagnosis and prognosis [10,11,12].
In this review, we report the clinical and experimental evidence sustaining the role of platelets and their released products (soluble mediators and vesicles) in the development of cancer, its malignant progression leading to tumor cell spreading to other distant organs. Finally, the possible evaluation of platelets and their microparticle (MP) molecular signatures for the detection of cancer and monitoring of anticancer treatments is discussed.
1 Clinical evidence of the role played by platelets in cancer
The evidence that daily low-dose aspirin may help combat cancer derives from the results of observational case-control studies and their meta-analysis [5, 13], meta-analysis from randomized controlled trials (RCTs) in subjects with sporadic colorectal adenomas [14], an RCT in Lynch syndrome patients with a post-trial follow-up [15, 16], and a meta-analysis of data from individual patients of 51 RCTs, in which aspirin was used for cardiovascular (CV) prevention [4]. The analysis of these studies is reported in excellent reviews that have been recently published [6, 7, 9, 17]. Thus, it is only briefly covered here.
The most convincing mechanism of aspirin action is related to its capacity to cause the irreversible inactivation of cyclooxygenase (COX)-1 and COX-2 by acetylating a specific serine residue located in the COX active site, at positions 529 and 516, respectively [18,19,20]. The data of aspirin pharmacology show that, when used at low doses (75–100 mg daily), it acts by a preferential inhibition of platelet COX-1 activity which translates into a virtually complete inhibition of platelet thromboxane (TX)A2 [17, 21, 22]. This lipid mediator acts as an amplifier of the response to primary platelet agonists, such as thrombin and collagen [23]. It contributes to the release of ADP from platelets, which is another pro-aggregatory stimulus for platelets by activating the receptor P2Y12 [23]. This receptor is the target of the class of antiplatelet agents which include clopidogrel [23, 24].
Similarly to the prevention of CV disease, the anticancer effects of aspirin seem to be maximal at low doses given once daily [9]. Thus, it has been hypothesized that CRC and atherothrombosis share a common mechanism of disease, i.e., platelet activation in response to epithelial (in tumorigenesis) and endothelial (in tumorigenesis and atherothrombosis) injury [8, 9].
The evidence for a chemopreventive effect of low-dose aspirin against CRC recently leads the U.S. Preventive Services Task Force “to recommend the use of the drug for primary prevention of CV disease and CRC in adults of 50–59 years old with a 10% or greater 10-year CV disease risk, who are not at increased risk for bleeding, have a life expectancy of at least 10 years, and are willing to take low-dose aspirin daily for at least 10 years” [25].
2 Platelet-related mechanisms of intestinal tumorigenesis
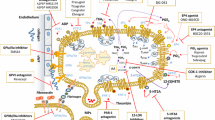
In the early stages of colorectal tumorigenesis, environmental and lifestyle factors, such as western style dietary habits and lack of physical activities, may contribute in activating platelets, as consequence of intestinal epithelial damage/dysfunction (Fig. 1a) [26]. Platelet activation is a physiological response aimed at the mucosal damage repair and, if uncontrolled, it may result in the activation of stromal and immune cells, leading to a chronic inflammatory response (Fig. 1a) [26,27,28].

Role of platelets in tumorigenesis and metastasis. a) In the early stages of colorectal tumorigenesis, platelet activation is a physiological response in the repair of intestinal mucosal damage. Activated platelets release mediators such as prostanoids [i.e., thromboxane (TX)A2, prostaglandin (PG)E2], ADP, growth and angiogenic factors, IL-1β and microvesicles [microparticles (MPs) and exosomes], which trigger an inflammatory response. Cellular components of stroma release different mediators and induce cyclooxygenase (COX)-2 expression in stromal cells. Activated stroma releases various mediators, including cytokines and growth factors. Enhanced levels of prostanoids (TXA2, PGE2) occur via the upregulation of COX-2. Altogether, these events contribute to the induction of epithelial-mesenchymal transition (EMT); low-dose aspirin may act upstream by affecting platelet COX-1 and, in turn, the cascade of these events; coxibs work downstream by inhibiting the activity of COX-2 in stromal cells and epithelial cells. b) During metastasis formation, activated platelets interact with circulating cancer cells and promote the aberrant expression of COX-2 and upregulation of mesenchymal markers, such as vimentin, Twist1, and SNAIL, coupled with the downregulation of E-cadherin. Mesenchymal tumor cells are characterized by an increased capacity to extravasate and to colonize distant organs. AA, arachidonic acid
In this scenario, the persistent activation of platelets and stromal cells promotes, in intestinal epithelial cells, a process known as epithelial-mesenchymal transition (EMT) [29], characterized by the reversible de-differentiation of epithelial cells into mesenchymal-like cells [30]. This process contributes to the generation of tumor-initiating cells [31] (Fig. 1a) and promotes migration and invasiveness of cancer cells [32, 33]. Interestingly, it has been reported that an inflammatory response plays a crucial role in EMT and the formation of tumor-initiating cells [34]. Furthermore, phenotypic changes induced in epithelial cells contribute to enhancing their tumorigenicity [35]. These changes include the induction of aberrant expression of COX-2 in epithelial cells [35], which is classically considered a crucial event for the development and progression of different types of cancer [36]. Several lines of experimental and clinical evidence support the role of COX-2 in cancer due to the numerous biological responses induced by its principal product prostaglandin (PG)E2 [36, 37]. It is noteworthy that the use of selective COX-2 inhibitors, named coxibs, such as celecoxib and rofecoxib, is associated with the risk reduction of sporadic colorectal adenoma recurrence [38,39,40]. However, the administration of these drugs as chemopreventive agents is discouraged due to the enhanced risk of adverse CV effects associated with their inhibition of vascular COX-2-dependent prostacyclin (PGI2) biosynthesis [41]. It has been reported that COX-2 functionally regulates EGFR, a transmembrane receptor tyrosine kinase of the ErbB family, involved in the CRC etiology [42]. Moreover, COX-2 overexpression in epithelial cells is associated with (i) enhanced capacity to interact with extracellular matrix components, (ii) reduced expression of TGF-βII receptor and E-cadherin, (iii) increased resistance to apoptosis, and (iv) increased expression of anti-apoptotic protein BCL-2 [43].
The contribution of platelets to the early events of colorectal carcinogenesis is summarized in Fig. 1a. A model has been proposed where platelets, activated at sites of the intestinal mucosa damage, release several soluble mediators, including PGE2, TXA2, cytokines and chemokines, growth and angiogenic factors, and stromal cell-derived factor (SDF)-1α, together with microvesicles (MV) containing genetic material [mRNAs and microRNAs (miRNAs)]. These events lead to the activation of stromal cells that will contribute to the amplification of an inflammatory response by the further release of several mediators, including COX-2-dependent PGE2 biosynthesis (Fig. 1a). Altogether, these events culminate in the transformation of normal epithelial cells into a hyperproliferative phenotype and the formation of aberrant crypt foci [44]. In this context, the aberrant expression of epithelial COX-2 plays an essential role in the development of an adenoma and its progression to adenocarcinoma [36, 44]. In fact, COX-2-dependent PGE2 produced in epithelial cells translates into the inhibition of apoptosis which may contribute to the accumulation of mutations (such as APC, KRAS, and p53) [36].
This model explains the comparable efficacy of low-dose aspirin and coxibs, found in RCTs, to reduce the relative risk of the recurrence of adenomatous polyps [45, 46]. In fact, low-dose aspirin may act upstream by affecting platelet COX-1 and, in turn, the cascade of events triggered by activated platelets, including the overexpression of COX-2, in the stromal and epithelial cells. Differently, coxibs act downstream by inhibiting the activity of COX-2 in stromal cells and epithelial cells (Fig. 1a).
3 Platelet-related mechanisms of metastasis formation
The role of platelets in the metastatic dissemination of cancer cells is supported by the reduced cancer incidence and mortality associated with the interference of metastasis formation by aspirin [3,4,5]. Moreover, numerous clinical observations support a complex relationship between platelet activation, the coagulation system, and dissemination of tumor cells via the bloodstream [47]. The crosstalk between cancer cells and platelets promotes metastasis formation through well-known mechanisms [47]. They include the capacity of platelet aggregates to encircle tumor cells, thus providing protection from immune elimination and improving their survival and the adhesion to the endothelium allowing the arrest of tumor cells and their extravasation [47] (Fig. 1b). Recently, it has been shown that platelets cause the induction of EMT in cancer cells through to the release of several mediators, such as PDGF, TGF-β, and PGE2 [32, 33, 35]. First, cancer cells activate platelets through a direct interaction involving the participation of different molecular partners (Table 1). Then, platelet-derived products induce the acquisition of a mesenchymal phenotype in cancer cells (as demonstrated by the expression of mesenchymal markers, including vimentin, fibronectin, the transcription factors Twist1, Snail, and Zeb) and the downregulation of epithelial marker expression, such as E-cadherin [32, 33, 35] (Fig. 1b). The release of TGF-β1 and the activation of TGF-β/Smad signaling has been identified as one of the mechanisms involved in the capacity of platelets to induce EMT in murine colon and breast cancer cells, promoting their capacity to colonize distant organs [32]. Platelets also induce COX-2 expression in the human adenocarcinoma cell line HT-29 associated with enhanced PGE2 production which affects the expression of proteins involved in cell cycle progression (such as the downregulation of p21(waf1/cip1 and the upregulation of cyclin B1) [35]. The overexpression of COX-2 requires direct contact between cancer cells and platelets through the interaction of galectin (Gal)-3 (expressed in cancer cells) and the collagen receptor glycoprotein (GP)VI (expressed in platelets) [35] (Table 1). Moreover, the release of PDGF from platelets participated in enhanced COX-2 levels in cancer cells via the post-transcriptional regulation of COX-2 expression mediated by the translocation of Hur, a known stability protein for COX-2 mRNA [35]. The platelet-dependent induction of COX-2 in HT-29 cells was affected by Gal-3 functional blockers and revacept [35]. Revacept is a novel antiplatelet agent in clinical development, which prevents the binding of platelet collagen receptors (including GPVI) at sites of vascular lesions [59]. In fact, revacept is a dimeric Fc fusion protein with the IgG part and the extracellular domain of the human GPVI [59]. Gal 3, a member of a family of carbohydrate-binding proteins, which uniquely consists of a C-terminal carbohydrate recognition domain(CRD), a collagen-like internal R-domain, and a N-terminal domain, is highly elevated in malignancies including colon cancer [60]. It is localized inside the cells but also on the cell surface where it mediates cell-cell and cell-matrix interactions by binding to glycoconjugates that contain β-galactosides via the CRD. It was hypothesized that platelet collagen receptors interact with cancer cells Gal 3 via its “collagen-like” domain and this phenomenon is affected by revacept. In fact, the induction of COX-2 in HT-29 cells co-cultured with platelets was prevented by revacept [35]. Altogether, these results suggest that compounds targeting collagen binding sites, such as revacept [59], and Gal-3 inhibitors might prevent the development of colon cancer metastasis.
Guillem-Llobat et al. found that platelet-derived PGE2 and a direct platelet-tumor cell interaction synergize to promote EMT and migration through the induction of Twist1 [33]. Twist1 mediates the downregulation of E-cadherin and the upregulation of RAC1, a Rho GTPase involved in cancer cell motility control [61]. These events led to the enhanced migratory capacity of HT-29 cells and the acquisition of a pro-aggregatory phenotype for platelets [33]. Platelet-induced EMT and migration in vitro were prevented by the inhibition of platelet function by aspirin, an inhibitor of COX-1, and other antiplatelet agents (including ticagrelor an antagonist of P2Y12 or DG-041, an antagonist of the PGE2 receptor EP3). The mesenchymal-like phenotype of HT-29 cells, induced by the interaction with platelets in vitro, were characterized by an enhanced metastatic potential when injected into the circulation of immunodeficient mice (Fig. 1b) and these properties were prevented by the inhibition of platelet function by aspirin. Interestingly, platelet-induced EMT in cancer cells was associated with an enhanced prothrombotic phenotype and, when these cells were injected in mice, induced an increase in the systemic biosynthesis of PGE2 and TXA2. These responses were prevented by the administration of low-dose aspirin [33]. Overall, these results showed that the antiplatelet agent low-dose aspirin may prevent metastasis formation due to its capacity to control the stem cell mimicry of cancer cells and also their pro-aggregatory effects on platelets.
It is well-known that tumor cells can re-arrange their genetic heritage in response to several molecules that populate tumor microenvironment. In particular, through the expression of megakaryocytic genes, tumor cells can activate platelets and promote coagulation, a phenomenon described as “platelet mimicry” [62]. Moreover, some tumor cells can mimic endothelial cell function, thus contributing to neovascularization (“vasculogenic mimicry”) [63]. These processes are associated with the acquisition of novel phenotypic features by cancer cells, as consequences of their direct crosstalk with stromal cells and platelets and/or the release of specific mediators occurring during cell activation.
4 Platelets and cancer-related inflammation
As reported above, platelets play an important role in the events linking tissue damage/dysfunction and the inflammatory response, because they are crucial players involved in the tissue damage repair [27]. However, when their activation is not adequately controlled, they contribute to the development of the chronic inflammatory process associated with the development of various disease states, including atherothrombosis [27], cancer, and the appearance of metastases in organs distant from the primary tumor [26, 28]. In this scenario, platelets act via the release of numerous mediators and MVs.
Platelets may trigger the induction of the pro-inflammatory COX-2-dependent pathway in immune, endothelial cells and fibroblasts. In monocytes, activated platelets induce COX-2 through transcriptional and post-transcriptional mechanisms [64]. This effect may occur through the interaction of P-selectin (expressed on platelet surface) and P-selectin glycoprotein ligand-1 (PSGL-1) on the monocytes. Moreover, activated platelets induce COX-2 expression in adherent monocytes through the release of soluble factors, in particular TGF-β1, and the activation of p38 MAPK signaling [65].
The overexpression of pro-inflammatory COX-2 in activated stromal cells promotes the release of several growth and inflammatory mediators which are involved in the phenotypic changes of epithelial cells necessary for invasion and metastasis (Fig. 1). The activation of stromal cells (including fibroblasts, myofibroblasts, pericytes, and different inflammatory cells) has been shown to be associated with genetic and epigenetic changes of the adjacent epithelial cells [66] (Fig. 1a). In human colonic fibroblasts, the induction of COX-2 expression by IL-1β promotes cell proliferation and invasion [67]. In endothelial cells, the interaction with activated platelets causes COX-2 overexpression, which is associated to the increased biosynthesis of the vasodilator PGI2, through a mechanism which depends on platelet-derived TXA2 and involves the selective activation of the p44/42 MAPK pathway [68].
Recently, Servais and collaborators studied the role of platelet and platelet-derived proteins in inflammation-associated colorectal tumorigenesis [69]. Using a mouse model of colitis-associated cancer, the authors found that during the early inflammatory stage, platelet activation occurred by the release of protein mediators [including the protumoral serum amyloid A (SAA)] which promoted the recruitment and expansion of myeloid-derived suppressor cells (MDSCs) [69]. The inhibition of platelet function by the administration of the P2Y12 receptor antagonist, clopidogrel, prevented the elevation of plasmatic SAA, decreased colitis severity, and delayed the formation of dysplastic lesions and adenocarcinoma [69]. Also, clopidogrel inhibited the expansion and function of MDSCs and their infiltration into tumors, thus restoring the antitumor immunity response [69]. These results support a new role of platelets as modulators of immunosuppressive responses both in early tumor development and metastasis.
5 Platelets, immunity, and cancer
Within the bloodstream, the survival of circulating tumor cells before extravasation is crucial for the development of metastasis. Here, the recruitment of cells, such as monocytes, neutrophils, and also platelets, protects tumor cells from surveillance by killer cells, thus shattering the effects of immunotherapy and enabling metastasis formation [70]. This is a well-known, but not unique, mechanism through which platelets blunt the immune response to cancer. In fact, platelets exert immunological functions via their crosstalk with other players of the innate and adaptive immunity, including the T cells. Some evidence suggests that they participate in the initiation of acquired immune response (via an MHC class I-dependent manner) [71]. Interestingly, it has recently reported that platelets from patients with nonsmall cell lung cancer (NSCLC) cells express programmed cell death ligand (PDL-1) whose ligation to PD-1 induces T cell exhaustion [72]. Some tumors show high levels of PDL-1, thus leading to their immune escape [73], and inhibition of PDL-1/PD-1 interaction with blocking antibodies has been associated to long-term responses in patients with metastatic NSCLC [74]. Moreover, recent findings have shown a platelet-dependent mechanism for TGF-β activation which may contribute to an immunosuppressive effect. This occurred through the cell surface TGF-β-docking receptor glycoprotein A repetitions predominant (GARP) [75]. These findings may open the way to the use of antiplatelet agents to interfere with the capacity of cancer cells to escape the immuno-surveillance.
Recently, Sitia et al. unrevealed a central role of platelets in the immune pathogenesis of hepatocellular carcinoma (HCC), a life-threatening complication of chronic HBV infection [76]. HBV chronic infection is characterized by an inefficient response of HBV-specific CD8+ T cells, which are not able to eradicate the infection, thus promoting immune-mediated hepatocellular necrosis, regeneration, inflammation, and then HCC development [77]. Interestingly, the administration of two antiplatelet drugs, aspirin, and clopidogrel, was associated with a significant reduction of the number of HBV-specific CD8+ T cells and secondary nonspecific infiltrate, thus preventing liver injury and fibrosis and the development of HCC [76]. The mechanism by which platelets, through the interaction with T cells, promote their local accumulation and all the pathologic events in the setting of HBV remains to be clarified. One of the proposed mechanisms involves platelet CD154 (CD40 ligand) which may sustain lymphocyte function [78] and trigger an inflammatory response in endothelial cells [79]. These findings suggest the possible use of antiplatelet agents in the prevention of HCC. Moreover, recent evidence has shown that liver disease may be associated with increased thrombotic events [80], thus justifying the use of antiplatelet drugs in further RCTs designed to address their effects on the development of HCC in patients with chronic HBV infection.
The growing appreciation of the contribution of platelets in immunity has led to study their role in response to immune complexes (ICs) formed by antibodies and their antigens. ICs circulate in blood contributing to chronic and acute inflammation in several pathological conditions [81]. Cloutier and colleagues have recently found that, in mice, circulating ICs can induce a systemic shock through the activation of the platelet receptor Fcγ receptor IIA (FcγRIIA). This response was mediated by the release of serotonin from platelet granules. An interesting finding of this work is that, during shock, platelets were sequestered in the lungs and brain and, after degranulation, they re-circulate, thus suggesting that in adaptive immunity, which involves antibody responses, activated platelets have a longer lifespan [81]. This may provide a possible explanation for the altered platelet count in IC-related diseases.
6 Platelets as a source of biomarkers for cancer detection
The development of non-invasive biomarkers for the early detection of cancer is an important need to reduce cancer deaths. Recently, Coehen and colleagues developed a novel blood test (named CancerSEEK) that can detect eight common cancer types (ovarian, liver, stomach, pancreas, oesophageal, colorectal, lung, and breast cancers) [82]. CancerSEEK assesses circulating levels of eight known proteins and 16 genes (frequently mutated in different types of cancer) in cell-free DNA [82]. This test was used in more than 1000 patients, and it gave a positive result in approximately 70% of the eight cancer types. This test allows to identify the molecular “signature” of the tumors with a specificity > 99% and only 7 of 812 healthy controls were recorded as false-positive [82]. However, it is important to underline that about 80% of the cancers detected were at advanced stages (II or III stage) [82]. The detection rate was reduced in patients at earliest stages [82], thus suggesting that it could not be useful for the detection of the initial symptomatic stage of the disease or its pre-symptomatic phase.
The capacity of platelets to uptake and store in their granules different types of molecules from the environment (including bloodstream) (a process known as “platelet education”) makes these cells able to acquire distinct “signatures” which may reflect disease states. In cancer, this phenomenon may provide the opportunity to interrogate the signature of the “tumor-educated platelets” as a biomarker reflecting the evolution of the disease [10, 83]. In fact, platelets of cancer patients can be characterized by a different pattern of proteins, nucleic acids (including mRNAs and miRNAs). Thus, the assessment of their proteome or RNA profiles is considered a promising approach for non-invasive detection of cancer [10, 11, 82]. In fact, Best et al. have recently demonstrated that the assessment of tumor-educated platelet (TEP) RNA profiles is a reliable blood-based cancer diagnostic approach to identify patients with NSCLC also at the early stage of the disease [11]. In particular, through the use of particle-swarm optimization algorithms and RNA-seq of TEPs, they identified a panel of RNA biomarkers to discriminate NSCLC patients, healthy subjects, and, importantly, also individuals without cancer but with several inflammatory conditions not related to cancer. This approach also allowed identifying panels of RNA biomarkers for the classification of lung cancer [11].
7 Platelet microparticles as mediators of the intercellular communication
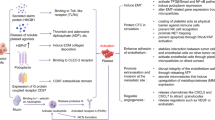
Upon activation, eukaryotic cells shed components from their cytoplasm through the release of tiny MVs. These include MPs (0.1–1 μm) of plasma membrane origin and exosomes (30–100 nm) of endosomal origin. The highest number of circulating MPs (70–90%) is released from platelets [84]. MPs are characterized by the inversion of membrane phospholipids and by the exposure on the outer membrane of phosphatidylserine (PS). They express integrins such as glycoprotein IIb/IIIa (CD41/CD61), Ib, IaIIa, P-selectin, and enzymes that include matrix metalloproteinases [85, 86]. Based on their ability to transfer part of their content to target cells, platelet-derived MPs play a pivotal role in the crosstalk between platelets and other cells [86] (Fig. 2).

Role of platelet-derived MPs in cell-cell communication. MPs released from activated platelets interact with other cells, such as monocytes, neutrophils, endothelial cells, tumor cells, and dendritic cells and promote phenotypic changes and novel functions in recipient cells through the delivery of several factors, including bioactive lipids, proteins, transcripts, and miRNAs. EMT, epithelial-mesenchymal transition; AA, arachidonic acid; S1P, sphingosine 1-phosphate; PAR-1, protease activated receptor 1; FBXW7, F-box/WD repeat-containing protein 7; EFNA1, glycosylphosphatidyl inositol-anchored receptor tyrosine kinase ligand; gamma-globin transcripts, HBG1/HBG2; HBA2, hemoglobin subunit alpha-2; HBA1, hemoglobin subunit alpha-1; 12-HETE, 12(S)-hydroxyeicosatetranoic acid; sPLA2IIA, secretory phospholipase A2-IIa. +, increase; − reduction
MPs may influence the biology of the target cells mainly through the following mechanisms: (i) the stimulation of target cells by forming “signaling complexes” with specific surface MP-expressed ligands, such as growth factors and bioactive lipids; (ii) the intercellular transfer of surface receptors, such as adhesion molecules, membrane receptors; (iii) delivery of proteins and bioactive lipids into the target cells and transfer of mRNA, transcription factors, which may cause the epigenetic reprogramming of the cells [87] (Fig. 2). Moreover, MPs may also act as vectors for the delivery of infectious particles (such as HIV, prions), and finally, they may be able to deliver to the cells intact organelles (such as mitochondria) [87].
Platelet-derived MPs have a surface that is 50 to 100 times more pro-coagulant than that of activated platelets. Several pieces of evidence suggest that platelet MPs are implicated in the transport and delivery of bioactive molecules, thus participating in hemostasis and thrombosis, but also in inflammation, tumorigenesis, angiogenesis, and immunity [86].
Platelet MPs can transfer membrane receptors expressed on their surface (i.e., CD41, CD61, CD62, CXCR4, and PAR-1) to normal and cancer cells, inducing their adhesion, for example to fibrinogen, proliferation, and survival [88] (Fig. 2). In addition to proteins, MPs are rich in bioactive lipids, such as sphingosine 1-phosphate (S1P) and arachidonic acid (AA). It has been reported that AA can be transferred from platelet MPs to monocytes and endothelial cells, thus causing the induction of COX-2 and prostanoid production [89, 90] (Fig. 2). MPs are rich in genetic material, such as mRNA and miRNA [86]. In fact, Risitano and colleagues reported that platelet MPs incubated with monocytic THP-1 cells and endothelial cells led to the transfer of mRNA molecules [91]. The microarray gene expression analysis of monocytic THP-1 cells showed an increase in the gamma-globin transcripts HBG1/HBG2 and the hemoglobin subunit alpha-2 (HBA2) and alpha-1 (HBA1) mRNAs that were directly related to the transfer [91] (Fig. 2). Also, platelet MPs contain a plethora of miRNAs which can regulate gene expression of cancer cells [92]. The work of Laffont and collaborators reported platelet-derived MPs transferred the miR-223 in endothelial cells and that the complex Ago2-miR-223 downregulated the expression of two genes: the FBXW7, an oncosuppressor protein, and EFNA1, a glycosylphosphatidyl inositol-anchored receptor tyrosine kinase ligand [93]. The transfer of miRNAs by platelet MPs to target cells has been reported to promote tumor progression. In fact, in ovarian cancer cells, platelet MPs delivered the miR-939 which induced the downregulation of E-cadherin and the upregulation of vimentin, a critical molecular events of EMT [94]. In this study, the authors found a key role of sPLA2-IIA in the promotion of platelet MP internalization by ovarian cancer cells [94]. Differently, Michael et al. reported that platelet MPs infiltrate solid tumors both in humans and mice and deliver miRNAs, especially miR-24, to tumor cells in vivo and in vitro inducing cell apoptosis and suppressing tumor growth [95] (Fig. 2).
In the inflammatory milieu, the release of platelet-derived MPs can either promote pro-inflammatory actions of macrophages [96,97,98] or inhibit their secretion of pro-inflammatory cytokines [96, 99, 100] (Fig. 2). The effects of platelets and platelet-derived MPs on monocyte/macrophage functions are mediated by several mechanisms which are not entirely clarified. They include the direct cellular interaction, the action of adhesion molecules, such as P-selectin, and, at least in part, the involvement of the release of platelet-derived mediators, such as platelet factor 4 (PF4) [96]. Serotonin (5-hydroxytryptamine) is another platelet-derived factor which may affect monocyte activity by promoting NF-κB activation, the enhancement of LPS-induced cytokine production, and also by reducing apoptosis, possibly through the alteration of Bcl-2 or Mcl-1 expression [101].
Platelet MPs can modulate the phenotype and the functions of neutrophils in an inflammatory milieu. Duchez and collaborators demonstrated that platelet MPs are internalized by activated neutrophils in inflammatory condition like arthritis, both in humans and in animal models [102].
The lipidomic analysis of these MPs revealed that the most abundant eicosanoid was the 12(S)-hydroxyeicosatetranoic acid [12(S)-HETE] which, in concert with sPLA2-IIA, promoted the MP internalization by neutrophils [102]. Also, MPs by transferring their cargo modified the levels of transcripts in neutrophils, thus potentially modulating the biological processes and functions of these target cells [102] (Fig. 2).
Platelet-derived MPs are implicated in the modulation of adaptive immunity through the transfer of CD145 to B cells, thus stimulating antigen-specific IgG production and modulating germinal center formation through the cooperative activity with CD4 positive T cells [103].
Shedding of platelet-derived MPs occurs not only upon activation but also upon aging by an apoptosis-like process (apoptosis-induced platelet MPs) [98]. It has been shown that these MPs are chemotactic for monocytes, able to bind these cells and promote their polarization into resident M2 monocytes, thus changing their behavior and activation state towards resident professional phagocytes, as demonstrated by the expression of the (ox)LDL receptors, CD36 and CD68, and the production of pro-inflammatory and immunomodulating cytokines by monocytes [98] (Fig. 2). Sadallah et al. have demonstrated that MPs shed by human platelets are able to modify the monocyte differentiation towards immature dendritic cells (iDC) and their subsequent maturation to DC, affecting the normal immune response through the reduction of their potential for inflammation and Ag presentation, because they express PS, known for its pro-coagulant function [99] (Fig. 2), but also for its immunosuppressive activity when expressed by apoptotic cells [99].
8 Microparticles as biomarkers of cancer disease
The capacity of platelets to be “educated” by cancer cells, thus acquiring a specific repertoire of proteins, mRNAs, and miRNAs related to the disease state of the individual, supports the idea that circulating platelet-derived MPs could be used as a biomarker for cancer diagnosis. Recently, several studies were performed to characterize the cellular origin and the heterogeneous cargo of circulating MPs for the development of disease-associated biomarkers. Kim et al. evaluated the number of circulating platelet MPs in patients with gastric cancer at different stages and demonstrated that the plasma levels of platelet MPs, together with VEGF, IL-6, and RANTES, were significantly increased in patients with stage IV disease, thus suggesting that the evaluation of platelet MPs in plasma might be useful for identifying patients with metastatic gastric cancer [104]. Mege and colleagues characterized the specific signature of circulating MPs in patients with CRC and pancreatic cancer in comparison to those with inflammatory bowel or pancreatic diseases and healthy subjects [105]. It has been found that platelet-derived MPs have a distinct signature in cancer patients depending on the progression of the disease and can be used as a complex biomarker reflecting the evolution of the disease [105]. Additional evidence on platelet MPs as a prognostic biomarker of cancer development derived from a recent study performed in NSCLC patients [106]. In these patients, the levels of four types of circulating MPs (mainly originated from endothelial cells and platelets) were higher in cancer patients than in control subjects, decreased after the therapy, and, interestingly, they predicted the 1-year prognostic clinical outcome in the advanced stage NSCLC patients [106]. These data suggested that the repetitive measurement of circulating platelet MPs can predict therapeutic response and prognostic outcomes in advanced lung cancer patients [106].
9 Conclusive remarks and future perspectives
Platelets promote tumor-related chronic inflammation and immune responses, tumor progression, and metastasis through complex and bidirectional communication with stromal, immune, and cancer cells [28, 35]. This cell-to-cell communication classically involves (i) the release of soluble factors [i.e., growth factors, cytokines, chemokines and bioactive lipids and genetic material (mRNA and miRNAs)] and (ii) intercellular adhesion contacts, which are mediated by sets of specific molecules [28, 35] (Table 1). A growing interest is now focused on the mechanisms of platelet communication with other cells through the release of MPs. In fact, a substantial percentage of the platelet-released factors or molecules, such as mRNA or miRNA, are associated with circulating MPs [86].
Recently, the scientists have made significant advances towards developing blood tests for cancer. However, further research has to be carried out to validate them and to improve their sensitivity and specificity. Since platelets actively endocytose plasma constituents, their granule contents may reflect a specific disease state. Thus, in the contest of cancer disease, the assessment of the molecular signature of “tumor-educated platelets” and/or circulating MPs may provide a complex of biomarkers potentially reflecting the evolution of the disease status. Moreover, novel knowledge of platelet MP-mediated intercellular crosstalk in the tumor microenvironment may open the way to the development of novel platelet-related therapies for the treatment of inflammation-dependent diseases, such as atherothrombosis and cancer.
Recent evidence supports the use of MVs to carry a wide variety of molecules, including macromolecules, such as DNA, RNA, and proteins, and small molecules, for example, doxorubicin, curcumin, or paclitaxel [107]. In fact, successful preclinical studies show that the use of autologous MVs derived from dendritic cells (Dex) pulsed with antigenic tumor peptides for the treatment of melanoma and NSCLC, respectively [108], is feasible and safe. Moreover, MVs can be engineered to deliver suicide gene mRNAs and proteins to target tissue, thus establishing a new strategy for cancer treatment [109]. The improvement of the knowledge on the biology of MPs and the mechanisms underlying their role as mediators of the intercellular communication will lead to the development of specific techniques for MP-mediated drug delivery to cancer cells. Further research is necessary to translate this knowledge into novel therapeutic approaches to fight cancer and metastasis. However, the rapid increase in the knowledge in this field represents a guarantee for success to improve cancer therapy, its prediction, and prognosis.
References
Flossmann, E., Rothwell, P. M., & for the British Doctors Aspirin Trial and the UK-TIA Aspirin Trial. (2007). Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet, 369(9573), 1603–1613.
Rothwell, P. M., Price, J. F., Fowkes, F. G., Zanchetti, A., Roncaglioni, M. C., Tognoni, G., Lee, R., Belch, J. F., Wilson, M., Mehta, Z., & Meade, T. W. (2011). Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet, 377(9759), 31–41.
Rothwell, P. M., Wilson, M., Price, J. F., Belch, J. F., Meade, T. W., & Mehta, Z. (2012). Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet, 379(9826), 1591–1601.
Rothwell, P. M., Price, J. F., Fowkes, F. G., Zanchetti, A., Roncaglioni, M. C., Tognoni, G., Lee, R., Belch, J. F., Wilson, M., Mehta, Z., & Meade, T. W. (2012). Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet, 379(9826), 1602–1612.
Algra, A. M., & Rothwell, P. M. (2012). Effects of regular aspirin on long-term cancer incidence and metastasis: a systemic comparison of evidence from observational studies versus randomised trials. The Lancet Oncology, 13(5), 518–527.
Thun, M. J., Jacobs, E. J., & Patrono, C. (2012). The role of aspirin in cancer prevention. Nature Reviews. Clinical Oncology, 9(5), 259–267.
Dovizio, M., Bruno, A., Tacconelli, S., & Patrignani, P. (2013). Mode of action of aspirin as a chemopreventive agent. Recent Results in Cancer Research, 191, 39–65.
Patrono, C., Patrignani, P., & García Rodríguez, L. A. (2001). Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. The Journal of Clinical Investigation, 108(1), 7–13.
Patrignani, P., & Patrono, C. (2016). Aspirin and Cancer. Journal of the American College of Cardiology, 68(9), 967–976.
Best, M. G., Sol, N., Kooi, I., Tannous, J., Westerman, B. A., Rustenburg, F., Schellen, P., Verschueren, H., Post, E., Koster, J., Ylstra, B., Ameziane, N., Dorsman, J., Smit, E. F., Verheul, H. M., Noske, D. P., Reijneveld, J. C., Nilsson, R. J. A., Tannous, B. A., Wesseling, P., & Wurdinger, T. (2015). RNA-Seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell, 28(5), 666–676.
Best MG, Sol N, In 't Veld SGJG, Vancura A, Muller M, Niemeijer AN, et al. (2017) Swarm intelligence-enhanced detection of non-small-cell lung cancer using tumor-educated platelets. Cancer Cell. 32(2):238–52.
Best, M. G., Vancura, A., & Wurdinger, T. (2017). Platelet RNA as a circulating biomarker trove for cancer diagnostics. Journal of Thrombosis and Haemostasis, 15(7), 1295–1306.
Kune, G. A., Kune, S., & Watson, L. F. (1988). Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Research, 48(15), 4399–4404.
Cole, B. F., Logan, R. F., Halabi, S., Benamouzig, R., Sandler, R. S., Grainge, M. J., Chaussade, S., & Baron, J. A. (2009). Aspirin for the chemoprevention of colorectal adenomas: meta-analysis of the randomized trials. Journal of the National Cancer Institute, 101(4), 256–266.
Burn, J., Bishop, D. T., Mecklin, J. P., Macrae, F., Möslein, G., Olschwang, S., Bisgaard, M. L., Ramesar, R., Eccles, D., Maher, E. R., Bertario, L., Jarvinen, H. J., Lindblom, A., Evans, D. G., Lubinski, J., Morrison, P. J., Ho, J. W., Vasen, H. F., Side, L., Thomas, H. J., Scott, R. J., Dunlop, M., Barker, G., Elliott, F., Jass, J. R., Fodde, R., Lynch, H. T., Mathers, J. C., & CAPP2 Investigators. (2008). Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. The New England Journal of Medicine, 359(24), 2567–2578.
Burn, J., Gerdes, A. M., Macrae, F., Mecklin, J. P., Moeslein, G., Olschwang, S., Eccles, D., Evans, D. G., Maher, E. R., Bertario, L., Bisgaard, M. L., Dunlop, M. G., Ho, J. W., Hodgson, S. V., Lindblom, A., Lubinski, J., Morrison, P. J., Murday, V., Ramesar, R., Side, L., Scott, R. J., Thomas, H. J., Vasen, H. F., Barker, G., Crawford, G., Elliott, F., Movahedi, M., Pylvanainen, K., Wijnen, J. T., Fodde, R., Lynch, H. T., Mathers, J. C., Bishop, D. T., & CAPP2 Investigators. (2011). Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet, 378(9809), 2081–2087.
Patrono, C. (2015). The multifaceted clinical readouts of platelet inhibition by low-dose aspirin. Journal of the American College of Cardiology, 66(1), 74–85.
Simmons, D. L., Botting, R. M., & Hla, T. (2004). Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacological Reviews, 56(3), 387–437.
Loll, P. J., Picot, D., & Garavito, R. M. (1995). The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nature Structural Biology, 2(8), 637–643.
Lecomte, M., Laneuville, O., Ji, C., DeWitt, D. L., & Smith, W. L. (1996). Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. The Journal of Biological Chemistry, 269(18), 13207–13215.
Patrignani, P., & Patrono, C. (2015). Cyclooxygenase inhibitors: from pharmacology to clinical read-outs. Biochimica et Biophysica Acta, 1851(4), 422–432.
Patrono, C., Baigent, C., Hirsh, J., & Roth, G. (2008). Antiplatelet drugs: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest, 133(6 Suppl), 199S–233S.
Davì, G., & Patrono, C. (2007). Platelet activation and atherothrombosis. The New England Journal of Medicine, 357(24), 2482–2494.
Patrono, C., Coller, B., FitzGerald, G. A., Hirsh, J., & Roth, G. (2004). Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest, 126(Suppl 3), 234S–264S.
Bibbins-Domingo, K., & Preventive Services Task Force, U. S. (2016). Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Annals of Internal Medicine, 164(12), 836–845.
Dovizio, M., Sacco, A., & Patrignani, P. (2017). Curbing tumorigenesis and malignant progression through the pharmacological control of the wound healing process. Vascular Pharmacology, 89, 1–11.
Gawaz, M., Langer, H., & May, A. E. (2005). Platelets in inflammation and atherogenesis. The Journal of Clinical Investigation, 115(12), 3378–3384.
Contursi, A., Sacco, A., Grande, R., Dovizio, M., & Patrignani, P. (2017). Platelets as crucial partners for tumor metastasis: from mechanistic aspects to pharmacological targeting. Cellular and Molecular Life Sciences, 74(19), 3491–3507.
Wang, H., Fang, R., Wang, X. F., Zhang, F., Chen, D. Y., Zhou, B., Wang, H. S., Cai, S. H., & Du, J. (2013). Stabilization of Snail through AKT/GSK-3β signaling pathway is required for TNF-α-induced epithelial-mesenchymal transition in prostate cancer PC3 cells. European Journal of Pharmacology, 714(1–3), 48–55.
Kalluri, R., & Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. The Journal of Clinical Investigation, 119(6), 1420–1428.
Mani, S. A., Guo, W., Liao, M. J., Eaton, E. N., Ayyanan, A., Zhou, A. Y., Brooks, M., Reinhard, F., Zhang, C. C., Shipitsin, M., Campbell, L. L., Polyak, K., Brisken, C., Yang, J., & Weinberg, R. A. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 133(4), 704–715.
Labelle, M., Begum, S., & Hynes, R. O. (2011). Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell, 20(5), 576–590.
Guillem-Llobat, P., Dovizio, M., Bruno, A., Ricciotti, E., Cufino, V., Sacco, A., Grande, R., Alberti, S., Arena, V., Cirillo, M., Patrono, C., FitzGerald, G., Steinhilber, D., Sgambato, A., & Patrignani, P. (2016). Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget, 7(22), 32462–32477.
Jing, Y., Han, Z., Zhang, S., Liu, Y., & Wei, L. (2011). Epithelial-mesenchymal transition in tumor microenvironment. Cell & Bioscience. https://doi.org/10.1186/2045-3701-1-29.
Dovizio, M., Maier, T. J., Alberti, S., Di Francesco, L., Marcantoni, E., Münch, G., John, C. M., Suess, B., Sgambato, A., Steinhilber, D., & Patrignani, P. (2013). Pharmacological inhibition of platelet-tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Molecular Pharmacology, 84(1), 25–40.
Dixon, D. A., Blanco, F. F., Bruno, A., & Patrignani, P. (2013). Mechanistic aspects of COX-2 expression in colorectal neoplasia. Recent Results in Cancer Research, 191, 7–37.
Zelenay, S., van der Veen, A. G., Böttcher, J. P., Snelgrove, K. J., Rogers, N., Acton, S. E., Chakravarty, P., Girotti, M. R., Marais, R., Quezada, S. A., Sahai, E., & Reis e Sousa, C. (2015). Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell, 162(6), 1257–1270.
Arber, N., Eagle, C. J., Spicak, J., Rácz, I., Dite, P., Hajer, J., Zavoral, M., Lechuga, M. J., Gerletti, P., Tang, J., Rosenstein, R. B., Macdonald, K., Bhadra, P., Fowler, R., Wittes, J., Zauber, A. G., Solomon, S. D., Levin, B., & PreSAP Trial Investigators. (2006). Celecoxib for the prevention of colorectal adenomatous polyps. The New England Journal of Medicine, 355(9), 885–895.
Bertagnolli, M. M., Eagle, C. J., Zauber, A. G., Redston, M., Solomon, S. D., Kim, K., Tang, J., Rosenstein, R. B., Wittes, J., Corle, D., Hess, T. M., Woloj, G. M., Boisserie, F., Anderson, W. F., Viner, J. L., Bagheri, D., Burn, J., Chung, D. C., Dewar, T., Foley, T. R., Hoffman, N., Macrae, F., Pruitt, R. E., Saltzman, J. R., Salzberg, B., Sylwestrowicz, T., Gordon, G. B., Hawk, E. T., & APC Study Investigators. (2006). Celecoxib for the prevention of sporadic colorectal adenomas. The New England Journal of Medicine, 355(9), 873–884.
Baron, J. A., Sandler, R. S., Bresalier, R. S., Quan, H., Riddell, R., Lanas, A., Bolognese, J. A., Oxenius, B., Horgan, K., Loftus, S., Morton, D. G., & APPROVe Trial Investigators. (2006). A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology, 131(6), 1674–1682.
Grosser, T., Fries, S., & FitzGerald, G. A. (2006). Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. The Journal of Clinical Investigation, 116, 4–15.
Li, H., Zhu, F., Boardman, L. A., Wang, L., Oi, N., Liu, K., Li, X., Fu, Y., Limburg, P. J., Bode, A. M., & Dong, Z. (2015). Aspirin prevents colorectal cancer by normalizing EGFR expression. EBioMedicine, 2(5), 447–455.
Tsujii, M., & DuBois, R. N. (1995). Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell, 83(2), 493–501.
Jänne, P. A., & Mayer, R. J. (2000). Chemoprevention of colorectal cancer. The New England Journal of Medicine, 342(26), 1960–1968.
Patrono, C., & Rocca, B. (2008). Aspirin: promise and resistance in the new millennium. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(3), s25–s32.
Dovizio, M., Tacconelli, S., Sostres, C., Ricciotti, E., & Patrignani, P. (2012). Mechanistic and pharmacological issues of aspirin as an anticancer agent. Pharmaceuticals (Basel, Switzerland), 5(12), 1346–1371.
Gay, L. J., & Felding-Habermann, B. (2011). Contribution of platelets to tumour metastasis. Nature Reviews. Cancer, 11(2), 123–134.
Felding-Habermann, B., Habermann, R., Saldívar, E., & Ruggeri, Z. M. (1996). Role of beta3 integrins in melanoma cell adhesion to activated platelets under flow. The Journal of Biological Chemistry, 271(10), 5892–5900.
Lonsdorf, A. S., Krämer, B. F., Fahrleitner, M., Schönberger, T., Gnerlich, S., Ring, S., Gehring, S., Schneider, S. W., Kruhlak, M. J., Meuth, S. G., Nieswandt, B., Gawaz, M., Enk, A. H., & Langer, H. F. (2012). Engagement of αIIbβ3 (GPIIb/IIIa) with ανβ3 integrin mediates interaction of melanoma cells with platelets: a connection to hematogenous metastasis. The Journal of Biological Chemistry, 287(3), 2168–2178.
Mitrugno, A., Williams, D., Kerrigan, S. W., & Moran, N. (2014). A novel and essential role for FcγRIIa in cancer cell-induced platelet activation. Blood, 123(2), 249–260.
Mammadova-Bach, E., Zigrino, P., Brucker, C., Bourdon, C., Freund, M., De Arcangelis, A., Abrams, S. I., Orend, G., Gachet, C., & Mangin, P. H. (2016). Platelet integrin α6β1 controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight, 1(14), e88245.
Mannori, G., Crottet, P., Cecconi, O., Hanasaki, K., Aruffo, A., Nelson, R. M., Varki, A., & Bevilacqua, M. P. (1995). Differential colon cancer cell adhesion to E-, P-, and L-selectin: role of mucin-type glycoproteins. Cancer Research, 55(19), 4425–4431.
Gong, L., Cai, Y., Zhou, X., & Yang, H. (2012). Activated platelets interact with lung cancer cells through P-selectin glycoprotein ligand-1. Pathology Oncology Research, 18(4), 989–996.
Alves, C. S., Burdick, M. M., Thomas, S. N., Pawar, P., & Konstantopoulos, K. (2008). The dual role of CD44 as a functional P-selectin ligand and fibrin receptor in colon carcinoma cell adhesion. American Journal of Physiology. Cell Physiology, 294(4), C907–C916.
Larrucea, S., Butta, N., Rodriguez, R. B., Alonso-Martin, S., Arias-Salgado, E. G., Ayuso, M. S., & Parrilla, R. (2007). Podocalyxin enhances the adherence of cells to platelets. Cellular and Molecular Life Sciences, 64(22), 2965–2974.
Boukerche, H., Berthier-Vergnes, O., Tabone, E., Doré, J. F., Leung, L. L., & McGregor, J. L. (1989). Platelet-melanoma cell interaction is mediated by the glycoprotein IIb-IIIa complex. Blood, 74(2), 658–663.
Suzuki-Inoue, K., Kato, Y., Inoue, O., Kaneko, M. K., Mishima, K., Yatomi, Y., Yamazaki, Y., Narimatsu, H., & Ozaki, Y. (2007). Involvement of the snake toxin receptor CLEC-2, in Podoplanin-mediated platelet activation, by cancer cells. The Journal of Biological Chemistry, 282(36), 25993–26001.
Chang, Y. W., Hsieh, P. W., Chang, Y. T., Lu, M. H., Huang, T. F., Chong, K. Y., Liao, H. R., Cheng, J. C., & Tseng, C. P. (2015). Identification of a novel platelet antagonist that binds to CLEC-2 and suppresses podoplanin-induced platelet aggregation and cancer metastasis. Oncotarget, 6(40), 42733–42748.
Ungerer, M., Rosport, K., Bültmann, A., Piechatzek, R., Uhland, K., Schlieper, P., Gawaz, M., & Münch, G. (2011). Novel antiplatelet drug revacept (dimeric glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation, 123(17), 1891–1899.
Yang, R. Y., Rabinovich, G. A., & Liu, F. T. (2008). Galectins: structure, function and therapeutic potential. Expert Reviews in Molecular Medicine, 10, e17. https://doi.org/10.1017/S1462399408000719.
Yang, W. H., Lan, H. Y., Huang, C. H., Tai, S. K., Tzeng, C. H., Kao, S. Y., Wu, K. J., Hung, M. C., & Yang, M. H. (2012). RAC1 activation mediates Twist1-induced cancer cell migration. Nature Cell Biology, 14(4), 366–374.
Tímár, J., Tóvári, J., Rásó, E., Mészáros, L., Bereczky, B., & Lapis, K. (2005). Platelet-mimicry of cancer cells: epiphenomenon with clinical significance. Oncology, 69(3), 185–201.
Qiao, L., Liang, N., Zhang, J., Xie, J., Liu, F., Xu, D., Yu, X., & Tian, Y. (2015). Advanced research on vasculogenic mimicry in cancer. Journal of Cellular and Molecular Medicine, 19(2), 315–326.
Dixon, D. A., Tolley, N. D., Bemis-Standoli, K., Martinez, M. L., Weyrich, A. S., Morrow, J. D., Prescott, S. M., & Zimmerman, G. A. (2006). Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. The Journal of Clinical Investigation, 116(10), 2727–2738.
Eligini, S., Barbieri, S. S., Arenaz, I., Tremoli, E., & Colli, S. (2007). Paracrine up-regulation of monocyte cyclooxygenase-2 by platelets: role of transforming growth factor-beta1. Cardiovascular Research, 74(2), 270–278.
Achyut, B. R., Bader, D. A., Robles, A. I., Wangsa, D., Harris, C. C., Ried, T., & Yang, L. (2013). Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-beta signaling. PLoS Genetics, 9, e1003251. https://doi.org/10.1371/journal.pgen.1003251.
Zhu, Y., Zhu, M., & Lance, P. (2012). IL1β-mediated stromal COX-2 signaling mediates proliferation and invasiveness of colonic epithelial cancer cells. Experimental Cell Research, 318(19), 2520–2530.
Caughey, G. E., Cleland, L. G., Gamble, J. R., & James, M. J. (2001). Up-regulation of endothelial cyclooxygenase-2 and prostanoid synthesis by platelets. Role of thromboxane A2. The Journal of Biological Chemistry, 276, 37839–37845.
Servais, L., Wéra, O., Dibato Epoh, J., Delierneux, C., Bouznad, N., Rahmouni, S., Mazzucchelli, G., Baiwir, D., Delvenne, P., Lancellotti, P., & Oury, C. (2018). Platelets contribute to the initiation of colitis-associated cancer by promoting immunosuppression. Journal of Thrombosis and Haemostasis, 16(4), 762–777.
Kitamura, T., Qian, B. Z., & Pollard, J. W. (2015). Immune cell promotion of metastasis. Nature Reviews. Immunology, 15(2), 73–86.
Ali, R. A., Wuescher, L. M., & Worth, R. G. (2015). Platelets: essential components of the immune system. Current Trends in Immunology, 16, 65–78.
Hinterleitner, C., Strähle, J., Wirths, S., Bugl, S., Malenke, E., Mueller, M. R., Kanz, L., & Kopp, H.-G. (2017). Platelet programmed cell death ligand 1 (pPDL-1) is a prognostic marker in advanced lung cancer. Blood, 130, 3610.
Juneja, V. R., McGuire, K. A., Manguso, R. T., LaFleur, M. W., Collins, N., Haining, W. N., Freeman, G. J., & Sharpe, A. H. (2017). PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. The Journal of Experimental Medicine, 214(4), 895–904.
Dang, T. O., Ogunniyi, A., Barbee, M. S., & Drilon, A. (2016). Pembrolizumab for the treatment of PD-L1 positive advanced or metastatic non-small cell lung cancer. Expert Review of Anticancer Therapy, 16(1), 13–20.
Rachidi, S., Metelli, A., Riesenberg, B., Wu, B. X., Nelson, M. H., Wallace, C., Paulos, C. M., Rubinstein, M. P., Garrett-Mayer, E., Hennig, M., Bearden, D. W., Yang, Y., Liu, B., & Li, Z. (2017). Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Science Immunology, 2(11), eaai7911. https://doi.org/10.1126/sciimmunol.aai7911.
Sitia, G., Aiolfi, R., Di Lucia, P., Mainetti, M., Fiocchi, A., Mingozzi, F., Esposito, A., Ruggeri, Z. M., Chisari, F. V., Iannacone, M., & Guidotti, L. G. (2012). Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proceedings of the National Academy of Sciences of the United States of America, 109(32), E2165–E2172.
Guidotti, L. G., & Chisari, F. V. (2006). Immunobiology and pathogenesis of viral hepatitis. Annual Review of Pathology, 1, 23–61.
Elzey, B. D., Tian, J., Jensen, R. J., Swanson, A. K., Lees, J. R., Lentz, S. R., Stein, C. S., Nieswandt, B., Wang, Y., Davidson, B. L., & Ratliff, T. L. (2003). Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity, 19(1), 9–19.
Henn, V., Slupsky, J. R., Gräfe, M., Anagnostopoulos, I., Förster, R., Müller-Berghaus, G., & Kroczek, R. A. (1998). CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature, 391(6667), 591–594.
Tripodi, A., & Mannucci, P. M. (2011). The coagulopathy of chronic liver disease. The New England Journal of Medicine, 365(2), 147–156.
Cloutier, N., Allaeys, I., Marcoux, G., Machlus, K. R., Mailhot, B., Zufferey, A., Levesque, T., Becker, Y., Tessandier, N., Melki, I., Zhi, H., Poirier, G., Rondina, M. T., Italiano, J. E., Flamand, L., McKenzie, S. E., Cote, F., Nieswandt, B., Khan, W. I., Flick, M. J., Newman, P. J., Lacroix, S., Fortin, P. R., & Boilard, E. (2018). Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proceedings of the National Academy of Sciences of the United States of America, 115(7), E1550–E1559.
Cohen, J. D., Li, L., Wang, Y., Thoburn, C., Afsari, B., Danilova, L., Douville, C., Javed, A. A., Wong, F., Mattox, A., Hruban, R. H., Wolfgang, C. L., Goggins, M. G., Dal Molin, M., Wang, T. L., Roden, R., Klein, A. P., Ptak, J., Dobbyn, L., Schaefer, J., Silliman, N., Popoli, M., Vogelstein, J. T., Browne, J. D., Schoen, R. E., Brand, R. E., Tie, J., Gibbs, P., Wong, H. L., Mansfield, A. S., Jen, J., Hanash, S. M., Falconi, M., Allen, P. J., Zhou, S., Bettegowda, C., Diaz Jr., L. A., Tomasetti, C., Kinzler, K. W., Vogelstein, B., Lennon, A. M., & Papadopoulos, N. (2018). Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science, 359(6378), 926–930.
Nilsson, R. J., Balaj, L., Hulleman, E., van Rijn, S., Pegtel, D. M., Walraven, M., Widmark, A., Gerritsen, W. R., Verheul, H. M., Vandertop, W. P., Noske, D. P., Skog, J., & Würdinger, T. (2011). Blood platelets contain tumor-derived RNA biomarkers. Blood, 118(13), 3680–3683.
Burnouf, T., Goubran, H. A., Chou, M. L., Devos, D., & Radosevic, M. (2014). Platelet microparticles: detection and assessment of their paradoxical functional roles in disease and regenerative medicine. Blood Reviews, 28(4), 155–166.
Janowska-Wieczorek, A., Wysoczynski, M., Kijowski, J., Marquez-Curtis, L., Machalinski, B., Ratajczak, J., & Ratajczak, M. Z. (2005). Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. International Journal of Cancer, 113(5), 752–760.
Italiano Jr., J. E., Mairuhu, A. T., & Flaumenhaft, R. (2010). Clinical relevance of microparticles from platelets and megakaryocytes. Current Opinion in Hematology, 17(6), 578–584.
Ratajczak, J., Wysoczynski, M., Hayek, F., Janowska-Wieczorek, A., & Ratajczak, M. Z. (2006). Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia, 20(9), 1487–1495.
Baj-Krzyworzeka, M., Majka, M., Pratico, D., Ratajczak, J., Vilaire, G., Kijowski, J., Reca, R., Janowska-Wieczorek, A., & Ratajczak, M. Z. (2002). Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Experimental Hematology, 30(5), 450–459.
Barry, O. P., Kazanietz, M. G., Praticò, D., & FitzGerald, G. A. (1999). Arachidonic acid in platelet microparticles up-regulates cyclooxygenase-2-dependent prostaglandin formation via a protein kinase C/mitogen-activated protein kinase-dependent pathway. The Journal of Biological Chemistry, 274(11), 7545–7556.
Barry, O. P., Pratico, D., Lawson, J. A., & FitzGerald, G. A. (1997). Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. The Journal of Clinical Investigation, 99(9), 2118–2127.
Risitano, A., Beaulieu, L. M., Vitseva, O., & Freedman, J. E. (2012). Platelets and platelet-like particles mediate intercellular RNA transfer. Blood, 119(26), 6288–6295.
Diehl, P., Fricke, A., Sander, L., Stamm, J., Bassler, N., Htun, N., Ziemann, M., Helbing, T., El-Osta, A., Jowett, J. B., & Peter, K. (2012). Microparticles: major transport vehicles for distinct microRNAs in circulation. Cardiovascular Research, 93(4), 633–644.
Laffont, B., Corduan, A., Plé, H., Duchez, A. C., Cloutier, N., Boilard, E., & Provost, P. (2013). Activated platelets can deliver mRNA regulatory Ago2•microRNA complexes to endothelial cells via microparticles. Blood, 122(2), 253–261.
Tang, M., Jiang, L., Lin, Y., Wu, X., Wang, K., He, Q., Wang, X., & Li, W. (2017). Platelet microparticle-mediated transfer of miR-939 to epithelial ovarian cancer cells promotes epithelial to mesenchymal transition. Oncotarget, 8(57), 97464–97475.
Michael, J. V., Wurtzel, J. G. T., Mao, G. F., Rao, A. K., Kolpakov, A., Sabri, A., Hoffman, N. E., Rajan, S., Tomar, D., Madesh, M., Nieman, M. T., Yu, J., Edelstein, L. C., Rowley, J. W., Weyrich, A. S., & Goldfinger, L. E. (2017). Platelet microparticles infiltrating solid tumors transfer miRNAs that suppress tumor growth. Blood, 130(5), 567–580.
Linke, B., Schreiber, Y., Picard-Willems, B., Slattery, P., Nüsing, R. M., Harder, S., Geisslinger, G., & Scholich, K. (2017). Activated platelets induce an anti-inflammatory response of monocytes/macrophages through cross-regulation of PGE(2) and cytokines. Mediators of Inflammation, 2017, 1–14. https://doi.org/10.1155/2017/1463216.
Boilard, E. N., Larabee, P. A., Watts, K., et al. (2010). Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science, 29(5965), 580–583.
Vasina, E. M., Cauwenberghs, S., Feijge, M. A., Heemskerk, J. W., Weber, C., & Koenen, R. R. (2011). Microparticles from apoptotic platelets promote resident macrophage differentiation. Cell Death & Disease, 2, e211. https://doi.org/10.1038/cddis.2011.94.
Sadallah, S., Eken, C., Martin, P. J., & Schifferli, J. A. (2011). Microparticles (ectosomes) shed by stored human platelets downregulate macrophages and modify the development of dendritic cells. Journal of Immunology, 186(11), 6543–6552.
Laffont, B., Rousseau, M., Duchez, A. C., Lee, C. H., Boilard, E., & Provost, P. (2016). Platelet microparticles reprogram macrophage gene expression and function. Thrombosis and Haemostasis, 115(2), 311–323.
Soga, F., Katoh, N., Inoue, T., & Kishimoto, S. (2007). Serotonin activates human monocytes and prevents apoptosis. The Journal of Investigative Dermatology, 127(8), 1947–1955.
Duchez, A. C., Boudreau, L. H., Naika, G. S., Bollinger, J., Belleannée, C., Cloutier, N., Laffont, B., Mendoza-Villarroel, R. E., Lévesque, T., Rollet-Labelle, E., Rousseau, M., Allaeys, I., Tremblay, J. J., Poubelle, P. E., Lambeau, G., Pouliot, M., Provost, P., Soulet, D., Gelb, M. H., & Boilard, E. (2015). Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proceedings of the National Academy of Sciences of the United States of America, 112, E3564–E3573. https://doi.org/10.1073/pnas.1507905112.
Sprague, D. L., Elzey, B. D., Crist, S. A., Waldschmidt, T. J., Jensen, R. J., & Ratliff, T. L. (2008). Platelet-mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood, 111(10), 5028–5036.
Kim, H. K., Song, K. S., Park, Y. S., Kang, Y. H., Lee, Y. J., Lee, K. R., Kim, H. K., Ryu, K. W., Bae, J. M., & Kim, S. (2003). Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: possible role of a metastasis predictor. European Journal of Cancer, 39(2), 184–191.
Mege, D., Panicot-Dubois, L., Ouaissi, M., Robert, S., Sielezneff, I., Sastre, B., Dignat-George, F., & Dubois, C. (2016). The origin and concentration of circulating microparticles differ according to cancer type and evolution: a prospective single-center study. International Journal of Cancer, 138(4), 939–948.
Wang, C. C., Tseng, C. C., Chang, H. C., Huang, K. T., Fang, W. F., Chen, Y. M., Yang, C. T., Hsiao, C. C., Lin, M. C., Ho, C. K., & Yip, H. K. (2017). Circulating microparticles are prognostic biomarkers in advanced non-small cell lung cancer patients. Oncotarget, 8(44), 75952–75967.
Fais, S., O’Driscoll, L., Borras, F. E., Buzas, E., Camussi, G., Cappello, F., Carvalho, J., Cordeiro da Silva, A., Del Portillo, H., El Andaloussi, S., Ficko Trček, T., Furlan, R., Hendrix, A., Gursel, I., Kralj-Iglic, V., Kaeffer, B., Kosanovic, M., Lekka, M. E., Lipps, G., Logozzi, M., Marcilla, A., Sammar, M., Llorente, A., Nazarenko, I., Oliveira, C., Pocsfalvi, G., Rajendran, L., Raposo, G., Rohdem, E., Siljander, P., van Niel, G., Vasconcelos, M. H., Yáñez-Mó, M., Yliperttula, M. L., Zarovni, N., Zavec, A. B., & Giebel, B. (2016). Evidencebased clinical use of nanoscale extracellular vesicles in nanomedicine. ACS Nano, 10(4), 3886–3899.
Escudier, B., Dorval, T., Chaput, N., André, F., Caby, M. P., Novault, S., Flament, C., Leboulaire, C., Borg, C., Amigorena, S., Boccaccio, C., Bonnerot, C., Ohellin, O., Movassagh, M., Piperno, S., Robert, C., Serra, V., Valente, N., Le Pecq, J. B., Spatz, A., Lantz, O., Tursz, T., Angevin, E., & Zitvogel, L. (2005). Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. Journal of Translational Medicine, 3(1), 10.
Kim, S. M., & Kim, H. S. (2017). Engineering of extracellular vesicles as drug delivery vehicles. Stem Cell Investig. https://doi.org/10.21037/sci.2017.08.07.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dovizio, M., Bruno, A., Contursi, A. et al. Platelets and extracellular vesicles in cancer: diagnostic and therapeutic implications. Cancer Metastasis Rev 37, 455–467 (2018). https://doi.org/10.1007/s10555-018-9730-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-018-9730-4