
Abstract
After decades of limited success in the treatment of castration-resistant prostate cancer (CRPC), five novel therapeutics were granted Food and Drug Administration regulatory approval in the last 4 years based on several randomized phase III studies that have reported a survival benefit. Among them, two drugs targeting the androgen receptor pathway, namely abiraterone acetate and enzalutamide, have demonstrated that targeting androgen signalling following progression to classical androgen blockade continues to be an effective strategy despite the emergence of resistance mechanisms to sequential treatments. In addition to these two approved drugs, several other promising agents that block steroidogenesis interact with the androgen receptor or modulate post-receptor signal transduction that are undergoing clinical evaluation. This issue reviews the current data and the state of development of novel androgen receptor-targeting drugs and further discusses how this revolution in therapeutic armamentarium for the treatment of CRPC has raised challenges for clinicians about the optimal usage of these compounds.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
By the end of the nineteenth century, W. White observed a correlation between castration and size of the prostate gland in dogs, proposing surgical castration as a potential treatment for urinary flow obstructions [1]. These findings arrived four decades after the first histological description of prostate cancer [2] and were followed in 1941 by a report from C. Huggins regarding a series of patients with prostate cancer treated with surgical (orchiectomy) or chemical (with oestrogen injection) castration [3]. His conclusion that prostate cancer is hormone dependant continues to be the basis for treating incurable advanced disease.
Luteinizing hormone-releasing hormone (LHRH) analogues became the first line of medical treatment for advanced prostate cancer based on their ability to suppress gonadal androgen production [4, 5], with leuprolide becoming the first drug in this class to obtain Food and Drug Administration (FDA) approval in 1985. Since then, alternative formulations such as modified release preparations have been developed.
The androgen receptor (AR) was characterized in the late 1960s, leading to the development of synthetic antiandrogens that bind to the AR and competitively inhibit its interaction with testosterone and dihydrotestosterone [6–8]; this led to flutamide being approved in 1989, followed by bicalutamide and nilutamide. Combination therapy with LHRH agonist was the next logical step and persists as the most common approach for treating unconfined prostate cancer [9, 10].
Even though initial responses to LHRH analogues and antiandrogens will result in remissions lasting 1 to 3 years, nearly all patients with prostate cancer will eventually develop castration-resistant disease, which eventually becomes fatal in the majority of patients [11]. Over the past 4 years, the therapeutic landscape of prostate cancer has dramatically evolved with the approval, among other drugs, of abiraterone acetate (Zytiga, Cougar Biotechnology, NJ/Johnson & Johnson, NJ) [12, 13] and enzalutamide (Xtandi, Medivation, CA/Astellas, IL) [14]. Successful results of these novel AR pathway-impacting drugs have illustrated the value of further targeting of the AR axis in prostate cancer.
In this review, we as summarize the current state of the development of novel agents exploring this strategy, either by targeting steroidogenesis (ligand-dependant mechanisms), altering AR activation (receptor-dependant mechanisms) or disrupting post-receptor signal transduction processes (targeting intracellular chaperones or receptor-DNA interaction). We furthermore discuss challenges of optimally utilizing these therapies in clinical practice.
2 Androgen receptor signalling in prostate cancer
The androgen-AR axis has important homeostatic roles in normal prostate cell biology under physiological conditions, and it is also a major effector of prostate cancer genesis, growth and survival [15]. Androgens derive from cholesterol and their levels are regulated by the hypothalamic-pituitary-adrenal/gonadal axis (HPAG) via a negative feedback loop. The intermittent secretion of gonadotropin-releasing hormone (GnRH) and corticotropin-releasing hormone (CRH) by the hypothalamus stimulates the pituitary gland to secrete follicle-stimulating hormone (FSH) and luteinizing hormone (LH) and adrenocorticotropic hormone (ACTH). FSH and LH act on the testes to produce androgens, and ACTH acts on the adrenal glands to produce and secrete corticosteroids; the principal steroid hormones with androgenic effect are 5α-dihydrotestosterone (DHT), testosterone (T) and androstenedione (Fig. 1) [16, 17].

Schematic representation of the androgen-androgen receptor axis. LHRH luteinizing hormone-releasing hormone, ACTH adrenocorticotropic hormone, DHEA dehydroepiandrosterone, A.R. androgen receptor, HSP heat-shock protein, L ligand
Androgens are primarily produced in the testicular Leydig cells and in the adrenal glands. Two critical steps in the androgen synthesis pathway are the conversion of pregnenolone-like steroids into androgens via cytochrome P450 17A1 (CYP17) and the conversion of T into DHT by 5α-reductase, both of which can be chemically targeted. Only a small fraction of free, non-protein bound circulating T (1–2 %) is able to diffuse into peripheral tissues to be irreversibly converted into DHT by 5α-reductase. Indeed, DHT is a more potent and stable ligand for the AR than T [18].
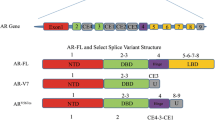
AR is a steroidal nuclear receptor encoded by the AR gene, located on the X chromosome (q11-12). Also named NR3C4 (nuclear receptor subfamily 3, group C, member 4), the main function of the AR is to modulate genomic transcription following binding of activating ligands through direct interaction with DNA. Both a full-length protein and shorter N-terminal truncated isoforms of AR have been described. It is structurally composed of a ligand-binding domain (LBD) separated from the DNA-binding domain by a small hinge region. The large N-terminal domain possesses regulatory functions, and it is of relevance for ligand-independent mechanisms of AR activation [19, 20]. Mutations in the LBD are also described and are associated with resistance to classical antiandrogens [21].
In physiological conditions, the AR resides in the cytoplasm of prostatic epithelial and stromal cells in close association with chaperones of the heat-shock protein (HSP) family [22]. Ligand binding displaces HSPs and induces a series of conformational changes, leading to the nuclear translocation of activated AR. Within the nucleus, AR binds to DNA and recruits various activators to modulate transcription of gene regulatory of several aspects of prostate cell metabolism, known as “androgen-responsive genes” [23, 24].
The testes are only responsible for 80–90 % of total androgen production [25]; therefore, surgical orchiectomy may be insufficient to achieve complete androgen deprivation. In fact, adrenalectomy and hypophisectomy were investigated for patients progressing through initial hormonal manoeuvres; however, their use is not widespread due to the associated morbidity [26]. LHRH agonists will deplete circulating androgens to undetectable levels but the adrenal gland may still produce androgens independent of LH or FSH signalling. Intracrine production of androgens within primary and metastatic tumours results in ongoing AR signalling despite undetectable circulating androgens [27, 28]. Selective pressure from treatments renders the development of a prostate cancer phenotype able to grow via stimulation by low concentrations of androgens from non-gonadal sources [29]. In effect, depletion of circulating testosterone cannot be equated to complete androgen ablation in the tumour microenvironment [30].
Additionally, it is now evident that AR develops different evolutionary aberrations as a result of exposure to sequential treatments [31]. Such alterations may favour resistance to antiandrogen therapies and include increased AR gene copy number [32], predominance of splice variants of AR which permit ligand-independent activation [33, 34], overexpression of mRNA [35] or mutations in the AR ligand-binding domain that can alter the response to other ligands such as glucocorticoids or AR antagonists such as flutamide [36, 37].
Prostate cancer may therefore become resistant and continues to proliferate following classical hormonal treatments while maintaining significant reliance on the androgen pathway. Consequently, further targeting of the androgen-AR axis remains a valid strategy to treat castration-resistant prostate cancer (CRPC). A number of novel compounds have been developed following this principle to further impact on androgen signalling (Fig. 2).

Outline of compounds in current development, indicating their target in the androgen-androgen receptor (AR) axis. Stars identify drugs already approved by FDA and EMA (abiraterone acetate and enzalutamide). HSP heat-shock proteins, ASO AR-mRNA antisense oligonucleotide designed against androgen receptor mRNA]
3 Targeting steroidogenesis
CYP17, or 17a-hydroxylase/17,20-lyase, is a multifunctional enzyme that plays a key role in steroidogenesis and is encoded by a single gene in chromosome X. CYP17 hydroxylase activity converts pregnenolone and progesterone to 17a-hydroxypregnenolone and 17a-hydroxyprogesterone, while CYP17 lyase activity converts these products to dehydroepiandrosterone (DHEA) and androstenedione, respectively (Fig. 1) [38]. In addition, 17a-hydroxypregnenolone and 17a-hydroxyprogesterone serve as precursors for cortisol synthesis; therefore, inhibition of CYP17 hydroxylase activity leads to depletion of glucocorticoid precursors, upregulation of ACTH and secondary mineralocorticoid excess which is clinically managed by either oral mineralocorticoid inhibition by eplerenone or concomitant exogenous corticosteroids [39]. Selective targeting of CYP17 lyase activity could theoretically bypass this requirement of concomitant steroid or mineralocorticoid antagonist administration.
The concept of targeting non-gonadal steroidal synthesis was initially tested by trials of ketoconazole, which blocked CYP17 17,20 lyase activity as well as hydroxylase activity at higher concentrations [40]; however, the use of ketoconazole is limited due to its unfavourable safety profile with an increased risk of thromboembolic events, hepatotoxicity and adrenal insufficiency.
Several novel drugs inhibiting CYP17 are at different stages of clinical development:
3.1 A. Abiraterone acetate
Abiraterone is a small molecule that irreversibly inhibits CYP17 with higher potency and selectivity than ketoconazole but with no antagonism of CYP11B1 and CYP11B2 which contribute to glucocorticoid and mineralocorticoid synthesis, respectively [41, 42]. The compound is orally bioavailable as its prodrug, abiraterone acetate (AA) was selected for clinical development out of several potent inhibitory 17-(3-pyridil) steroids tested [43, 44].
Phase I dose-escalation trials demonstrated substantial suppression of circulating T levels in parallel with objective tumour responses and falls in circulating tumour cell counts. Modulation of T increased with escalating doses until achieving a plateau at doses of 1,000 mg orally once daily, and this dose was selected as the recommended dose for further studies [45]. Declines in prostate-specific antigen (PSA) were observed at all dose levels tested and were frequently associated with symptomatic improvement, reduction in serum alkaline phosphatase levels, normalization of elevated lactate dehydrogenase levels and radiological responses.
Interestingly, initial studies demonstrated significant inhibition of T in both castrate and non-castrate patients; however, concomitant treatment with LHRH agonists is necessary to avoid the reactive LH rise observed in non-castrate males which rapidly counteracted the effects of AA [46].
Tolerability was carefully assessed in dose-escalation studies where hypokalaemia (55–88 %), hypertension (17–40 %) and fluid overload (15–31 %) were the most common adverse events [45, 47]. Concomitant use of the selective aldosterone antagonist eplerenone or low-dose glucocorticoid can ameliorate these effects associated with mineralocorticoid excess. Spironolactone is not recommended for this indication as it has been shown to activate AR [48]. Specific studies are currently trying to better define the optimal concomitant steroid therapy with AA, particularly due to some evidence that glucocorticoid receptors may be relevant as a mechanism of resistance [49]. Other adverse events described included hot flushes (10 %) and asymptomatic transaminase elevation (5 % grade 3; 2 % grade 2). Implementation of concomitant steroids from the onset of treatment in clinical trials improved tolerability; interestingly, patients pretreated with ketoconazole achieved fewer responses compared to their ketoconazole naïve counterparts [50].
Phase II data of AA in combination with prednisolone 5 mg twice daily demonstrated excellent tolerability, with much less hypokalaemia (5 % grade 1) and non-significant hypertension [51], with fatigue being the only grade 3 event described. PSA response rates (decline in PSA of at least 50 %) across different studies ranged between 36 and 67 %, with reports of RECIST-based responses in 18–38 % of patients with measurable disease [45, 47, 51].
An international double-blinded and randomized phase III study demonstrated overall survival benefit in the castration-resistant, post-chemotherapy setting with AA 1,000 mg once daily in combination with prednisone 5 mg twice daily vs placebo (14.8 vs. 10.9 months; hazard ratio (HR), 0.65; p < 0.001) [12]. Recruitment was stopped at an interim analysis when the positive results exceeded the preplanned criteria for study termination. All secondary endpoints favoured the treatment arm including time to PSA progression, progression free survival and PSA response rate. Incidence of fluid retention and hypokalaemia was significantly higher in the group receiving AA plus prednisone (31 vs. 22 %, p = 0.04; 17 vs. 8 %, p < 0.001, respectively). A secondary study also demonstrated significant benefits in pain relief, delayed pain progression and prevention of skeletal-related events [52]. A study evaluating AA in combination with prednisone vs placebo in the pre-chemotherapy setting also confirmed benefit of AA, being the study terminated at an interim analysis after showing significant improvement in radiographic progression-free survival (16.5 vs. 8.3 months; HR, 0.53; p < 0.001). This study demonstrated a trend towards OS benefit (median not reached vs. 27.2 months; HR, 0.75; p = 0.01) at interim analysis [13]. Based on results from the aforementioned studies, abiraterone has been approved by the US FDA and European Medicines Agency in both the pre and post-chemotherapy setting.
3.2 B. Drugs in current development
3.2.1 Orteronel
Several compounds were designed based on a substrate-mimic strategy in a screening program for synthetic next-generation CYP17 inhibitors, looking for high selectivity in targeting the C17,20-lyase component over the hydroxylase function and therefore avoiding the need for concomitant steroids.
Among several chemical compounds tested, 6-[(7S)-7-hydroxy-6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-7-yl]-N-methyl-2-naphthamide (TAK-700, later named Orteronel; Takeda Pharmaceutical Company Limited, Japan) was found to inhibit CYP17 17,20-lyase activity with over 200-fold higher potency than for the 17-hydroxylase activity in rat models. In higher species models, the IC50 value of orteronel for cortisol was about triple that for DHEA [53–55].
Orteronel is currently under clinical development. Several phase I/II studies were initiated with or without concomitant steroids as well as in combination with docetaxel. Patients treated with 400 mg twice daily had significant falls in circulating T and DHEA-sulfate [56]. However, despite its predicted selectivity for C17,20-lyase inhibition, cases of grade 3 hypertension and hypokalaemia have been reported in the phase I/II study [57]. Ongoing clinical trials evaluating antitumor efficacy against placebo in both pre-chemotherapy (NCT01193244) and post-chemotherapy (NCT01193257) settings are administering orteronel with concomitant steroids.
3.2.2 Galeterone
Galeterone (Tokai Pharmaceuticals, Cambridge, MA), formerly known as TOK-001 or VN/124-1, is a 17-benzoimidazole selected from a drug-screening program at the University of Maryland for synthetic potent CYP17 inhibitors and found also to directly inhibit AR signalling [58, 59].
The postulated advantage of galeterone over other similar compounds is the ability to disrupt several nodes of the androgen receptor axis simultaneously: (1) inhibiting 17A-hydroxylase/17,20-lyase, with a lower IC50 (300 vs. 800 nM) and higher specificity to inhibit the lyase activity than abiraterone (300 vs. 800 nM) [58, 60]; (2) competitively binding to both mutant and wild-type androgen receptor for a pure antagonist effect and (3) inducing degradation of androgen receptor protein, opposite to classic antiandrogens which cause an upregulation of AR expression [59].
Overall, preclinical studies demonstrated dose-dependent down-regulation of AR signalling in prostate cancer cell lines insensitive to bicalutamide and significant antitumor effect in PC mouse models, being superior to abiraterone acetate in inducing regression of LAPC-4 tumour in xenografts [61, 62].
A phase I/II dose-finding study (“Androgen Receptor Modulation Optimized for Response—ARMOR-1”) enrolled 49 chemotherapy-naïve CRPC patients to receive galeterone at doses ranging between 650 and 2,600 mg once daily. Eleven out of 49 (22 %) patients had a >50 % decline in PSA after 12 weeks of treatment, and it is noteworthy that at the maximum administered dose of 2,600 mg, 5/12 (42 %) achieved a >50 % PSA decline. No maximum tolerated dose was identified. While the majority of patients reported non-severe toxicities, grade ≥2 transaminase elevations were reported in 15 (30 %) patients. One case of grade 4 rhabdomyolysis and acute renal failure was described. No signs of mineralocorticoid excess were described even though no concomitant steroid was used, in consistence with the proposed mechanism of action [63]. A new tablet formulation is being evaluated in a further study (ARMOR-2, NCT01709734)
3.2.3 Other compounds in early stages of clinical development
A compound named VT-464 (Viamet 464) has entered phase I trials; it has a 60-fold greater specificity for C17,20-lyase over 17a-hydroxylase and has been shown to achieve significant effect in animal models without upstream steroid upregulation [64].
4 Targeting the androgen receptor
4.1 A. Enzalutamide
Enzalutamide (Xtandi, Medivation, CA/Astellas IL), previously known as MDV-3100, is a novel orally available androgen receptor antagonist which impacts androgen signalling by: (1) inhibiting androgen binding to AR, with a much higher affinity for the ligand-binding domain than bicalutamide; (2) reducing the efficiency of AR complex nuclear translocation (reducing the ratio nuclear: cytoplasmic AR by fivefold compared with bicalutamide) and (3) preventing the binding of the AR complex to DNA and the recruitment of its coactivators [65]. Unlike abiraterone, its use does not require concomitant steroids administration.
It was selected after preclinical screening of nearly 200 non-steroidal compounds because of its high AR-binding affinity in competition assays using 16β-[18 F]fluoro-5α-DHT (18-FDHT) and its antitumor activity in both in vitro and in vivo models. A better understanding of the crystal structure of AR and its interaction with bicalutamide supported the design of this compound, which has up to eightfold higher affinity for the AR ligand-binding domain, and presents other favourable characteristics when compared with bicalutamide: it does not act as an agonist in the context of increased AR expression, it does not induce androgen-regulated genes and it remains an antagonist to W741C mutant AR. However, recent studies of resistance mechanisms to antiandrogens have described how in the context of AR F876L mutation, enzalutamide may function as an agonist of AR. Interestingly, cell lines with this mutation remain sensitive to bicalutamide [66].
A phase I-II trial selected 160 mg QD (four capsules of 40 mg each per day) as the recommended dose after treating over 100 patients at doses ranging from 30 to 600 mg daily, based on the tolerability profile and observation of PSA declines and other pharmacodynamics markers at doses over 150 mg [67]. Fatigue was the main toxicity at higher doses and three patients (2 %) at doses of 360 mg or higher presented seizures.
A double-blinded randomized phase III trial (AFFIRM) recruited 1,199 patients to enzalutamide (160 mg QD) or placebo (2:1 randomization favouring the experimental arm). All of them were enrolled having previously progressed on docetaxel. Having difference in overall survival as the primary objective, the trial was interrupted after a planned interim analysis (after 520 events), demonstrating significant benefit for the experimental arm (HR, 0.63; 95 % CI, 0.53–0.73; p < 0.001). Enzalutamide was also clearly superior to placebo in all secondary endpoints including PSA declines over 50 %, soft-tissue responses rate, time to PSA progression, time to first skeletal event and time to radiological progression as well as in quality-of-life-related endpoints. Based on these results, enzalutamide was approved in 2012 for the treatment of CRPC after progression on taxane-based chemotherapy.
Similar to the phase I-II study, five cases of seizures (0.6 %) were observed in the registration trial, all of them in patients receiving enzalutamide. Despite potential predisposing factors like dural and brain metastasis or concomitant medications being identified among many of these patients, these events have influenced the selection criteria for further trials of enzalutamide and other novel antiandrogens. Dose-dependent convulsive episodes were observed in animal toxicology studies; the drug concentration in the central nervous system together with the off-target inhibition of the g-aminobutyric acid (GABA)-gated chloride channel has been postulated as a potential cause for seizures [68].
To further characterize the antitumor activity of enzalutamide and the optimal time of treatment, results from a randomized phase III study in the pre-chemotherapy setting for enzalutamide are awaited (PREVAIL, NCT01212991)
4.2 B. Drugs in current development
4.2.1 ARN-509
ARN-509 (Aragon Pharmaceuticals, CA) is a similar AR antagonist to enzalutamide. This compound has seven to tenfold greater affinity than bicalutamide to compete for the ligand-binding pocket of the receptor and is highly selective in binding AR over other hormone receptors. It has also been shown to reduce efficiency of AR nuclear translocation and to impair AR binding to androgen-response elements of DNA. The in vitro, the activity of ARN-509 was similar to enzalutamide, but lower doses were required to achieve similar inhibition, in murine in vivo models. The intratumoral/plasma concentration ratio for ARN-509 was higher, theoretically offering a higher therapeutic index [69].
A dose-finding trial recruited 30 patients with CRPC. The doses administered ranged from 30–480 mg QD [70]. Most common side effects described were fatigue (47 % patients) and gastrointestinal events (30 % abdominal pain, 30 % nausea, 23 % diarrhoea) but these events were very rarely severe. No episodes of seizures have been described in patients taking ARN-509, while this compound has similar affinity for GABAa receptors, penetration of the blood-brain barrier is minimal. However, clinical trials of ARN-509 prospectively selected a population with no predisposing factors to seizures, so direct comparison of safety data with the AFFIRM trial is not possible.
Imaging assessments by 18 F-fluoro-5a-dihydrotestosterone positron-emission tomography provided evidence of AR blockade at different dose levels; the final recommended phase II dose was 160 mg QD based on the safety data and on achievement of the desired exposure expected according to preclinical models; pharmacokinetics were linear and dose-dependent.
A phase II trial is evaluating the antitumor effect of ARN-509 in patients with either locally advanced or metastatic CRPC who have not received prior chemotherapy. The primary endpoint of the study is PSA response after 12 weeks of treatment. Preliminary results reported that the majority of patients with no prior AA exposure achieved the predefined response (91 % of patients with non-metastatic disease and 88 % of patients with metastatic disease). For the subgroup pretreated with AA, the PSA response at 12 weeks was 29 % [71, 72].
4.2.2 AZD3514
AZD3514 is an AR antagonist with additional properties as a down-regulator of AR expression. It decreases the presence of intranuclear AR in LNCaP cells in the absence of androgens [73]. In vitro, it inhibited growth in VCaP (wild-type AR) and LNCaP (T877A mutated AR) PC cell lines [74].
A phase I trial has been completed showing modest antitumor activity. Although there were no formal dose-limiting toxicities observed, the prevalence of mild nausea (80 %), vomiting (49 %) and thrombocytopenia was high if compared with the tolerability profile of drugs such as abiraterone and enzalutamide. Clinical development of the compound has been discontinued [75].
4.2.3 ODM-201
ODM-201 is an AR antagonist that, unlike other antiandrogens, does not cross the blood-brain barrier. After showing superior binding affinity to wild-type AR than enzalutamide in preclinical studies, a phase I/II trial of ODM-201 in patients with CRPC (AREDES) showed a good tolerability profile. PSA response was achieved in 17 (81 %) of 21 patients evaluable at 12 weeks, in groups of chemotherapy-naïve (92 %) and post-chemotherapy (86 %) patients [76]. Based on the phase I results, further evaluation is ongoing.
4.2.4 ASP9521
ASP9521 inhibits the enzyme AKR1C3 (aldo-keto reductase family 1, member C3), also named 17β-hydroxysteroid dehydrogenase type 5 (17βHSD5), an enzyme mediating conversion of adrenal molecules such as androstenedione, progesterone and estrone to testosterone. This pathway is coined the “backdoor pathway” and is postulated to be a mechanism for prostate cancer progression to androgen independence [27]. A phase I/II study has been completed to evaluate safety and preliminary antitumor activity of this compound.
4.2.5 BMS641988
BMS641988 is an AR antagonist with in vivo antitumor effect in AR-mutated models. Initial clinical data declines in PSA upon discontinuation of the drug, suggesting partial agonistic activity and leading to the closure of the study [77, 78].
5 Targeting post-receptor events and other strategies
Heat-shock proteins (HSP) are chaperone molecules that exhibit increased expression to help cells overcome multiple environmental stresses. They are involved in structural and functional stability of other proteins, including the folding, trafficking and transcriptional activity of steroid receptors such as AR [79]. AR is naturally bound to HSP in the cytoplasm, and dissociation is a key event to permit nuclear internalization of AR following ligand binding.
HSP90 is an adenosine triphosphate (ATP)-dependent chaperone protein that plays an important role in the heat-shock response (HSR) by interacting with a complex of other molecules, including HSP70, co-chaperones and tetratricopeptide repeat (TPR)-containing proteins [80]; this HSR complex interacts with AR as well as other proteins involved in prostate cancer. Several HSP90 inhibitors are currently under preclinical or clinical evaluation.
In vitro, the HSP-90 inhibitor tanespimycin (17-N-allylamino-17-demethoxygeldanamycin, 17-AAG) inhibits PSA expression and disrupts ligand-independent nuclear localization of AR as well as inhibiting expression of IL-6, a cytokine involved in prostate cancer progression [81, 82]. Clinical trials of retaspimycin were discontinued due to lack of activity and unacceptable toxicity including gastrointestinal, liver function alteration, fatigue and ophthalmological events [83]. More recently, there has been interest in evaluating combinatory regimens: a phase I-II study is currently recruiting men with CRPC to receive the HSP90 inhibitor AT13387 alone or in combination with abiraterone acetate (AA) after progression on AA (NCT01685268).
HSP27 (heat-shock protein beta-1, HSPB1) is an ATP-independent cytoprotective chaperone protein classed as a small HSP (sHSP). It has been shown to enhance AR stability, shuttling and transcriptional activity via a feedforward loop involving cooperative interactions between ligand-activated AR and HSP27 [84]. Its expression becomes highly upregulated by cellular stresses caused by hormone therapy or chemotherapy and inhibits treatment-induced apoptosis [85, 86]. HSP27 is also a key element of epithelial-to-mesenchymal transition (EMT) in prostate cancer [87]. OGX-427, an antisense oligonucleotide targeting HSP27, entered randomized phase II trials after demonstrating to have a satisfactory safety profile and, interestingly, induced reductions in CTC counts. Preliminary results reported a 71 % rate of progression-free survival after 12 weeks of treatment compared to 40 % with prednisone [88].
Phase III trials combining OGX-011, an antisense oligonucleotide-targeting clusterin (a cytoprotective chaperone which promotes of cell survival) in combination with taxanes, are ongoing based on results from previous studies [89, 90]. Combining AR-targeting drugs with docetaxel or cabazitaxel is of special interest because of the inhibitory effect of taxanes over AR nuclear translocation through affecting microtubules stabilization [91].
Based on the principle of antisense oligonucleotides (ASO) or RNA silencers like OGX-427 or OGX-011, an alternative strategy is to modulate androgen signalling with specific antisense ASO designed against AR mRNA, aiming to induce its degradation and diminish AR protein transcription [92, 93]. Although over the last two decades several ASO have been evaluated against different tumour types, clinical success has been limited and drugs were associated with significant toxicities. A new generation of compounds have introduced modifications in the chemistry structure to increase stability and favouring RNA binding.
Recently reported clinical studies have shown little preliminary antitumor effect with EZN-4176 (Enzon Pharmaceuticals, NJ), a locked nucleic acid ASO targeting androgen receptor mRNA at the hinge region. It was shown to down-regulate AR expression when transfected to AR prostate cancer cell lines and consequently inhibiting growth selectively in AR-positive cell lines. Dose-dependent reduction in AR mRNA expression was observed in animal models following intravenous administration of ENZ-4176 [94, 95].
6 Current challenges
6.1 Optimal use of novel agents: combinations, rational sequences
Despite significant benefits in overall survival being achieved with recently approved therapies for CRPC, clinical benefit continues to be temporary and disease progression inexorably appears over time. One challenge will be to define the optimal sequence and/or combination of novel agents to counteract resistances.
Progression of prostate cancer on these novel therapies targeting the AR pathway is often reflected by rising PSA levels, suggesting the persistence of AR signalling as the biological driver of the disease [49]. Such observations and some encouraging preclinical data support therapeutic strategies of further targeting this pathway [96]; indeed, several trials are evaluating specific combinations of enzalutamide and AA (NCT01650194) or ARN-509 and AA (NCT01792687). Interestingly, some data has recently emerged, suggesting some cross-resistance among these compounds; two separate publications analysed the response to AA in patients previously exposed to enzalutamide in the AFFIRM phase III trial [14]. These series reported on 37 patients treated at European centres [97] and 30 from four Canadian institutions [98]. In both subgroups, the benefit obtained from treatment with abiraterone was smaller than expected, according to the data from randomized trials for abiraterone acetate in the post-docetaxel setting. Only 15 % of the combined population from these two studies achieved a PSA response >30 %, whereas up to 66 % of the same population had such response to prior treatment with enzalutamide. Median progression-free survival on abiraterone was also shorter than expected from previous publications on abiraterone (2.7 months in the Canadian study and 15.4 weeks in the European report). Similar data can be inferred from a phase II trial of ARN-509, which recruited patients with metastatic CRPC in two separate cohorts based on prior exposure to Abiraterone. The rate of >50 % PSA declines at 12 week was 88 % in the Abi-naïve group vs only 29 % in those patients who received prior abiraterone [72]. Prospective randomized trials assessing the optimal sequence of these drugs are now needed, exploring predefined sequences of treatments or the addition of second or third drugs upon progression.
Among other signalling pathways potentially contributing to progression, the role of PI3K-PTEN-AKT pathway may be particularly important in many prostate cancers due to the existence of signalling feedback between this pathway and the AR [99–101]. Several mechanisms are involved; among them, functional loss of PTEN (as a result of point mutations, microRNA expression changes, post-translational modifications or epigenetic silencing mechanisms) is present in 50–60 % of PC [102] and has been related to promotion of androgen independence in animal models [103]. Preclinical studies have demonstrated promising effect when combining the inhibition of both pathways in PTEN-deficient prostate cancer models [104]. Initiation of clinical studies exploring this strategy is therefore justified, either testing combinatory blockade of both pathways from the onset of treatment or evaluating the addition of a PI3K/AKT inhibitor when progressing to AR pathway modulation to counteract the eventual reciprocal feedback loops.
6.2 Will these new drugs have a role in earlier stages of prostate cancer?
Another challenge will be to understand whether using AA or enzalutamide in earlier stages of the disease to achieve a stronger androgen blockade might maximize the benefit derived from such drugs [105]. Enzalutamide has been shown to induce rapid and dramatic responses in non-castrated patients as a single agent, with 90 % of patients maintaining PSA declines over 90 % after 25 weeks of treatment but with the expected upregulation of serum LH and testosterone [106]. Preclinical data also support the clinical evaluation of ARN-509 in non-castrate men [69]. However, to properly assess the role of novel compounds in the castration-sensitive scenario, a trial studying combinations of abiraterone and/or enzalutamide with the current standards for chemical castration is needed. A randomized, double-blind study of standard androgen deprivation therapy with or without abiraterone plus prednisone has been recently initiated (NCT01715285).
Other therapeutic scenarios in which these novel compounds may also play a relevant role are the neoadjuvant and adjuvant settings. Adjuvant hormonal treatment after definitive treatment has been proven to improve outcome in patients with locally advanced and/or high-risk prostate cancer in several randomized studies, especially before or after definitive radiotherapy [107–109]. The best timing and duration of this adjuvant treatment remains to be elucidated, but response to neoadjuvant hormonal therapy is a long-term predictor of benefit [110]. Therefore, clinical trials are being conducted to evaluate if the addition of AA and enzalutamide may optimize the effects of neoadjuvant-adjuvant treatments (NCT01547299) [111].
7 Conclusions
The successful development of abiraterone acetate and enzalutamide and other non-androgen axis-related treatments has opened a wide spectrum of possibilities for patients with castration-resistant prostate cancer. Biological evidence that AR signalling remains relevant in prostate cancer despite serum androgen depletion justifies why the androgen-AR axis continues to be a valid target. Overall, a new generation of androgen targeting therapies have shown significant responses with a relatively low-toxicity profile and potential for combination. Current compounds in development will bring even more specificity of action and may potentially improve results and tolerability as well as facilitate drug delivery by avoiding concomitant use of steroids.
Significant achievements in the last 5 years justify the increasing interest of the scientific community in prostate cancer. Despite this, responses to novel treatments continue to be limited in time and the development of adaptive mechanisms of resistance remains, so far, inevitable. While the therapeutic scenario becomes more complex with the approval of further agents, the development of optimal sequences and combination of these treatments demands bespoke clinical trial designs, able to answer biologically relevant questions about reversal of resistances and optimal timeline of treatment.
Moreover, most of the drugs approved over the last 5 years have demonstrated survival benefit in comparison to placebo, which may now not longer be an acceptable comparison, making the search for significant overall survival differences in randomized clinical trials more challenging; therefore, validation of biological intermediate endpoints as surrogate biomarkers will be essential. Serum-based biomarkers would be of special interest as they permit non-invasive multiple sampling over time.
Achieving the goal of delivering precision medicine in CRPC will depend on more and better drugs becoming available, an increasing understanding of the relation among prostate cancer biology and drug efficacy and the development of biomarkers to guide individual selection of optimal sequence and combination of treatments.
8 Key unanswered questions
-
What treatment should be used first: abiraterone acetate or enzalutamide?
-
What biomarkers can guide clinicians in deciding the optimal sequence of treatment for each patient?
-
How much cross-resistance exists among novel drugs targeting the AR pathway?
-
Would using these drugs as first-line treatment extend duration of responses?
-
Would these drugs increase the cure rate of patients with high-risk localized disease?
-
How do we combine these drugs with non-AR-targeting emerging drugs?
-
Can we define combinations of drugs to counteract resistances as they appear?
References
White W. (1893). Surgical removal of the hypertrophied prostate. Annals of Surgery, 18, 152–158.
Adams, J. (1853). The case of scirrhous of the prostate gland with corresponding affliction of the lymphatic glands in the lumbar region and in the pelvis. Lancet, 1, 393.
Huggins, C., & Hodges, C. (1941). The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prosta. Cancer Research, 1, 293–297.
Sandow, J., Von Rechenberg, W., Jerzabek, G., & Stoll, W. (1978). Pituitary gonadotropin inhibition by a highly active analog of luteinizing hormone-releasing hormone. Fertility and Sterility, 30, 205–209.
The Leuprolide Study Group. (1984). Leuprolide versus diethylstilbestrol for metastatic prostate cancer. The Leuprolide Study Group. New England Journal of Medicine, 311, 1281–1286.
Mainwaring, W. I. (1969). A soluble androgen receptor in the cytoplasm of rat prostate. Journal of Endocrinology, 45, 531–541.
Anderson, K. M., & Liao, S. (1968). Selective retention of dihydrotestosterone by prostatic nuclei. Nature, 219, 277–279. 20.
Bruchovsky, N., & Wilson, J. D. (1968). The intranuclear binding of testosterone and 5-alpha-androstan-17-beta-ol-3-one by rat prostate. Journal of Biological Chemistry, 243, 5953–5960. 25.
Crawford, E. D., Eisenberger, M. A., McLeod, D. G., Spaulding, J. T., Benson, R., Dorr, F. A., et al. (1989). A controlled trial of leuprolide with and without flutamide in prostatic carcinoma. New England Journal of Medicine, 321, 419–424. 17.
Schellhammer, P. F., Sharifi, R., Block, N. L., Soloway, M. S., Venner, P. M., Patterson, A. L., et al. (1996). A controlled trial of bicalutamide versus flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy, in patients with advanced prostate carcinoma. Analysis of time to progression. CASODEX Combination Study Group. Cancer, 78, 2164–2169. 15.
Pienta, K. J., & Bradley, D. (2006). Mechanisms nnderlying the development of androgen-independent prostate cancer. Clinical Cancer Research, 12, 1665–1671. 15.
De Bono, J. S., Logothetis, C. J., Molina, A., Fizazi, K., North, S., Chu, L., et al. (2011). Abiraterone and increased survival in metastatic prostate cancer. New England Journal of Medicine, 364, 1995–2005. 26.
Ryan, C. J., Smith, M. R., De Bono, J. S., Molina, A., Logothetis, C. J., De Souza, P., et al. (2013). Abiraterone in metastatic prostate cancer without previous chemotherapy. New England Journal of Medicine, 368, 138–148. 10.
Scher, H. I., Fizazi, K., Saad, F., Taplin, M.-E., Sternberg, C. N., Miller, K., et al. (2012). Increased survival with enzalutamide in prostate cancer after chemotherapy. New England Journal of Medicine, 367, 1187–1197. 27.
Cunha, G. R., Ricke, W., Thomson, A., Marker, P. C., Risbridger, G., Hayward, S. W., et al. (2004). Hormonal, cellular, and molecular regulation of normal and neoplastic prostatic development. Journal of Steroid Biochemistry and Molecular Biology, 92, 221–236.
Auchus, M. L., & Auchus, R. J. (2012). Human steroid biosynthesis for the oncologist. Journal of Investigative Medicine, 60, 495–503.
Sharifi, N., & Auchus, R. J. (2012). Steroid biosynthesis and prostate cancer. Steroids, 77, 719–726.
Miller, W. L., & Auchus, R. J. (2011). The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrine Reviews, 32, 81–151.
Brinkmann, A. O., Klaasen, P., Kuiper, G. G., Van der Korput, J. A., Bolt, J., De Boer, W., et al. (1989). Structure and function of the androgen receptor. Urological Research, 17, 87–93.
Klokk, T. I., Kurys, P., Elbi, C., Nagaich, A. K., Hendarwanto, A., Slagsvold, T., et al. (2007). Ligand-specific dynamics of the androgen receptor at its response element in living cells. Molecular and Cellular Biology, 27, 1823–1843.
Taplin, M.-E., Bubley, G. J., Shuster, T. D., Frantz, M. E., Spooner, A. E., Ogata, G. K., et al. (1995). Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. New England Journal of Medicine, 332, 1393–1398.
Pratt, W. B., & Toft, D. O. (1997). Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocrine Reviews, 18, 306–360.
Velasco, A. M., Gillis, K. A., Li, Y., Brown, E. L., Sadler, T. M., Achilleos, M., et al. (2004). Identification and validation of novel androgen-regulated genes in prostate cancer. Endocrinology, 145, 3913–3924.
Agoulnik, I. U., & Weigel, N. L. (2009). Coactivator selective regulation of androgen receptor activity. Steroids, 74, 669–674.
Attard, G., Belldegrun, A. S., & De Bono, J. S. (2005). Selective blockade of androgenic steroid synthesis by novel lyase inhibitors as a therapeutic strategy for treating metastatic prostate cancer. BJU International, 96, 1241–1246.
Brendler, H. (1973). Adrenalectomy and hypophysectomy for prostatic cancer. Urology, 2, 99–102.
Stanbrough, M., Bubley, G. J., Ross, K., Golub, T. R., Rubin, M. a., Penning, T. M., et al. (2006). Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Research, 66, 2815–2825.
Montgomery, R. B., Mostaghel, E. a., Vessella, R., Hess, D. L., Kalhorn, T. F., Higano, C. S., et al. (2008). Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Research, 68, 4447–4454.
Isaacs, J. T., & Coffey, D. S. (1981). Adaptation versus selection as the mechanism responsible for the relapse of prostatic cancer to androgen ablation therapy as studied in the Dunning R-3327-H adenocarcinoma. Cancer Research, 41, 5070–5075.
Page, S. T., Lin, D. W., Mostaghel, E. a., Hess, D. L., True, L. D., Amory, J. K., et al. (2006). Persistent intraprostatic androgen concentrations after medical castration in healthy men. Journal of Clinical Endocrinology and Metabolism, 91, 3850–3856.
Donovan, M. J., Osman, I., Khan, F. M., Vengrenyuk, Y., Capodieci, P., Koscuiszka, M., et al. (2010). Androgen receptor expression is associated with prostate cancer-specific survival in castrate patients with metastatic disease. BJU International, 105, 462–467.
Bubendorf, L., Kononen, J., Koivisto, P., Schraml, P., Moch, H., Gasser, T. C., et al. (1999). Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Research, 59, 803–806. 15.
Watson, P. a., Chen, Y. F., Balbas, M. D., Wongvipat, J., Socci, N. D., Viale, A., et al. (2010). Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proceedings of the National Academy of Sciences of the United States of America, 107, 16759–16765. 28.
Dehm, S. M., Schmidt, L. J., Heemers, H. V., Vessella, R. L., & Tindall, D. J. (2008). Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Research, 68, 5469–5477.
Waltering, K. K., Helenius, M. A., Sahu, B., Manni, V., Linja, M. J., Jänne, O. A., et al. (2009). Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Research, 69, 8141–8149. 15.
Taplin, M.-E., Rajeshkumar, B., Halabi, S., Werner, C. P., Woda, B. a., Picus, J., et al. (2003). Androgen receptor mutations in androgen-independent prostate cancer: cancer and leukemia group B study 9663. Journal of Clinical Oncology, 21, 2673–2678. 15.
Matias, P. M., Carrondo, M. A., Coelho, R., Thomaz, M., Zhao, X.-Y., Wegg, A., et al. (2002). Structural basis for the glucocorticoid response in a mutant human androgen receptor (AR(ccr)) derived from an androgen-independent prostate cancer. Journal of Medicinal Chemistry, 45, 1439–1446. 28.
Miller, W. L., Auchus, R. J., & Geller, D. H. (1997). The regulation of 17,20 lyase activity. Steroids, 62, 133–142.
Attard, G., Reid, A. H. M., Auchus, R. J., Hughes, B. A., Cassidy, A. M., Thompson, E., et al. (2012). Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. Journal of Clinical Endocrinology and Metabolism, 97, 507–516.
Pont, A., Williams, P. L., Azhar, S., Reitz, R. E., Bochra, C., Smith, E. R., et al. (1982). Ketoconazole blocks testosterone synthesis. Archives of Internal Medicine, 142, 2137–2140.
Haidar, S., Ehmer, P. B., Barassin, S., Batzl-Hartmann, C., & Hartmann, R. W. (2003). Effects of novel 17alpha-hydroxylase/C17, 20-lyase (P450 17, CYP 17) inhibitors on androgen biosynthesis in vitro and in vivo. Journal of Steroid Biochemistry and Molecular Biology, 84, 555–562.
Reid, A. H., Attard, G., Barrie, E., & De Bono, J. S. (2008). CYP17 inhibition as a hormonal strategy for prostate cancer. Nature Clinical Practice Urology, 5, 610–620.
Barrie, S. E., Potter, G. A., Goddard, P. M., Haynes, B. P., Dowsett, M., & Jarman, M. (1994). Pharmacology of novel steroidal inhibitors of cytochrome P450(17) alpha (17 alpha-hydroxylase/C17-20 lyase). Journal of Steroid Biochemistry and Molecular Biology, 50, 267–273.
Potter, G. A., Barrie, S. E., Jarman, M., & Rowlands, M. G. (1995). Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. Journal of Medicinal Chemistry, 38, 2463–2471. 23.
Attard, G., Reid, A. H. M., A’Hern, R., Parker, C., Oommen, N. B., Folkerd, E., et al. (2009). Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. Journal of Clinical Oncology, 27, 3742–3748. 10.
O’Donnell, A., Judson, I., Dowsett, M., Raynaud, F., Dearnaley, D., Mason, M., et al. (2004). Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. British Journal of Cancer, 90, 2317–2325. 14.
Reid, A. H. M., Attard, G., Danila, D. C., Oommen, N. B., Olmos, D., Fong, P. C., et al. (2010). Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. Journal of Clinical Oncology, 28, 1489–1495. 20.
Luthy, I. A., Begin, D. J., & Labrie, F. (1988). Androgenic activity of synthetic progestins and spironolactone in androgen-sensitive mouse mammary carcinoma (Shionogi) cells in culture. Journal of Steroid Biochemistry, 31, 845–852.
Attard, G., Cooper, C. S., & De Bono, J. S. (2009). Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell, 16, 458–462. 8.
Ryan, C. J., Smith, M. R., Fong, L., Rosenberg, J. E., Kantoff, P., Raynaud, F., et al. (2010). Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. Journal of Clinical Oncology, 28, 1481–1488. 20.
Danila, D. C., Morris, M. J., De Bono, J. S., Ryan, C. J., Denmeade, S. R., Smith, M. R., et al. (2010). Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. Journal of Clinical Oncology, 28, 1496–1501. 20.
Logothetis, C. J., Basch, E., Molina, A., Fizazi, K., North, S. A., Chi, K. N., et al. (2012). Effect of abiraterone acetate and prednisone compared with placebo and prednisone on pain control and skeletal-related events in patients with metastatic castration-resistant prostate cancer: exploratory analysis of data from the COU-AA-301 randomised trial. Lancet Oncology, 13, 1210–1217.
Matsunaga, N., Kaku, T., Ojida, A., Tanaka, T., Hara, T., Yamaoka, M., et al. (2004). C(17,20)-lyase inhibitors. Part 2: design, synthesis and structure-activity relationships of (2-naphthylmethyl)-1H-imidazoles as novel C(17,20)-lyase inhibitors. Bioorganic & Medicinal Chemistry, 12, 4313–4336.
Yamaoka, M., Hara, T., Hitaka, T., Kaku, T., Takeuchi, T., Takahashi, J., et al. (2012). Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. The Journal of Steroid Biochemistry and Molecular Biology, 129, 115–128.
Hara, T., Kouno, J., Kaku, T., Takeuchi, T., Kusaka, M., Tasaka, A., et al. (2013). Effect of a novel 17,20-lyase inhibitor, orteronel (TAK-700), on androgen synthesis in male rats. The Journal of Steroid Biochemistry and Molecular Biology, 134, 80–91.
Dreicer, R., Agus, D. B. , Bellmunt, J., De Bono, J.S., Petrylak, D., Tejura, B., et al. (2012). A phase III, randomized, double-blind, multicenter trial comparing the investigational agent orteronel (TAK-700) plus prednisone (P) with placebo plus P in patients with metastatic castration-resistant prostate cancer (mCRPC) that has progressed during or following docetaxel-based chemotherapy [abstract]. Journal of Clinical Oncology, 30, 2012 (suppl; abstr TPS4693).
Agus, D. B., Stadler, W. M., Shevrin, D. H., Hart, L., Macvicar, G. R., Hainsworth, J. D., et al. (2012). Safety, efficacy, and pharmacodynamics of the investigational agent orteronel (TAK-700) in metastatic castration-resistant prostate cancer (mCRPC): updated data from a phase I/II study. [abstract]. Journal of Clinical Oncology, 50, 50–51. 30, 2012 (suppl 5; abstr 98).
Handratta, V. D., Vasaitis, T. S., Njar, V. C. O., Gediya, L. K., Kataria, R., Chopra, P., et al. (2005). Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. Journal of Medicinal Chemistry, 48, 2972–2984. 21.
Vasaitis, T., Belosay, A., Schayowitz, A., Khandelwal, A., Chopra, P., Gediya, L. K., et al. (2008). Androgen receptor inactivation contributes to antitumor efficacy of 17{alpha}-hydroxylase/17,20-lyase inhibitor 3beta-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Molecular Cancer Therapeutics, 7, 2348–2357.
DeVore, N. M., & Scott, E. E. (2012). Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature, 482(7383), 116–119.
Bruno, R., Lu, J.-F., Sun, Y.-N., & Claret, L. (2011). A modeling and simulation framework to support early clinical drug development decisions in oncology. Journal of Clinical Pharmacology, 51, 6–8.
Schayowitz, A., Sabnis, G., Njar, V. C. O., & Brodie, A. M. H. (2008). Synergistic effect of a novel antiandrogen, VN/124-1, and signal transduction inhibitors in prostate cancer progression to hormone independence in vitro. Molecular Cancer Therapeutics, 7, 121–132.
Montgomery, R. B., Eisenberger, M. a., Rettig, M., Chu, F., Pili, R., Stephenson, J., et al. (2012). Phase I clinical trial of galeterone (TOK-001), a multifunctional antiandrogen and CYP17 inhibitor in castration resistant prostate cancer (CRPC). [abstract]. Journal of Clinical Oncology, 30, 2012 (suppl; abstr 4665). 2012.
Eisner, J. R., Aboott, D., I.M. B, Rafferty, S. W., Schotzinger, R. J. (2012). Assessment of Steroid Hormones Upstream of P450c17 (CYP17) in Chemically Castrate Male Rhesus Monkeys Following Treatment with the CYP17 Inhibitors VT‐464 and Abiraterone Acetate (AA) [abstract]. Endocrine Reviews, 2012; Vol 33 (03_MeetingAbstracts): Sat 266.
Tran, C., Ouk, S., Clegg, N. J., Chen, Y., Watson, P. A., Wongvipat, J., et al. (2009). Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science, 324, 787–790.
Balbas, M. D., Evans, M. J., Hosfield, D. J., Wongvipat, J., Arora, V. K., Watson, P. a., et al. (2013). Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife, 2, e00499.
Scher, H. I., Beer, T. M., Higano, C. S., Anand, A., Taplin, M.-E., Efstathiou, E., et al. (2010). Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet, 375, 1437–1446.
Treiman, D. M. (2001). GABAergic mechanisms in epilepsy. Epilepsia, 42(Suppl 3), 8–12.
Clegg, N. J., Wongvipat, J., Joseph, J. D., Tran, C., Ouk, S., Dilhas, A., et al. (2012). ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Research, 72, 1494–1503.
Rathkopf, D. E., Morris, M. J., Danila, D. C., Slovin, S., Steinbrecher, J., Arauz, G., et al. (2012). A phase I study of the androgen signaling inhibitor ARN-509 in patients with metastatic castration-resistant prostate cancer (mCRPC) [abstract]. Journal of Clinical Oncology, 30, 2012 (suppl; abstr 4548). 2012.
Smith, M., Antonarakis, E. S., Ryan, C. J., Berry, W., Shore, N., Liu, G., et al. (2012) Arn-509 in men with high risk non-metastatic castration-resistant prostate cancer [abstract]. Annals of Oncology, 23 (suppl 9) abst 920.
Rathkopf, D. E., Antonarakis, E. S., Shore, N., Tutrone, R., Amlukai, J., Ryan, C. J., et al. (2012). Arn-509 in men with metastatic castration-resistant prostate cancer (CRPC) [abstract]. Annals of Oncology, 23 (suppl 9) abst 964.
Bradbury, R. H., Acton, D. G., Broadbent, N. L., Brooks, a. N., Carr, G. R., Hatter, G., et al. (2013). Discovery of AZD3514, a small-molecule androgen receptor downregulator for treatment of advanced prostate cancer. Bioorganic & Medicinal Chemistry Letters, 23, 1945–1948.
Loddick, S. a., Bradbury, R., Broadbent, N., Campbell, H., Gaughan, L., Growcott, J., et al. (2012). Preclinical profile of AZD3514: A small molecule-targeting androgen receptor function with a novel mechanism of action and the potential to treat castration-resistant prostate cancer [abstract]. Cancer Research, 72, 3848–3848.
Omlin, A., Jones, R. J., vn der Noll, R., Graham, J., Ong, M., Schellens, J. H. M., et al. (2013). A first-in-human study of the oral selective androgen receptor down-regulating drug (SARD) AZD3514 in patients with castration-resistant prostate cancer (CRPC) [abstract]. Journal of Clinical Oncology, 31, 2013 (suppl; abstr 4511).
Massard, C., James, N., Culine, S., Jones, R., Vuorela, A., Mustonen, M., et al. (2012). ARADES trial: a first- in-man, open-label, phase I/II safety, pharmacokinetic, and proof-of-concept study of ODM-201 in patients (pts) with progressive metastatic castration-resistant prostate cancer (mCRPC) [abstract]. Annals of Oncology, 23 (suppl 9) abst LBA25.
Attar, R. M., Jure-Kunkel, M., Balog, A., Cvijic, M. E., Dell-John, J., Rizzo, C. A., et al. (2009). Discovery of BMS-641988, a novel and potent inhibitor of androgen receptor signaling for the treatment of prostate cancer. Cancer Research, 69, 6522–6530.
Rathkopf, D., Liu, G., Carducci, M. A., Eisenberger, M. A., Anand, A., Morris, M. J., et al. (2011). Phase I dose-escalation study of the novel antiandrogen BMS-641988 in patients with castration-resistant prostate cancer. Clinical Cancer Research, 17, 880–887.
Georget, V., Térouanne, B., Nicolas, J.-C., & Sultan, C. (2002). Mechanism of antiandrogen action: key role of hsp90 in conformational change and transcriptional activity of the androgen receptor. Biochemistry, 41, 11824–11831.
Prescott, J., & Coetzee, G. A. (2006). Molecular chaperones throughout the life cycle of the androgen receptor. Cancer Letters, 231, 12–19.
Saporita, A. J., Ai, J., & Wang, Z. (2007). The Hsp90 inhibitor, 17-AAG, prevents the ligand-independent nuclear localization of androgen receptor in refractory prostate cancer cells. Prostate, 67, 509–520.
Tsui, K.-H., Feng, T.-H., Lin, C.-M., Chang, P.-L., & Juang, H.-H. (2008). Curcumin blocks the activation of androgen and interlukin-6 on prostate-specific antigen expression in human prostatic carcinoma cells. Journal of Andrology, 29, 661–668.
Oh, W. K., Galsky, M. D., Stadler, W. M., Srinivas, S., Chu, F., Bubley, G., et al. (2011). Multicenter phase II trial of the heat shock protein 90 inhibitor, retaspimycin hydrochloride (IPI-504), in patients with castration-resistant prostate cancer. Urology, 78, 626–630.
Zoubeidi, A., Zardan, A., Beraldi, E., Fazli, L., Sowery, R., Rennie, P., et al. (2007). Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Research, 67, 10455–10465.
Rocchi, P., Beraldi, E., Ettinger, S., Fazli, L., Vessella, R. L., Nelson, C., et al. (2005). Increased Hsp27 after androgen ablation facilitates androgen-independent progression in prostate cancer via signal transducers and activators of transcription 3-mediated suppression of apoptosis. Cancer Research, 65, 11083–11093.
Andrieu, C., Taieb, D., Baylot, V., Ettinger, S., Soubeyran, P., De-Thonel, A., et al. (2010). Heat shock protein 27 confers resistance to androgen ablation and chemotherapy in prostate cancer cells through eIF4E. Oncogene, 29, 1883–1896.
Shiota, M., Bishop, J. L., Nip, K. M., Zardan, A., Takeuchi, A., Cordonnier, T., et al. (2013). Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Research, 73, 3109–3119.
Chi, K. N., Hotte, S. J., Ellard, S. L., Gingerich, J., Joshua, A., Yu, E. Y., et al. (2012). A randomized phase II study of OGX-427 plus prednisone (P) versus P alone in patients (pts) with metastatic castration resistant prostate cancer (CRPC). [abstract]. Journal of Clinical Oncology, 30, 2012 (suppl; abstr 4514). 2012.
Chi, K. N., Hotte, S. J., Yu, E. Y., Tu, D., Eigl, B. J., Tannock, I., et al. (2010). Randomized phase II study of docetaxel and prednisone with or without OGX-011 in patients with metastatic castration-resistant prostate cancer. Journal of Clinical Oncology, 28, 4247–4254.
Chi, K. N., Siu, L. L., Hirte, H., Hotte, S. J., Knox, J., Kollmansberger, C., et al. (2008). A phase I study of OGX-011, a 2′-methoxyethyl phosphorothioate antisense to clusterin, in combination with docetaxel in patients with advanced cancer. Clinical Cancer Research, 14, 833–839.
Darshan, M. S., Loftus, M. S., Thadani-Mulero, M., Levy, B. P., Escuin, D., Zhou, X. K., et al. (2011). Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Research, 71, 6019–6029.
Cook, P. D. (1999). Making drugs out of oligonucleotides: a brief review and perspective. Nucleosides & Nucleotides, 18, 1141–1162.
Gleave, M. E., & Monia, B. P. (2005). Antisense therapy for cancer. Nature Reviews Cancer, 5, 468–479.
Zhang, Y., Castaneda, S., Dumble, M., Wang, M., Mileski, M., Qu, Z., et al. (2011). Reduced expression of the androgen receptor by third generation of antisense shows antitumor activity in models of prostate cancer. Molecular Cancer Therapeutics, 10, 2309–2319.
Bianchini, D., Omlin, A., Pezaro, C., Mukherji, D., Lorente Estelles. D., Zivi, A., et al. (2013). First-in-human phase I study of EZN-4176, a locked nucleic acid antisense oligonucleotide (LNA-ASO) to androgen receptor (AR) mRNA in patients with castration-resistant prostate cancer (CRPC) [abstract]. Journal of Clinical Oncology, 31 (suppl; abstr 5052).
Richards, J., Lim, A. C., Hay, C. W., Taylor, A. E., Wingate, A., Nowakowska, K., et al. (2012). Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: a rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Research, 72, 2176–2182.
Loriot, Y., Bianchini, D., Ileana, E., Sandhu, S., Patrikidou, A., Pezaro, C., et al. (2013). Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100). Annals of Oncology, 24(7), 1807–1812.
Noonan, K. L., North, S., Bitting, R. L., Armstrong, a. J., Ellard, S. L., & Chi, K. N. (2013). Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Annals of Oncology, 24(7), 1802–1807.
Manning, B. D., & Cantley, L. C. (2007). AKT/PKB signaling: navigating downstream. Cell, 129, 1261–1274.
Mediwala, S. N., Sun, H., Szafran, A. T., Hartig, S. M., Sonpavde, G., Hayes, T. G., et al. (2013). The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. Prostate, 73, 267–277.
Trotta, A. P., Need, E. F., Selth, L. A., Chopra, S., Pinnock, C. B., Leach, D. A., et al. (2013). Knockdown of the co-chaperone SGTA results in the suppression of androgen and PI3K/AKT signaling and inhibition of prostate cancer cell proliferation. International Journal of Cancer, 133(12), 2812–2823.
Yoshimoto, M., Cunha, I. W., Coudry, R. A., Fonseca, F. P., Torres, C. H., Soares, F. A., et al. (2007). FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. British Journal of Cancer, 97, 678–685.
Gao, H., Ouyang, X., Banach-Petrosky, W. A., Shen, M. M., & Abate-Shen, C. (2006). Emergence of androgen independence at early stages of prostate cancer progression in Nkx3.1; Pten mice. Cancer Research, 66, 7929–7933.
Carver, B. S., Chapinski, C., Wongvipat, J., Hieronymus, H., Chen, Y., Chandarlapaty, S., et al. (2011). Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell, 19, 575–586.
Mostaghel, E. a., Page, S. T., Lin, D. W., Fazli, L., Coleman, I. M., True, L. D., et al. (2007). Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Research, 67, 5033–5041.
Smith, M., Borre, M., Rathenborg, P., Werbrouck, P., Van Poppel, H., Heidenreich, A., et al. (2013). Efficacy and safety of enzalutamide (ENZA) monotherapy in hormone-naive prostate cancer (HNPC). [abstract]. Journal of Clinical Oncology, 31, 2013 (suppl; abstr 5001).
Kumar, S., Shelley, M., Harrison, C., Coles, B., Wilt, T. J., Mason, M. D. (2006). Neo-adjuvant and adjuvant hormone therapy for localised and locally advanced prostate cancer. Cochrane Database Systematic Reviews. doi: 10.1002/14651858.CD006019.pub2
Pilepich, M. V., Winter, K., Lawton, C. A., Krisch, R. E., Wolkov, H. B., Movsas, B., et al. (2005). Androgen suppression adjuvant to definitive radiotherapy in prostate carcinoma—long-term results of phase III RTOG 85–31. International Journal of Radiation Oncology Biology and Physics, 61, 1285–1290.
Bolla, M., De Reijke, T. M., Van Tienhoven, G., Oddens, J., Poortmans, P. M. P., Gez, E., et al. (2009). Duration of androgen suppression in the treatment of prostate cancer. New England Journal of Medicine, 360, 2516–2527.
McGuire, S. E., Lee, A. K., Cerne, J. Z., Munsell, M. F., Levy, L. B., Kudchadker, R. J., et al. (2013). PSA response to neoadjuvant androgen deprivation therapy is a strong independent predictor of survival in high-risk prostate cancer in the dose-escalated radiation therapy era. International Journal of Radiation Oncology Biology and Physics, 85, e39–e46.
Taplin, M.-E., Montgomery, R. B., Logothetis, C. J., Bubley, G. J., Richie, J. P., Dalkin, B. L., et al. (2012). Effect of neoadjuvant abiraterone acetate (AA) plus leuprolide acetate (LHRHa) on PSA, pathological complete response (pCR), and near pCR in localized high-risk prostate cancer (LHRPC): Results of a randomized phase II study. [abstract]. Journal of Clinical Oncology, 30, 2012 (suppl; abstr 4521).
Acknowledgments
The Drug Development Unit of the Royal Marsden NHS Foundation Trust and The Institute of Cancer Research is supported in part by a program grant from Cancer Research UK. Support was also provided by the Experimental Cancer Medicine Centre (to The Institute of Cancer Research) and the National Institute for Health Research Biomedical Research Centre (jointly to the Royal Marsden NHS Foundation Trust and The Institute of Cancer Research).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mateo, J., Smith, A., Ong, M. et al. Novel drugs targeting the androgen receptor pathway in prostate cancer. Cancer Metastasis Rev 33, 567–579 (2014). https://doi.org/10.1007/s10555-013-9472-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-013-9472-2