
Abstract
Maspin, a non-inhibitory member of the serine protease inhibitor superfamily, has been characterized as a tumor suppressor gene in multiple cancer types. Among the established anti-tumor effects of Maspin are the inhibition of cancer cell invasion, attachment to extracellular matrices, increased sensitivity to apoptosis, and inhibition of angiogenesis. However, while significant experimental data support the role of Maspin as a tumor suppressor, clinical data regarding the prognostic implications of Maspin expression have led to conflicting results. This highlights the need for a better understanding of the context dependencies of Maspin in normal biology and how these are perturbed in the context of cancer. In this review, we outline the regulation and roles of Maspin in normal and developmental biology while discussing novel evidence and emerging theories related to its functions in cancer. We provide insight into the immense therapeutic potential of Maspin and the challenges related to its successful clinical translation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The ultimate goal in cancer research is that the information gleaned from studying a specific pathway, gene, or interaction be translated for the diagnosis and prevention of cancer as well as the betterment of patient survival through more directed therapeutic approaches. Whether the latter goal is accomplished by development of a novel therapy or improvement to an existing treatment is complicated by cell and tissue context dependencies, signaling pathway interactions, and a milieu of microenvironmental influences [1–3]. Understanding the mechanisms of tumor suppressor genes and how to effectively incorporate this knowledge to the clinic has been a focus of cancer research and translational development. This approach has led to noteworthy news of therapies that have increased the survival rates of patients living with specific types of cancer.
When evaluating studies related to cancer-associated genes with the goal of translating these findings to the clinic, several key points must be considered, such as which reported effects are correlative and which are causative to the suppression of tumorigenicity and/or metastasis. More specifically, while experiments using in vivo models are essential prior to clinical trials, these data do not always reflect the clinical course of disease in humans—sometimes leading to unexpected or discouraging results. It is, therefore, a priority to continue to improve and develop in vivo models which more closely resemble and recapitulate cancer progression in humans to avoid the generation of in vivo data that are artifactual consequences of species variation [4–6].
Collectively, these complexities highlight the fact that while important paradigms have been established, cancer does not always progress in a dogmatic or linear fashion. This is frequently demonstrated by conflicting clinical data related to the expression or absence of a given gene in patient tissue. In addition, the genetic instability of human cancer, epigenetic influences, host background, and heterogeneous stromal interactions not only confounds experimental interpretation but also enables cancer to remain a moving target for therapy, often leading to resistance of effective treatments [7–10].
This review focuses on the tumor suppressor gene Maspin (mammary serine protease inhibitor; also SerpinB5), which has been shown to inhibit both tumor growth and metastasis in multiple models and cancer types [11–17], though data from the clinic have both supported and conflicted with experimental results, highlighting gaps in our knowledge of this gene across various cancers. As discussed in this review, Maspin possesses a multitude of functions that suggest great therapeutic potential including alterations to cancer cell motility, apoptosis, angiogenesis, and adhesion [11, 16, 18–27]. Delineation of these relationships and how each relates to context-specific aspects of cancer progression are keys to successful translational studies for Maspin. Equally important is the evaluation of how these effects can be legitimately implemented for patient care and treatment.
2 Key features of Maspin
Maspin is an unusual member of the serine protease inhibitor (serpin) superfamily which includes inhibitory members that target proteinases, as well as non-inhibitory members that possess a diverse array of functions. Inhibitory serpins are characterized by the ability to deactivate proteases at a 1:1 stoichiometric ratio [28, 29]. A characteristic feature of serpin structure is the reactive center loop (RCL; also referred to as reactive site loop), a flexible region exposed toward the top of the molecule. For inhibitory serpins, this loop acts as bait for target proteases. The RCL, in part, designates the protease specificity for inhibitory serpin members, dependent on the amino acid present at the scissile bond P1 site (P1–P1′) which is cleaved by the protease. Upon cleavage, the protease is trapped and the RCL is inserted into β-sheet A contained within the serpin. This reaction is non-reversible rendering the protease inactive and increasing the stability of the serpin. The associated conformational change is known as the “stressed-to-relaxed” transition. Maspin, however, contains a relatively short, hydrophobic RCL not capable of undergoing this transition [30, 31]. Inhibitory serpins also contain a highly conserved sequence proximal to the RCL known as the hinge region. This region is involved in proper structure/function relationships related to target proteases binding to the RCL and subsequent inhibition. Non-inhibitory serpins, however, often contain divergent hinge regions as is the case with Maspin [31, 32]. The Maspin hinge region is composed of amino acids with larger side chains, and the insertion site within Maspin β-sheet A exists in a closed conformation [32].To date, there have been no proteases found that are directly inhibited by Maspin through classical serpin mechanisms. Collectively, these properties place Maspin into the non-inhibitory category of the large serpin superfamily and shift the focus away from attempting to identify a target protease as explanation for the biological activities of Maspin. Rather, Maspin possesses a diversity of functions, unique structural characteristics, and several context dependencies that have made its study both complicated and interesting.
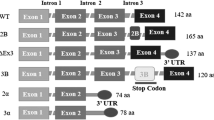
The human Maspin gene is located within a serpin cluster at 18q21.3 which includes PI8, PI10, SCCA1, SCCA2, Headpin, PAI2, and Megsin [33–36]. The gene extends just over 28 kb spanning seven exons and six introns with translation beginning in exon 2. The Maspin protein spans 375 amino acids (1,128 bp CDS) with a molecular weight of 42 kDa containing three β-sheets (A, B, C) and nine alpha-helices (A–I) [32, 37, 38] (Fig. 1). Studies using recombinant Maspin (rMaspin) demonstrated that the protein is stable at 37 °C/pH 7.0 and remains monomeric at pH 7.0 with concentrations as high as 20 μM [31, 39]. Lower pH levels (pH 5.0) induce self-association and precipitation of rMaspin, while temperatures above 40 °C result in denaturation [31]. Additionally, Maspin has been characterized from a cancer biology perspective in other species such as rat and mouse [40, 41] after isolation from cDNA libraries and shown to have 88 and 89 % sequence homology with human Maspin, respectively. As with human Maspin, these homologs are expressed in normal murine and rat mammary cells, downregulated in murine and rat cancer cell lines, and re-expression inhibited invasion, migration, tumorigenicity, and metastasis in multiple models [14, 22, 40, 41], demonstrating conservation of function among species.

2.1 Reactive center loop
Despite the RCL of Maspin having no protease inhibitory activity, it remains a vitally important component required for many of Maspin’s functional abilities including effects on invasion [11, 23, 41, 42], increased apoptosis [24, 43], and inhibition of urokinase plasminogen activator (uPA) and tissue plasminogen activator [44–46]. Ngamkitidechakul et al. reported that changing the RCL of Maspin with that of ovalbumin, a non-inhibitory serpin with homology to Maspin, imparted Maspin-like functional activities including adhesion to extracellular matrix [47]. In fact, in this study, the Maspin RCL itself was capable of inducing cell adhesion and inhibiting invasion. In contrast, however, the RCL is not required for the anti-angiogenic functions of Maspin as will be discussed later [25]. Additionally, Maspin’s RCL is sensitive to cleavage by trypsin-like proteases [42], demonstrating a mechanism for Maspin functions to be post-translationally inhibited. It is possible that the short length of the RCL may impart structure/function relationships that would be hindered by the longer RCL regions of inhibitory serpins. The precise mechanisms related to how all of these interactions are mediated by the RCL remain to be determined.
2.2 G-helix
One of the most intriguing aspects of the three-dimensional structure of Maspin pertains to the G α-helix (G-helix). Law et al. demonstrated that the Maspin G-helix is capable of an “open and closed” conformational change inducing redistribution of charged residues within the molecule [38] and suggested that this G-helix-dependent transition may mediate cofactor binding. This was the first report of conformational change in the G-helix of a serpin and highlights the unique nature of Maspin’s structure. What cellular events induce this G-helix switch, either intracellularly or extracellularly at the plasma membrane, have not yet been revealed. However, Ravenhill et al. demonstrated that Maspin-null DU145 cells transfected with Maspin containing a mutated G-helix did not possess the ability to inhibit migration compared to wild type. Maspin mutations in other regions of the protein, including the RCL, did not cause loss of inhibitory capability on migration [48]. While the G-helix on Maspin was found to be critical for the modulation of cell migration, a sequence comprised of a 15-mer peptide spanning the sequence of the G-helix (residues 236–250) was used to determine if this isolated portion of Maspin could exert the same effect. Intriguingly, the G-helix 15-mer peptide successfully mimicked full length Maspin’s ability to inhibit cell migration, demonstrating the G-helix is both essential and sufficient for this function. Furthermore, this effect was shown to involve the direct binding of the G-helix 15-mer peptide to the β1 integrin subunit resulting in the inactivation of β1 integrins (see section below).
Understanding the key structural and stability features of Maspin and how these are related to both normal and cancer biology will be important to unraveling the molecular details of how Maspin may be translated therapeutically. To address the functional implications of Maspin, this review will summarize the multiple modes of Maspin regulation and highlight its functions in both normal and cancer biology.
3 Regulation of Maspin
3.1 Transcriptional
Seminal work regarding promoter analysis, transcriptional, and cell-type-specific regulation of Maspin expression was performed by Zhang et al. These studies demonstrated that a 1-kb region upstream from the transcription start site was sufficient to induce the expression of Maspin [49]. This 1-kb region contained multiple transcriptional regulatory regions including Ets and AP1 sites, as well as a hormone response element (HRE). Through deletion analysis, it was determined that the proximal Ets site in the Maspin promoter was the primary inducer of Maspin expression, while dual activity of a downstream AP1 site increased Maspin levels synergistically compared to the Ets site alone. The activity of these elements was high in the normal mammary cell line 70N but decreased in tumorigenic and metastatic 21NT and MDA-MB-231 cell lines, suggesting that transcriptional elements may be altered during breast cancer progression and act to reduce expression of Maspin. Interestingly, Maspin was found to be regulated at the transcriptional level differently in prostate cells. While the proximal Ets site continued to serve as a positive regulator of Maspin expression, binding of the androgen receptor to the HRE negatively regulated Maspin transcription [50]. These findings highlighted both positive and negative transcriptional regulation of Maspin. Collectively, these data provide not only mechanistic information but also insight into how Maspin expression may be potentially regulated in different cells depending on the production of tissue and organ-specific hormones.
Additional research studying the Maspin promoter identified a consensus p53 site which induced expression of Maspin upon binding of p53 [51]. Following adenoviral mediated expression of wild-type p53 in Maspin-null prostate and breast cancer cell lines, Maspin expression was increased. This was also observed following induction of cellular stress and p53-responsive pathways while mutant p53 in this study failed to induce the expression of Maspin. Purified p53 protein bound to regions within the promoter from −297 bp and p53 antibody supershifted Maspin bands. In support of these data, a later study (using tissue microarray analysis) reported an inverse relationship between mutated p53 and Maspin in human tumors [52]. The implication of Maspin involvement in the p53 pathway demonstrated a potential hierarchy of tumor suppressor pathways. It also suggested that Maspin may act as an effector molecule downstream of the p53 stress-induced pathway. Interestingly, other proteins and signaling pathways related to Maspin regulation were reported that are dependent on p53. Transforming growth factor β (TGFβ) was also found to increase Maspin expression and required wild-type p53 activity [53]. This work demonstrated two p53 binding sites in the Maspin promoter that were either in close proximity or overlapped with a Smad binding element leading to recruitment of Smad2/3 and p53 to the Maspin promoter following TGFβ signaling. In addition, Smad 2/3 increased the binding of p53 to the Maspin promoter demonstrating transcriptional co-regulation. In another example, when glioblastoma cells were grown under hypoxic conditions, PTEN associated with p53 in the nucleus to induce Maspin expression [54]. Other factors regulating Maspin expression, however, have proven to be independent of p53. Expression of the antioxidant manganese superoxide dismutase in breast and prostate cells led to an increase in stability of Maspin mRNA, and this effect persisted in the presence of wild-type or mutant p53 [55]. Additionally, the activating transcription factor (ATF-2) was shown to induce Maspin expression independently of p53 by binding to a CRE-like sequence downstream of the transcription start site [56]. How all of these transcriptional regulators cooperate in different cell types and in response to various stimuli and p53 activation is unclear but further promote the idea of Maspin as a multi-functional protein tightly regulated by a variety of factors.
Other members of the p53 family, specifically the p63 isoform TAp63γ, induce expression of Maspin by binding to the same consensus p53 promoter element and can substitute for activation in the absence of p53 [57, 58]. Although mechanistic explanations for these observations can be attributed to protein homology between members of the p53 family, including similarities in the DNA binding domains and functional overlap between the proteins, the significance for regulation by both p53 and p63 is not known.
3.2 Epigenetic
Work by Domann and colleagues reported that methylation of the Maspin promoter inhibits expression and that while methylation was not present in normal human mammary epithelial cells, it was found to be common in breast cancer cell lines [59]. This methylation and reduced expression was also associated with a closed chromatin structure further reducing transcriptional accessibility. Treatment of Maspin-null cancer cell lines with 5-aza-2′-deoxycytidine resulted in the induction of Maspin expression. Furthermore, promoter methylation was found to serve as a mechanism for tissue and cell-specific expression of Maspin. Examination of cells positive for Maspin revealed unmethylated promoter regions and an acetylated histone structure in contrast to Maspin-null cells which contained methylated promoter regions and lacked acetylation of histones [60]. In support of this, additional studies showed that treatment of breast and prostate cancer cell lines with trichostatin A, a histone deacetylase (HDAC) inhibitor, led to induction of Maspin mRNA [61, 62]. Re-expression of Maspin using demethylating agents and HDAC inhibitors has been confirmed in additional studies [63–65], and in some cases, Maspin expression was only induced by trichostatin A regardless of promoter methylation status, highlighting that chromatin condensation alone may determine transcriptional activity [66, 67]. In melanoma cells, silencing of protease activated receptor-1 (PAR-1) was shown to induce expression of Maspin [68], and this increase was attributed to Ets-1 and the AP1 subunit, c-Jun, binding to the Maspin promoter. The study demonstrated that PAR-1 upregulated p38, an inhibitor of CBP/p300 (a histone acetyl transferase), thus making chromatin less accessible to the transcriptional activators. Upregulation of Maspin in these cells led to decreases in their invasive abilities by reducing MMP-2 activity, consistent with other studies examining Maspin in melanoma [27], and these results are in line with observations that PAR-1 is often overexpressed in malignant melanoma cell lines. These reports underscore the significance of chromatin remodeling and histone acetylation in addition to promoter methylation of Maspin.
Building on in vitro work in cancer cell lines, analysis of ductal carcinoma in situ (DCIS) breast cancer samples found that methylation of the Maspin promoter in neoplastic lesions was a frequent event in early stages of breast cancer [69]. Additional studies in breast cells, as well as other cancer cell types including thyroid, pancreatic, bladder, and ovarian, have confirmed the epigenetic regulation of Maspin [67, 70–79].
Taken together, these results offer further support for the development of therapies using demethylating agents and (HDAC) inhibitors. The effects of normal cellular processes must be taken into consideration, however, when using non-specific methods to induce Maspin. Interestingly, the use of ATFs targeting 18-bp regions in the Maspin promoter increased levels of Maspin in multiple breast cancer cell lines [80]. These ATFs could also induce Maspin expression in MDA-MB-231 cells which possess a methylated Maspin promoter. In fact, ATFs targeting Maspin worked synergistically with both demethylating and HDAC compounds, suggesting a benefit of co-treatment therapies [81]. Further evidence for a therapeutic approach based on these observations was demonstrated in non-small cell lung carcinoma cells. Maspin ATFs were shown to independently reprogram promoter methylation and decrease cancer cell aggressiveness in vitro and metastasis in vivo using an athymic murine model [82]. These results provide a more specific means to regulate Maspin at the transcriptional level even in cells with methylated promoters.
3.3 Nitric oxide
A link between Maspin expression and nitric oxide (NO) was discovered when experiments using MCF-7 breast cancer cells revealed that increasing NO levels in growth media by NO-donor molecules could induce Maspin expression [83]. Furthermore, transfection of endothelial nitric oxide synthase (eNOS) into eNOS-null MCF-7 cells led to increases in Maspin protein levels; however, reciprocal experiments which induced Maspin expression did not lead to eNOS induction suggesting that Maspin expression remains downstream of NO. In support of these data, acetylsalicylic acid (aspirin) has been shown to induce levels of NO. Additionally, increasing NO production through aspirin treatment increased Maspin levels in tissue and cells isolated from breast cancer patients [84] as well as plasma [85] and reduced metastasis [86]. Although the precise mechanism(s) related to how NO regulates Maspin expression remain unclear, they appear to be downstream of NO-induced cyclic GMP production [84] and are p53 dependent [87].
3.4 Tamoxifen
While both direct transcriptional and epigenetic regulation of Maspin has been established, an interesting observation has been reported for a role of tamoxifen treatment. When myoepithelial cells were treated with the estrogen receptor inhibitor tamoxifen, an increase in the secretion of Maspin resulted without a concomitant increase in mRNA levels [88]. In this study, 17β-estradiol inhibited the effects of tamoxifen suggesting that tamoxifen transcriptional events were responsible for the increase in Maspin secretion. These effects were attributed to estrogen receptor (ER)β expressed in myoepithelial cells since they do not express ERα. Inducible nitric oxide synthase also increased following tamoxifen treatment leading to higher NO levels and may have influenced Maspin expression in cooperation with tamoxifen. Further work using normal mammary epithelial and breast cancer cells demonstrated that Maspin expression could be increased by tamoxifen treatment in cells expressing ERα [89, 90]. Binding of tamoxifen to ERα was sufficient to induce Maspin promoter activity by binding in the promoter region between −97 and +87 bp containing Sp1 and Ap-1 elements [90]. However, mutation of the HRE site in the Maspin promoter decreased the effect of tamoxifen suggesting additional involvement of this transcriptional element in the tamoxifen response [89].
3.5 Fatty acids
A study by Jiang and colleagues revealed differential expression patterns of Maspin mRNA and protein expression following treatment of cancer cells with essential fatty acids (EFAs). Addition of omega-6 EFAs arachidonic acid and α-linolenic acid had no effects on the expression of Maspin, while treatment with γ-linolenic acid led to rapid increases in Maspin mRNA [91]. Consumption of γ-linolenic acid has been linked to many beneficial effects in humans including anti-inflammatory properties, reduced hypertension, and increased response to tamoxifen in breast cancer patients. Interestingly, in the same study, another omega-6 EFA, linoleic acid, resulted in decreased Maspin expression. This study highlighted specific effects of EFAs on Maspin that may have significant implications in cancer biology as well as a role for Maspin in lipid signaling and processing. Supporting this possibility, peroxisome proliferator-activated receptor-γ (PPAR-γ), a nuclear transcription factor activated in part through binding of prostaglandins and leukotrienes processed from fatty acids, increased Maspin expression in breast cancer cells [92]. Activation of PPAR-γ and upregulation of Maspin in these cells led to a more differentiated phenotype and reduced growth rates, consistent with previous reports of Maspin expression in breast cancer cells.
3.6 Post-translational
Maspin is a non-glycosylated protein [93]; however, phosphorylated forms have been identified and detected [94, 95]. Early studies using normal mammary and breast cancer cells found tyrosine phosphorylation in both endogenously expressed Maspin from mammary cells and after induction of Maspin in transfected breast cancer cells [94]. Although the kinases responsible for this phosphorylation have yet to be identified, incubation of rMaspin with the TKD38 EGFR kinase domain led to tyrosine phosphorylation in a cell-free system. The crystal structure of native Maspin suggests that the five tyrosine residues present within the molecule are not spatially available for incorporation of a phosphate group or accessible to a kinase; however, the flexibility of regions within Maspin may allow phosphorylation to occur [38]. The indication from these studies is that varying cellular or culture conditions may permit or inhibit Maspin tyrosine phosphorylation. More recently, serine and threonine phosphorylation sites have been identified on Maspin secreted from cornea cells using a mass spectrometry approach [95]. Unlike normal mammary cells, tyrosine phosphorylation was not detected, suggesting that the pattern of Maspin phosphorylation may potentially be due to cell-type-specific kinases and phosphatases. While much work remains to identify and demonstrate the relevance of these novel phosphorylation sites on Maspin, this is an area of investigation likely to aid in our molecular understanding of how Maspin is regulated and functions.
In addition to phosphorylation studies, Maspin also contains eight cysteine residues; however, intramolecular disulfide bonding had not previously been observed nor predicted from Maspin crystallography evidence [38]. Nonetheless, when MCF10A cells (a non-tumorigenic breast cell line) were grown under oxidative stress, Maspin adopted an oxidized, disulfide-bonded structure when analyzed under non-reducing conditions [96]. In this state, Maspin was no longer able to bind glutathione S-transferase (GST), a binding partner for Maspin, which suggested a potential difference in protein functionality. While the significance of this novel Maspin structure remains unknown, it may possess a role in cellular response to oxidative stress.
4 Role in normal biology
To understand the role of how a tumor suppressor functions in cancer, it is often helpful to understand its role in non-pathologic conditions and cellular processes. While many aspects related to Maspin’s functions under normal conditions have yet to be fully elucidated, the biological function(s) of Maspin in a few normal and developmental states have been characterized and are briefly described below.
4.1 Mammary gland
Since Maspin was initially discovered to have tumor suppressing activity in human mammary epithelial cells [11], initial efforts were made to understand its function in mammary biology. Murine Maspin was further evaluated in the developing mammary gland during pregnancy by placing it under the control of the whey acidic protein promoter in a transgenic model [97]. This promoter is activated within mammary cells during mid-pregnancy and early lactation, allowing evaluation of the effects of Maspin upregulation during these stages. Higher expression of Maspin resulted in abnormal development of the mammary gland as evidenced by alveolar defects including fewer and smaller alveolar structures, in addition to closed lumens. This also resulted in reduced milk protein production. Altered rates of apoptosis and proliferation of mammary cells were also noted at various stages during pregnancy and lactation compared to wild-type mice. Since these effects were noted when Maspin expression increased, this study provided primary evidence that Maspin levels are finely tuned within the mammary gland during pregnancy, and perturbations result in severe complications.
Additional support for the role of Maspin in mammary gland morphogenesis stems from the discovery that Maspin binds to a member of the interferon regulatory factor (IRF) family, IRF6. This association was first discovered using a yeast two-hybrid system to identify Maspin-interacting proteins [98]. RNA and protein expression analysis of IRF6 in human cancer cell lines exhibited similar patterns to that of Maspin—expression was reduced or absent in tumorigenic and aggressive cancer cells while strongly expressed in immortalized normal mammary cells. Data from in vitro studies were strengthened when staining of normal and invasive human breast cancer tissue revealed strong expression of IRF6 in normal, but not invasive samples. Interestingly, IRF6 existed in a phosphorylated and non-phosphorylated state, and Maspin predominantly immunoprecipitated with the phosphorylated form of IRF6. While the precise role(s) of IRF6 are largely not well understood, in quiescent cells IRF6 is phosphorylated via an unknown kinase in response to growth signals which subsequently target IRF6 to the proteasome [99]. This suggests that IRF6 is downregulated during the cells’ entry into the G1 phase of mitosis. Expression of IRF6 in IRF6-null breast cancer cell lines reduced proliferation, and this effect was further increased when Maspin was co-expressed, suggesting that IRF6 and Maspin may cooperate to maintain cells in a G0 differentiated phase. While the significance of Maspin interacting with the phosphorylated form of IRF6 is still being determined, it is known that this association is localized to the cytosol and Maspin may sequester a portion of IRF6 from nuclear translocation and/or proteosomal degradation. When mammary glands from C57/Black6 mice were examined for Maspin and IRF6 expression during pregnancy and lactation, IRF6 was expressed at lower levels during pregnancy and significantly upregulated during lactation, followed by progressive loss through involution. By comparison, Maspin expression was higher during pregnancy and late involution with both Maspin and IRF6 being strongly expressed at lactation. During this differentiation phase, polarized lobuloalveolar cells exhibit localization of IRF6 to the luminal compartment while Maspin remained throughout the cell [100]. IRF6 was also found secreted into the milk, although the significance of this is not clear. These studies demonstrated that similar to Maspin, IRF6 expression is tightly regulated during pregnancy, parturition, and involution. However, since the expression and cellular localization patterns of Maspin and IRF6 do not overlap at all stages, both independent and cooperative roles of these genes and their respective proteins may exist during mammary morphogenesis.
4.2 Development
Important additional information concerning the role of Maspin in normal cellular processes was revealed when Maspin knockout and heterozygous mice were developed and characterized in detail [101]. Maspin knockout proved to be embryonic lethal, which was not reported in other serpin knock out models, demonstrating a crucial role for Maspin in early embryonic development. It was found that Maspin −/− embryos successfully implanted into the uterine wall; however, these embryos died shortly afterward. Characterization of the embryos in vitro revealed that trophoblast differentiation was not affected by the absence of Maspin, but there were significant defects in the outgrowth of the inner cell mass. When embryonic stem cells from both normal and Maspin knockout mice were induced to form embryoid bodies, severe deficits were noted in lumen formation and overall structural organization in the absence of Maspin. These effects were partially rescued by transduction with a Maspin-expressing adenovirus. This work helped to define a critical role for Maspin in development, specifically at the stage of embryonic ectoderm formation. Combined with the roles for Maspin in mammary gland morphogenesis, it is clear that Maspin is involved during specific stages of early development and transiently during pregnancy. However, since Maspin remains expressed in multiple adult cell types (such as myoepithelial cells of the breast), Maspin is not strictly an embryonic gene, but rather possesses diverse, context-dependent functions maintained under tight regulation throughout the life of the organism.
Maspin expression has also been evaluated in the developing human placenta. Following blastocyst implantation into the uterine wall, cytotrophoblast cells from the inner trophoblast layer invade the uterine epithelium, anchoring the developing blastocyst and remodeling maternal arteries in this region. These events are part of normal placental development, and invasion of cytotrophoblasts during this process is regulated throughout pregnancy. Changes in Maspin expression within the placenta were observed and increased progressively from first to third trimester with localization to the cytotrophoblasts. These observations correlated to the invasive in vitro qualities of subsequently isolated cytotrophoblasts in that cells isolated in later terms expressed higher levels of both Maspin mRNA and protein and exhibited decreased invasive capabilities, similar to later stages of pregnancy in vivo [102]. When rMaspin was applied to isolated cytotrophoblasts in vitro, invasion of these cells was significantly decreased. It was later determined that this pattern of Maspin expression in the placenta was regulated at the chromatin level through modulation of histones, rather than changes in promoter methylation [67, 103]. These data suggest that Maspin expression is temporally regulated throughout pregnancy to control the invasion of cytotrophoblasts during placental development, providing the first evidence for a developmental role of Maspin in humans; however, additional studies are warranted to delineate a more comprehensive analysis of Maspin’s role throughout developmental stages.
4.3 Non-epithelial functions
While early research on Maspin focused on its role in epithelial cells, work from the laboratory of Sally Twining was the first to demonstrate that Maspin was produced by cells of non-epithelial origin. Examination of cells from human cornea revealed that Maspin is expressed in corneal epithelial and endothelial cells, as well as keratocytes [19], specialized fibroblasts present within the corneal stroma. Application of exogenous rMaspin increased stromal cell attachment to multiple proteins found within the extracellular matrix, including fibronectin, laminin, and collagen types I and IV [19]. This work was followed by observations showing both corneal epithelial and stromal cells secrete Maspin, and during the transformation of stromal keratocytes into wound healing fibroblasts and myofibroblasts, expression of Maspin is downregulated through epigenetic mechanisms [65]. Collectively, this work has led to a wound healing model within the cornea—stromal cells near an injured location decrease their production of Maspin in order to migrate to the wounded area. Epithelial cells adjacent to the wound deposit Maspin into the extracellular environment, and this pre-deposited Maspin leads to increased attachment of stromal cells as the wound is repaired. Indeed, this model supports remarkable regulation of multiple cell-type-specific functions that are, in part, coordinated through the dynamic regulation of Maspin in response to local environmental cues.
5 Maspin and cancer
5.1 Subcellular localization, clinical significance, and mutations
Numerous reports have demonstrated that Maspin protein can be found localized to the cytoplasm and within the nucleus. As will be discussed later, this nuclear/cytoplasmic localization of Maspin has prognostic implications for how various types of cancer progress. Maspin has also been reported by multiple research groups to be secreted and localized at the plasma membrane where it interacts with other membrane proteins [11, 65, 104–108]. An initial report using human mammary epithelial cells (HMEC) showed by multiple approaches that Maspin was predominantly localized within the cytoplasm, with small amounts found in the endoplasmic reticulum and Golgi as well as on the plasma membrane. When the 45 hydrophobic amino acids at the N-terminal were removed, Maspin was no longer found within the Golgi, suggesting the amino-terminus of Maspin may be involved in trafficking of Maspin to the membrane. Khalkhali-Ellis and Hendrix readily detected Maspin secreted from HMEC in culture media, and Maspin was deposited in the extracellular matrix by these cells over the course of 1–2 weeks [109]. In another study, H460 lung carcinoma cells were treated with γ-irradiation to induce a p53 stress response. Upregulating p53 resulted in the secretion of multiple proteins into the culture media, including Maspin, without the induction of apoptosis [110]. However, the observation that Maspin can be secreted has recently been challenged. Using MCF10A breast cells and RWPE-1 prostate cells (both of which express endogenous Maspin), Teoh et al. demonstrated that Maspin was not detected at the cell surface via immunofluorescence or biotinylation techniques [93]. Secreted Maspin was also not detected in growth media of these cells, although media was not concentrated as in previous reports. Interestingly, when a hemagglutinin signal sequence was added to Maspin to facilitate secretion, Maspin was found retained within the endoplasmic reticulum, signifying that Maspin is not capable of progressing through the classical secretory pathway. These results suggest that Maspin is restricted to functioning intracellularly and that the reported effects at the plasma membrane are indirect. Whether these discrepancies represent differences between cell types, detection methods, or whether Maspin is secreted and/or localized to the plasma membrane via non-traditional trafficking pathways remains to be conclusively defined.
While a substantial amount of in vitro and in vivo data provides mechanistic support for the description of Maspin as a tumor suppressor, the clinical data related to Maspin expression in tumors has been challenging to interpret. It has become clear that Maspin expression may serve as a negative prognostic marker in some cancers; however, Maspin expression alone may not predict outcome or tumor progression. Many of the early clinical observations reported for Maspin in breast cancer supported the concept that Maspin is a tumor suppressor in breast tissue. Analysis of primary breast tumors from patients comprising multiple histological types demonstrated less invasion of Maspin-expressing cells from the primary tumor into surrounding stroma [111], characteristic of the numerous reports of Maspin inhibition of invasion and motility. These tumor tissues also exhibited less tumor vasculature when Maspin expression was present, supportive of Maspin’s anti-angiogenic effects, which was confirmed in other studies examining microvessel density and vascular endothelial growth factor (VEGF)-A expression [112, 113]. In another study, Maspin protein expression was found to progressively decrease from normal tissue to DCIS, to invasive carcinoma, and finally, metastasis to the lymph nodes [114], in agreement with earlier reports that Maspin expression is lost in aggressive breast cancers. Other data using reverse transcriptase polymerase chain reaction methods demonstrated that breast tumors with lower Maspin expression correlated with reduced disease-free survival and higher metastatic rates [115]. Still more studies provided evidence that Maspin expression led to better outcomes and increased disease-free survival [116–118]. However, it was also determined that these supportive clinical observations for Maspin as a tumor suppressor did not always hold true. In a study of patients with invasive ductal carcinoma who were followed over the span of 1–10 years, expression of Maspin correlated with increased relapse and decreased overall survival, in addition to increased tumor size and histologic grade [119]. This decrease in relapse-free survival related to Maspin expression has been supported by additional studies in breast cancer [120, 121]. Umekita and Yoshida provided evidence in invasive ductal carcinoma of the breast that Maspin expression was associated with a more aggressive phenotype [122]. Further studies have also correlated Maspin expression with poor prognosis and malignant cellular characteristics in breast cancer [123–125].
Conflicting data regarding Maspin expression on clinical outcomes and prognostic implications are not limited only to breast cancer, and confounding reports exist in cancers including thyroid [126–128], gastric [129–138], and colorectal [139–143]. Additionally, while space limitations do not allow for a full discussion of all clinical observations regarding Maspin expression, much data are evident both in support [27, 144–167] and against [73, 168–186] the tumor suppressive implications of Maspin in patient tissue, while still other studies have found no associations [187–189]. Based on this, it is clear that significant discrepancies exist within the literature relating to the therapeutic implications of Maspin expression. It is important to remember that the overwhelming majority of in vitro and in vivo experimental data using Maspin re-expression or rMaspin treatment validate the inhibition of cancer cell aggressiveness by the Maspin protein. While new data are emerging, there is currently a paucity of experimental results demonstrating that Maspin expression imparts characteristics which increase the aggressive phenotype in cancer cells. Only when Maspin expression is examined in clinical samples within the milieu of host and stromal influences, additional mutations, and other variable factors, do discrepancies between reports most often become apparent. The presence or absence of Maspin in tumor samples is informative and important; however, other parameters must be considered as will be discussed below.
One area of research which appears to address some of these disparate observations examines the nuclear localization of Maspin which may result in different patient outcomes for certain types of cancer. This topic has recently been covered in an excellent review [190] and will only briefly be discussed here in the context of breast cancer. Mohsin et al. provided evidence that when Maspin localizes to the nucleus in invasive breast cancer tissue samples, there was an association with good prognostic factors, including the expression of estrogen and progesterone receptors [191]. When Maspin was localized solely to the cytoplasm, markers related to poor prognosis were observed, including S-phase fraction, aneuploidy, estrogen, and progesterone receptor negativity. Recently, strong experimental evidence from Goulet et al. demonstrated that nuclear localization of Maspin was essential for the inhibition of tumor growth and metastasis in breast cancer cells [192] and that the presence of Maspin within the nucleus alters expression of key genes involved in tumor progression [190, 192]. To further confirm the importance of nuclear localization of Maspin in breast cancer, a recent report demonstrated that Maspin is frequently expressed in triple negative basal-like breast cancers correlating with high histologic grade. Maspin protein expression in these samples was predominantly localized to the cytoplasm and not the nucleus [185]. Clinical data from other cancers also support the role of nuclear Maspin as an indicator of better prognosis or improved survival [138, 163, 193–202]. These data regarding cellular localization of Maspin and its subsequent differential roles provide possible explanations for why there is a disparity between clinical data associated with Maspin tumor suppressive functions in breast cancer. The manner in which Maspin translocates to the nucleus, under what conditions, and what biological functions are associated with differing localizations (cytoplasm, nucleus, membrane) are still being examined in both normal and cancer biology. It is important to note, however, that many of the early mechanistic studies that clearly defined changes in tumor characteristics with Maspin expression did not show a dependence on Maspin’s localization to the nucleus and therefore is not required for all of Maspin’s functions. Furthermore, while better prognostic outcomes regarding nuclear Maspin are supported in multiple cancers, nuclear localization of Maspin was linked to angiogenesis in lung carcinoma [203] and in colorectal cancer was associated with higher tumor grade [204] and shorter overall survival [141, 205], although it should also be mentioned that nuclear Maspin led to a better response to 5-flurouracil therapy in stage III colon cancer [205]. These reports once again highlight cell and tissue context differences related to patient outcomes as a consequence of Maspin expression and localization.
While Maspin has been considered a class II tumor suppressor gene, variations in human Maspin DNA have been reported since the original published sequence from mammary cells. Whether these changes represent somatic mutations in the Maspin gene or polymorphisms in the germ line have not been definitively determined. Conservative changes were noted in Maspin isolated from human prostate tissue and revealed an isoleucine to valine change at amino acid 66 (I66V) and leucine to valine at 187 (L187V) [40]. The I66V sequence change has also been reported in corneal cells [19], and we have observed both I66V and L187V in the human melanoma cell line C8161 (unpublished observation). In addition, Maspin isolated from prostate cancer cell lines contained an I319V variation [40]. These changes, however, all represent substitutions of branched chain amino acids (Leu, Ile, Val), and therefore, predicted structural changes to the Maspin protein would be minimal. These branch chained differences are also observed when comparing the sequence of other serpins, such as equine leukocyte elastase inhibitor and ovalbumin, that share sequence similarity with human Maspin as well as Maspin isolated from other species, including mouse and rat.
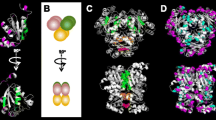
In contrast to this, a less conserved sequence change has been reported within exon 5 of Maspin at amino acid 176 constituting proline to serine (P176S). The differences between these amino acids suggest a higher possibility of alterations to the Maspin molecule that may affect functionality. Supporting this, Jang et al. showed that when Maspin containing serine at 176 was expressed in cancer cells, this resulted in decreased apoptosis, increased colony formation, and higher rates of tumorigenesis in a murine model when compared to proline at 176 [206]. Serine 176 was prevalent in both gastric and lung cancer cell lines and strikingly in this study was found in 89 % of gastric cancer tissue samples that expressed Maspin protein. This proposed the possibility that the P176S change may be involved in gastric cancer progression or may indicate a higher risk of gastric cancer incidence, although this remains to be determined. In addition to the original report in human cells, mouse, rat, and chicken also contain proline at this location. As Jang and colleagues pointed out, it is worth noting that the two reports for the crystal structure of Maspin contained different sequences, Val 66, Ser 176, Val 187 (in addition to eight cysteines mutated to serine for structure determination purposes) [32] and Ile 66, Pro 176, Leu 187 [38], the latter of which corresponds to the originally reported sequence in mammary cells. Importantly, comparison of the structures revealed changes within the G-helix. To address the effects of the P176S change on the inhibition of cancer cell invasion through three-dimensional matrices, a hallmark of Maspin function, we used recombinant forms of Maspin containing these sequence differences to evaluate the effects on cancer cell invasion through a collagen/laminin based matrix. While Pro 176 rMaspin inhibited invasion as previously reported, Ser 176 rMaspin surprisingly reversed the well-known inhibitory capability of rMaspin on invasion (Fig. 2), implying functional differences between these two recombinant forms. These results provide further experimental evidence for the sequence of Maspin related to its functionality in cancer biology. It may also provide reconciliation of clinical data in which sequence has not been examined.

Representative data from invasion experiments using rMaspin with proline or serine at amino acid 176. MDA-MB-231 were pre-treated with 30 μg/ml rMaspin containing a proline 176 or serine 176 point mutation for 24 h. Cells were then placed in a human laminin, collagen IV, gelatin matrix invasion chamber containing 10 μM pores for 24 h with additional rMas treatment (30 μg/ml). Cells invading through the matrix were counted, and percent invasion was compared to control. Proline 176 rMaspin inhibited invasion of cells as previously described. Serine 176 rMaspin lost the ability to inhibit invasion (error bars represent standard deviation, *p < 0.05)
Finally, Maspin expression alone is not indicative of its functional activities within a cell. For instance, Testisin, a serine protease often overexpressed in malignant cells, was shown to directly bind Maspin [207]. This association led to decreased ability of Maspin to induce cell death pathways through activation of caspase-3 in cervical cells. This binding also reduced Maspin’s effect on the inhibition of cancer cell invasion. Thus, the presence of Maspin protein expression within a cell does not necessarily indicate activity. The remainder of this review will focus on the molecular mechanisms of Maspin function that have been established and validated both in vitro and in vivo in cancer biology and will highlight the implications that many of these effects have on potential future therapies and how they may be applied in the clinic. The work of many research groups that have characterized Maspin as a tumor suppressor and as a negative indicator has greatly expanded our knowledge of Maspin in a relatively short time. Collectively these studies have sought to bring to fruition a therapeutic application for Maspin as well as increase our understanding of its role in cancer and its progression in various cell types.
5.2 Inhibition of cellular motility
One of the most consistent effects that Maspin exerts on cancer cells, whether endogenously expressed or exogenously applied, is the inhibition of cancer cell migration and invasion. These effects were first demonstrated by transfecting Maspin-null MDA-MB-435 breast carcinoma cells with a Maspin-expressing plasmid (for a discussion on the origin of the MDA-MB-435, please see [208]). When these transfectants were grown in Matrigel to recapitulate the three-dimensional microenvironment, the ability to invade was significantly inhibited [11]. When Maspin neutralizing antibodies were added to the culture conditions, the transfected cells regained their ability to invade, suggesting that Maspin may act at the plasma membrane, inducing changes that affect cytoskeletal networks. These observations on invasion were confirmed by other reports which utilized both breast and prostate cancer cells [23, 42]. Using time-lapse photomicroscopy, the migration of Maspin transfected cells in two-dimensional cultures was also reduced [11, 23], further supporting a role for Maspin on the inhibition of cell motility. The murine ortholog of Maspin was shown to possess similar characteristics by reducing both the migratory and invasive potentials of murine mammary tumor cells [41], and this was later corroborated using an orthotopic murine model. TM40D mammary tumor cells were implanted into the mammary fat pads of BALB/c mice, and the resulting tumors were highly invasive into surrounding tissue. These cells were also efficient in metastasizing to intestine and lung. When TM40D cells expressed Maspin, the tumors were less invasive and remained encapsulated within the primary tumor with no signs of metastasis [22]. In addition to this, re-expression of Maspin was also capable of inhibiting epidermal growth factor induced epithelial-to-mesenchymal transition in the esophageal cancer cell line EC109 [209].
Molecular data investigating Maspin effects on migration reported that treatment of MDA-MB-231 cells with rMaspin led to decreases in the activity of Rac1 and Cdc42, GTPases that are involved in cytoskeletal rearrangement and cell migration [210]. Confirming this effect, downstream signaling of Rac1 and Cdc42 through JNK activity was decreased following the addition of rMaspin to the culture media. These data help to provide mechanistic insight to the regulation of cellular motility by Maspin protein.
It is also interesting to note that within the context of breast tissue, Maspin is most strongly expressed in myoepithelial cells [11, 104, 211, 212], which have been proposed as a barrier for tumor growth [213–217]. Maspin’s ability to inhibit the invasive and migratory potential(s) of cancer cells has significant implications when considering the multi-step progression of the metastatic cascade. Cancer cells must migrate away from a primary tumor where they invade surrounding tissues or intravasate into nearby blood or lymphatic vessels, survive transport, and extravasate to distant tissue environments where they adapt and proliferate into a new tumor mass. Each step of the metastatic cascade is rate limiting, and thus, reducing the ability of tumor cells to invade local stroma and intravasate/extravasate holds significant potential to inhibit the process of metastasis. More extensive mechanistic data related to Maspin inhibition of cancer cell adhesion and invasion is discussed below.
5.3 Integrins
How tumor cells interact with their extracellular environment is a primary focus in studies of cancer progression and metastasis, and integrins—a family of transmembrane glycoproteins—have been shown to play a key role in the invasive and metastatic processes. Integrins are heterodimeric glycoproteins containing non-covalently linked α and β subunits with each subunit containing a single membrane-spanning helix and carboxy-terminal cytoplasmic domain of variable size. The large, extracellular segments contain the ligand binding sites composed of the N-terminal domains of the α and β subunits, and in general, the ligand specificity of the integrin is determined by the α subunit. While ligand-specific signals are conveyed to the cell via the cytoplasmic tail of some α subunits, ligand-independent clustering of integrins to focal adhesion sites results in their organizing at the ends of actin filaments where they associate with the proteins vinculin, talin, α-actinin, and kindlin through the cytoplasmic tail of the β-subunit. There are presently 18 α and 8 β subunits identified in vertebrates that can assemble into 24 different receptors with various binding properties in diverse tissues. Historically, integrins fall into three groups based on similar chain structures and/or ability to recognize similar protein or adhesion motifs: the β1-containing integrins, the β2-containing integrins, and the β3-(αv)−containing integrins. The integrin–ligand combination may be further clustered into four main classes based on their molecular interactions: All five αv integrins, two β1 (α5, α8) and αIIbβ3 integrins recognize ligands with an RGD tripeptide active site (e.g., found in fibronectin); the four members of the β2-subfamily, α4β1, α4β7, α9β1, and αEβ7 integrins, all recognize and bind to an acidic motif “LDV” (or its structural homolog) that is functionally related to RGD; the α1β1, α2β1, α10β1, and α11β1 integrins form a distinct laminin/collagen-binding subfamily; and α3β1, α6β1, α7β1, and α6β4 are highly selective laminin receptors [26, 218, 219].
Based on earlier observations that mammary carcinoma cells transfected with Maspin were significantly less invasive in vitro and less metastatic in nude mice [11] and that addition of rMaspin to human breast and prostate cancer cells acts at the cell membrane to inhibit invasion and motility [23], work from our laboratory was the first to examine whether integrins participate in the response of human breast carcinoma cells to exogenous rMaspin and/or in cells induced to express Maspin. Our work demonstrated that addition of rMaspin to human breast carcinoma cells resulted in an increase in the α5β1 (RGD or fibronectin receptor) and α3β1 (laminin receptor) integrins, decreases in the α2β1 (laminin/collagen receptor), α6-containing (laminin receptor) and αv-containing (RGD or fibronectin receptor) integrins, and an overall decrease in β1-containing integrins. The increase in expression of the α5β1 integrin was corroborated by Northern blot analysis that showed an increase in the cellular levels of the α5-subunit mRNA. Coincident with these changes was an increase in the cells’ adhesiveness to fibronectin (which could be abrogated by the addition of a function blocking antibody to the α5β1 integrin), together with little-to-no change in the cells’ adhesiveness to laminin, vitronectin, collagen IV or collagen I, and a decrease in the cells invasiveness through either a fibronectin- or laminin-enriched gelatin matrix in vitro. These studies further substantiated a prior observation that addition of an RGD peptide (known to block integrin function) could competitively reverse the effects of exogenously applied rMaspin. Phenotypically, rMaspin-treated cells assumed a more epithelial-like phenotype (compared to the fibroblastic phenotype of the untreated cells) and demonstrated an increase in the distribution of the α5β1 integrin on the cells. Transfecting the cells with Maspin resulted in a decrease in their invasive potential in vitro and resulted in a more epithelial-like phenotype with a broader distribution of the α5β1 integrin throughout a more extensive filopodial and lamellipodial network of projections. A change was also seen in the urokinase-type plasminogen activator receptor (uPAR) distribution pattern from a clustered to more uniform distribution in the perinuclear region of the cells [26, 220].
Since prior work from our laboratory showed that ligation and perturbation of integrins can result in changes in the activity and expression of matrix metalloproteinases that are involved in tumor cell invasion and metastasis, we also examined whether rMaspin could alter the cells’ expression and extracellular levels of matrix metalloproteinase-2 (MMP-2) coincident with the changes we saw in their integrin expression. We found that treating cells with rMaspin and plating them on a fibronectin-enriched gelatin matrix (compared to a matrix enriched with laminin, collagen IV, collagen I, or just Matrigel) resulted in a decrease in MMP-2 (pro-enzyme) activity in the conditioned medium as well as a decrease in the cellular level of MMP-2 mRNA. Furthermore, we found that addition of the RGD blocking peptide (but not an RGE non-blocking, control peptide), abrogated the decrease in MMP-2 activity which indicated that Maspin can modulate the expression of MMP-2 through an integrin signaling pathway, most probably via the α5β1 integrin [26].
While subsequent studies of Maspin and integrin interactions that followed this original research have made significant advances in our understanding of how Maspin functions via the integrins in normal compared to disease states, it can be noted that there appears, at least in some cases, to be a loss of a basic and underlying concept that integrins function as heterodimeric glycoproteins with the alpha subunit being a key component in ligand specificity leading to integrin function and activity (specifically, please see [222] and discussion below). While there is a substantial and growing body of research that focuses on a “β1 integrin,” in actuality, this “integrin” could be anywhere from one up to a combination of 12 different specific integrins with markedly different interactions and activities based on their α-subunit. In this regard, a first step to any study should be a profiling of which integrins are expressed by the cell line used in the study, as well as a clear understanding of what extracellular matrices and molecules are relevant to the cell line and particular condition(s) pertinent to the study.
A report that followed our original work used the non-transformed, immortalized human mammary epithelial cell line, MCF-10A (which expresses high levels of endogenous Maspin), and showed that addition of rMaspin acts at an early stage of cell adhesion at the cell surface through its association with an integrin belonging to the β1-subfamily of integrins (most probably α3β1 (termed “β1 integrin”), since MCF-10A cells deposit and predominantly adhere via the α3β1 integrin to an extracellular matrix composed mainly of laminin-5 [221]). This study found that Maspin physically and functionally associates with a “β1 integrin” where it co-localizes on the cell surface. Since Maspin was seen to also associate with cytoskeletal elements, it was suggested that Maspin may form part of a supramolecular structure of adhesion plaques. However, contrary to previous results from other laboratories working with tumor and stromal cells that showed Maspin’s effects are dependent on the RCL domain (essential for Maspin-mediated inhibition of cell invasion and migration), this study showed that treating MCF-10A cells with rMaspin increased their adhesiveness without involving the RCL. In this case, the rapid and increased adhesion of these cells in response to rMaspin was shown to be facilitated by amino acids 139–225 in the Maspin molecule. It is interesting to note that while an anti-“β1 integrin” [anti-β1-subunit] antibody could decrease the cell adhesiveness if the cells were first incubated with the antibody, this effect was nullified when the cells were incubated with rMaspin prior to the antibody treatment. In relation to our original work, it must be pointed out that the cells used in this study were not tumor cells, they normally express high levels of Maspin, they were grown and manipulated on their own deposited extracellular matrix (composed primarily of laminin-5), and rMaspin was added to an environment already rich in Maspin. Nonetheless, these different approaches arrived at a similar conclusion that Maspin can interact and act through integrins to affect how cells respond to their environment.
More recent work has extended these studies with MCF-10A cells and demonstrated that Maspin can form a bridge between uPA and its receptor uPAR with a “β1 integrin” (probably α3β1, see above) to form a mega-complex that regulates mammary epithelial cell adhesion [107]. Using competition peptides and mutation analyses, this study showed that two regions of Maspin, amino acid residues 190–202 and 260–275, facilitate the increase in MCF-10A cell adhesion in response to rMaspin. Furthermore, this work demonstrated that the uPA–uPAR complex is required for the localization and adhesion function of Maspin in MCF-10A cells.
Additional work using vascular smooth muscle cells (VSMC) demonstrated that rMaspin rapidly inhibits their ability to migrate by binding specifically to the surface of VSMC in the dedifferentiated, but not the differentiated phenotype [222]. Ligand blotting identified that of the Maspin binding proteins present, it was the integrin β1-subunit that was differentially expressed between the two VSMC phenotypes and co-immunoprecipitation with Maspin identified both the α3β1 and α5β1 integrins as these differentially expressed Maspin binding partners. Maspin affinity chromatography confirmed these integrins as Maspin binding partners in HT1080 cell lysates and direct binding of Maspin to the α5β1 integrin was shown using a recombinant α5β1-Fc fusion protein. Furthermore, rMaspin binding to VSMC was shown to lead to a decrease in the integrin activation state using a conformation-dependent anti-β1 antibody. More specifically, the functional involvement of the α5β1 integrin in binding rMaspin and inhibiting CHO cell migration was demonstrated in cells that were transfected with and overexpressed the human α5-integrin subunit, but not in cells lacking α5-subunit expression, and showed that the inhibition of CHO migration by rMaspin requires the α5-integrin subunit. This work clearly demonstrated that Maspin engages a limited number of integrins in specific interactions on VSMC which leads to the inactivation of these integrins and an inhibition of VSMC migration.
rMaspin has also been shown to affect endothelial cell adhesion and migration via an integrin signal transduction pathway [106]. Blood vessel endothelial cells and human umbilical vein endothelial cells (HUVEC) have been shown to express Maspin, and rMaspin enhanced HUVEC adhesion to various matrix proteins. The work in this study demonstrated that this effect depended on the activation of β1-containing integrin(s) vs. the work discussed above with VSMC where rMaspin’s interactions with specific integrins resulted in their inactivation and an inhibition of VSMC migration [222] and resulted in changes in the distribution pattern of vinculin and F-actin. Treating HUVECs with rMaspin increased integrin-linked kinase activity and phosphorylated FAK levels. It was also noted that HUVECs treated with bFGF to stimulate migration experienced a dramatic decrease in active Rac1 and cdc42 small GTPase levels within 30 min after the addition of rMaspin. The increase in FAK phosphorylation in HUVECs treated with rMaspin was correlated with a reduction in focal adhesion disassembly and inhibition of cell migration. Taken together, these results suggested that rMaspin inhibits HUVEC adhesion and migration through integrin signaling pathways.
5.4 Apoptosis
The initial link between Maspin and apoptosis was noted during mammary gland morphogenesis throughout pregnancy, as previously discussed, in which rates of apoptosis and proliferation were altered due to increased Maspin expression [97]. Further evidence supported a role for Maspin in apoptotic processes when Maspin transfected MDA-MB-435 breast carcinoma cells displayed increased sensitivity to staurosporine induced apoptosis [24]. However, exogenously applied rMaspin was not able to recapitulate this effect, demonstrating that endogenous expression and possible processing of Maspin was required. Additional supporting evidence using prostate cancer cells have also reported that Maspin induces apoptosis sensitivity to chemical reagents [223], as well as increased the rate of apoptosis related to inhibition of proteosomal pathways [224]. Maspin itself is not sufficient to induce apoptosis in all cell types considering the observation that Maspin protein expression is high in mammary cells without a concomitant high rate of apoptosis. Since these initial observations were made in cancer cells, it is possible that Maspin may restore apoptosis-sensitive pathways that are altered during neoplastic progression as well as coordinate the sensitivity of cells to stromal and context-dependent influences during development and mammary gland morphogenesis. In partial support of this, normal mammary epithelial cells are capable of inducing apoptosis in breast cancer cell lines in part, through secretion of Maspin [108].
The search for mechanisms linking Maspin to apoptosis sensitivity led to studies involving mitochondria. Maspin expression increased levels of the pro-apoptotic protein Bax in cancer cells with higher levels of Bax translocating to the mitochondria during chemical apoptosis induction [225]. When Bax levels were reduced, the effects of Maspin expression on apoptosis sensitivity were decreased. Additional work demonstrated that the Bax protein was stabilized by Maspin expression and not upregulated transcriptionally [226], although Maspin upregulated Bax expression and apoptosis sensitivity in the gastric cancer cell line SGC7901 [227], demonstrating dichotomy between cell types. Overexpression of Maspin in TM40D murine cancer cells induced higher rates of tumor cell apoptosis in mammary tumors. In vitro experiments employing subcellular fractionation of these TM40D cells revealed that Maspin protein was translocated to the inner mitochondrial membrane upon induction of apoptosis. This occurred through the mitochondrial permeability transition, and translocation was necessary for the increase in Maspin-induced sensitivity to apoptosis [43]. A recent paper indicates that the mechanism may involve increasing the acetylation of Ku70 [228]. Ku70 is a novel DNA repair protein that, when in a de-acetylated state, may also bind and sequester the pro-apoptotic protein Bax, which prevents its translocation to the mitochondrial membrane and activation of the apoptotic pathway. However, in the acetylated state, Ku70 dissociates from Bax leading to the induction of apoptosis, and it is thought that this may be a major mechanism by which Maspin prevents tumor development.
Clinical reports also support the observation that Maspin expression correlates with apoptosis levels when localized to the nucleus in ampullary [229], laryngeal [198], and head and neck squamous cell carcinomas [199]. However, Maspin expression appears to remain a negative prognostic indicator in colorectal cancer, and in the context of apoptosis, recent evidence has demonstrated that Maspin expression in colorectal cancer increases apoptosis resistance, rather than sensitivity [230]. In this study, prolonged treatment of colon cells to the carcinogen deoxycholate resulted in upregulated Maspin expression and apoptosis resistance. While an initial interpretation could be that Maspin is upregulated as an anti-tumor response to the carcinogenic insult, the authors demonstrated that when Maspin expression was reduced through siRNA, the cells exhibited increased apoptotic rates. These data provide initial mechanistic evidence regarding some of the clinical observations of Maspin in colorectal cancers [140, 141, 204] and highlight further evidence for the cell- and tissue-specific differential nature of Maspin function.
5.5 Angiogenesis
Definitive anti-angiogenic effects of Maspin were first demonstrated by Zhang and colleagues in endothelial cells. Increasing concentrations of rMaspin inhibited both the growth and migration of endothelial cells towards VEGF and bFGF in vitro. Furthermore, using a rat corneal pocket angiogenesis assay in which bFGF was added to avascular regions of rat corneas with or without rMaspin, neo-vasculogenesis was inhibited when rMaspin was present [25]. These data were coupled with in vivo experiments using LNCaP human prostate cancer cells in an immunodeficient xenograft model, LNCaP; tumor growth and neovascularization was reduced following rMaspin treatment. Subsequent reports using endogenous expression of Maspin corroborated the effects observed in the rMaspin studies. A chimeric bone cancer model in which human cancer cells were injected into human bone that had been implanted into scid/scid mice demonstrated that DU145 human prostate cancer cells transfected with Maspin exhibited less tumor neovascularization from murine endothelial cells compared to controls [231]. This effect was associated with a decrease in tumor growth and bone destruction. Another study demonstrated that conditioned media (CM) from Maspin-expressing human keratinocytes inhibited the ability of human endothelial cells to migrate toward angiogenic factors (VEGF, bFGF, IL8) in a dose-dependent manner [232]. When a Maspin neutralizing antibody was added to the CM, the cells resumed their ability to migrate, providing evidence for a paracrine anti-angiogenic role of Maspin. A wealth of clinical data has confirmed the inhibitory effects of Maspin on angiogenesis and microvessel density from colon, bladder, breast, laryngeal, head and neck squamous cell carcinomas, as well as melanoma [112, 142, 160, 194, 199, 233]. However, conflicting clinical data exist in ovarian carcinoma which both proposes an inhibitory as well as stimulatory role for Maspin on angiogenesis [161, 195, 234].
Li et al. provided more detailed molecular information on the anti-angiogenic effects of Maspin by using an adenoviral based strategy to induce endothelial cell expression of Maspin within tumor vasculature [235]. Following left ventricular cardiac injections of Maspin adenovirus, both mature blood vessels and developing tumor vasculature were infected by the virus. While existing blood vessels were not affected, tumor endothelial cells became leaky which suggested a breakdown in tumor vasculature. When endothelial cells were transduced with the virus in vitro, the resulting apoptosis suggested a possible mechanistic explanation for vessel leakiness, and Bax protein levels were found to be increased while Bcl-2 was reduced. Taken together, these results connect the apoptotic inductive effects of Maspin and further demonstrate the potent inhibitory capabilities of Maspin on angiogenesis as well as the therapeutic potential of Maspin for disrupting tumor vasculature. However, the role for Maspin inhibiting neovascularization under normal conditions, either during development or in adult cells, remains to be fully defined.
5.6 Protein binding partners of Maspin
While some of the direct protein–protein associations of Maspin have been discussed above, other proteins have also been shown to bind directly to Maspin within the cytosol and nucleus. Yeast two-hybrid approaches have identified possible protein–protein interactions with Maspin [98, 236, 237]. Included among these proteins are glutathione peroxidase, GST, heat shock protein 70, heat shock protein 90, and HDAC1. In particular, interactions of Maspin with GST and HDAC1 have been shown to have significant functional implications. For instance, breast and prostate cancer cells transfected with Maspin, or treated with rMaspin, exhibited increased levels of GST activity and a corresponding decrease in reactive oxygen species (ROS). This effect was reduced by either knockdown of Maspin or inhibition of GST. In addition, increasing the levels of ROS in these cells induced the interaction of Maspin and GST, suggesting that these proteins cooperate to reduce oxidative cellular stress [237]. Maspin has also been shown to decrease the activity of HDAC1 through a direct interaction and worked synergistically with HDAC inhibitors [238]. GST was found in the Maspin/HDAC1 complex, and a COOH-end truncation of the Maspin protein allowed for HDAC1, but not GST binding. When GST was not able to bind to Maspin, HDAC1 activity was not inhibited. It was determined that Maspin acts to recruit GST to this complex, and GST is required for HDAC1 inhibition. In addition, mutation of the Maspin RCL also decreased binding to GST, although not to the degree of C-terminal truncation. These studies help to highlight intricate relationships of Maspin at the protein level, possibly serving a scaffolding role in multi-protein complexes.
Yeast two-hybrid approaches continue to identify new protein binding relationships including interactions of Maspin with the RNA-binding protein KHDRBS3 [239] and FBX032 [239, 240], involved in ubiquitin protein ligase reactions. How these and subsequently discovered protein interactions with Maspin fit into our understanding of the molecular basis of Maspin function will be important.
6 Maspin and regulation of drug sensitivity
The ultimate goal of pharmacogenomics research is the prediction of patient response to drug therapy. This is specifically important in cancer treatment and would be instrumental in selecting the most effective chemotherapeutic agent tailored for individual patients. It is unfortunate that up until now, this selection remains largely arbitrary [241] and the administration of ineffective chemotherapeutic agents often results in adverse effects and diminishes the quality of life for many cancer patients.
Genome-wide expression profile analyses, such as cDNA microarray on panels of human cancer cell lines, have identified the genes associated with sensitivity to anticancer drugs [242, 243]. However, it is important to note that the majority of available breast cancer cell lines are derived from the pleural effusion of patients who have most probably undergone chemotherapy and thus developed some form of resistance. In addition, the very complex nature of the drug response in a multi-organ system compared to single cells renders the clinical application of such findings quite challenging.
Over a decade of research on tumor, suppressive properties of Maspin has resulted in valuable information on its role in the induction of chemosensitivity in different cancer cell lines. Most noteworthy, Maspin renders cancer cells more susceptible to chemotherapy-induced apoptosis. This is reported in MDA-MB-435 and MT40 (human and mouse mammary cancer cell lines, respectively) in response to staurosporine [24, 226]; in the prostate cancer cell line DU145 treated with doxazosin, or proteosome inhibitors [223, 224]; in lung cancer cell lines treated with doxorubicin and etoposide [244]; and in osteosarcoma cell lines U2OS and SAOS2 treated with doxorubicin and cisplatin [245]. Maspin’s sensitizing effect appears to be cell-specific (cancer-specific) and drug-dependent and occurs via distinct mechanisms. For example, in DU145 prostate cancer cells, application of proteasome inhibitor(s) induced Maspin expression through activation of p38MAPK, which resulted in proteasome-induced apoptosis. In lung cancer cells, Maspin-induced drug effects are prompted through the modulation of the AKT pathway. Interestingly, in osteosarcoma cell lines, the ability of Maspin to regulate the transcription factor E2F1 (and vice versa) rendered the cells, sensitive to doxorubicin and cisplatin [245].
Studies from our laboratory have also supported a significant role for Maspin in sensitizing breast cancer cells to the chemotherapeutic agent IFN-γ [246], and this effect involves the aspartyl endopeptidase cathepsin D (CatD). CatD was originally identified by a yeast two-hybrid approach as a Maspin binding partner [247], and contrary to Maspin, which is silenced in breast cancer [59], CatD is excessively produced and aberrantly secreted [248]. It exerts a mitogenic effect on cancer cells and the stroma of the tumor and thus is critical in tumor growth and extracellular matrix degradation [249].
Notably, IFN-γ has been a widely utilized chemotherapeutic agent in many types of cancer; however, the majority of breast cancer cells are refractory to this cytokine [250]. The nature of this non-conformity was studied in our laboratory and determined to be associated (at least in part) with the absence of Maspin and deregulated expression of CatD [246]. Our studies revealed that IFN-γ treatment of normal mammary epithelial cells results in reduced proliferation, changes in vacuolar pH, altered CatD processing, and the disruption of cell polarity, culminating in autophagic cell death. By comparison, breast cancer cells (which are commonly devoid of Maspin) are non-responsive to IFN-γ with respect to changes in vacuolar pH, CatD processing, and autophagy. However, Maspin transfection of these cell lines rendered them responsive to IFN-γ. Specifically, MCF-7 cells transfected with Maspin displayed disrupted cell polarity (comparable to that observed in normal mammary epithelial cells) and a modest increase in autophagy-associated gene beclin-1 when treated with IFN-γ. Based on our studies, Maspin transfection of breast cancer cell lines affects post-translational modification and changes in the processing of CatD.
Collectively, these studies suggest that Maspin-based therapeutic approaches have to be tailored to specific cancers and chemotherapeutic agents for maximal efficacy. While studies on Maspin’s role in the induction of chemosensitivity are yet at their infancy and need to be greatly expanded, detailed mechanistic analyses are required to dissect the crosstalk between Maspin and the molecules through which it works, in both a cell- and drug-specific context and under physiological conditions. Elucidation of such processes would further promote our insight into Maspin’s role in cancer development, progression and metastasis, and define another dimension in the discovery of more efficient Maspin-based therapeutic agents.
7 Human rMaspin as a viable anticancer drug candidate
A question that remains to be considered is whether rMaspin itself could be a viable anti-cancer candidate. Maspin clearly possesses different intracellular and extracellular activities, several binding partners, and different mechanisms of action depending on the cancer. Despite these data on intracellular Maspin function, it is clear that exogenously added rMaspin, or Maspin-derived peptide fragments, can act at the surface of numerous migratory cell types to suppress cell motility and movement by enhancing cellular adhesion to the extracellular matrix components laminin, fibronectin, and collagen. It is also clear that some or all of this Maspin is taken up by cells. In contrast to the intracellular pro-apoptotic activity of Maspin, these extracellular in vitro activities of Maspin appear to be cytostatic as cells regain motility and migratory activity when treatment with rMaspin is stopped. Furthermore, the extracellular effects appear to be dose-related and dependent on the nature of the rMaspin used since different results have been reported for wild-type rMaspin produced in yeast vs. GST-Maspin fusion proteins produced in E. coli. The wild-type yeast protein appears optimally active at 170 nM with decreasing activity at higher concentrations. In contrast, the GST-fusion protein is equally active at 170 nM as the wild-type yeast protein but appears to have maintained, if not enhanced inhibitory activity at higher concentrations. It is clear from these observations that these two forms of rMaspin are not the same and that fusing a GST molecule onto the N-terminal end of Maspin enhances Maspin’s inhibitory activity.
There have been as yet unpublished tumor xenograft mouse model studies performed using the breast MDA-MB-435 cancer cell line with yeast rMaspin administered intravenously to mice at different doses once tumors had been established (P. Pemberton, unpublished data). In brief, this rMaspin inhibited the growth of established breast tumors by up to 25 % compared to untreated controls but was dose-dependent and low-dose Maspin in combination with paclitaxel inhibited growth of MDA-MB-435 tumors by up to 41 %, proving superior to either Maspin or paclitaxel alone. In summary, it appears that an exogenously administered rMaspin may be a viable cancer therapeutic and has the potential to work more effectively as an adjunct therapy with certain other approved agents. Perhaps some of those agents might target the functions that intracellular Maspin normally controls, as future studies will hopefully illuminate.
8 Summary and perspectives
Our fundamental knowledge of the biological function(s) of Maspin has been significantly enhanced since its discovery in 1994. Studies related to the structure, biological activity, and translational promise of Maspin have generated new insights into the feasibility of administering rMaspin as a viable cancer therapy—either alone or in combination with other known anti-cancer agents. A summary of key, biologically relevant observations related to Maspin regulation, cellular localization, and function(s), presented in Fig. 3, offers a global perspective of the myriad effects of this unique serpin. The implications underlying the therapeutic potential of Maspin rest in the distinctive cell- and tissue-specific context of various cancers and their respective phenotypes. Indeed, the time is right to address our knowledge gaps and advance novel Maspin-based therapies based on the foundation of solid scientific discovery.

Summary of validated effects of Maspin in cancer biology. a Re-expression of Maspin or treatment with rMaspin decreases tumor growth and metastasis using in vivo models. b Cellular responses related to expression or treatment with Maspin in cancer cells
References
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144, 646–674.
Yuan, T. L., & Cantley, L. C. (2008). PI3K pathway alterations in cancer: variations on a theme. Oncogene, 27, 5497–5510.
Friedl, P., & Alexander, S. (2011). Cancer invasion and the microenvironment: plasticity and reciprocity. Cell, 147, 992–1009.
Frese, K. K., & Tuveson, D. A. (2007). Maximizing mouse cancer models. Nature Reviews Cancer, 7, 645–658.
Jacks, T. (2005). Modeling cancer in the mouse. Harvey Lectures, 101, 1–19.
Rangarajan, A., & Weinberg, R. A. (2003). Opinion: comparative biology of mouse versus human cells: modelling human cancer in mice. Nature Reviews Cancer, 3, 952–959.
Hunter, K. (2006). Host genetics influence tumour metastasis. Nature Reviews Cancer, 6, 141–146.
Anton, K., & Glod, J. (2009). Targeting the tumor stroma in cancer therapy. Current Pharmaceutical Biotechnology, 10, 185–191.
Eccles, S. A., & Welch, D. R. (2007). Metastasis: recent discoveries and novel treatment strategies. Lancet, 369, 1742–1757.
Sethi, N., & Kang, Y. (2011). Unravelling the complexity of metastasis—molecular understanding and targeted therapies. Nature Reviews Cancer, 11, 735–748.
Zou, Z., Anisowicz, A., Hendrix, M. J., et al. (1994). Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science, 263, 526–529.
Reddy, K. B., McGowen, R., Schuger, L., et al. (2001). Maspin expression inversely correlates with breast tumor progression in MMTV/TGF-alpha transgenic mouse model. Oncogene, 20, 6538–6543.
Streuli, C. H. (2002). Maspin is a tumour suppressor that inhibits breast cancer tumour metastasis in vivo. Breast Cancer Research, 4, 137–140.
Zhang, M., Shi, Y., Magit, D., et al. (2000). Reduced mammary tumor progression in WAP-TAg/WAP-maspin bitransgenic mice. Oncogene, 19, 6053–6058.
Hall, D. C., Johnson-Pais, T. L., Grubbs, B., et al. (2008). Maspin reduces prostate cancer metastasis to bone. Urologic Oncology, 26, 652–658.
Watanabe, M., Nasu, Y., Kashiwakura, Y., et al. (2005). Adeno-associated virus 2-mediated intratumoral prostate cancer gene therapy: long-term maspin expression efficiently suppresses tumor growth. Human Gene Therapy, 16, 699–710.
Shao, L. J., Shi, H. Y., Ayala, G., et al. (2008). Haploinsufficiency of the maspin tumor suppressor gene leads to hyperplastic lesions in prostate. Cancer Research, 68, 5143–5151.
Bass, R., Fernandez, A. M., & Ellis, V. (2002). Maspin inhibits cell migration in the absence of protease inhibitory activity. Journal of Biological Chemistry, 277, 46845–46848.
Ngamkitidechakul, C., Burke, J. M., O’Brien, W. J., et al. (2001). Maspin: synthesis by human cornea and regulation of in vitro stromal cell adhesion to extracellular matrix. Investigative Ophthalmology & Visual Science, 42, 3135–3141.
Abraham, S., Zhang, W., Greenberg, N., et al. (2003). Maspin functions as tumor suppressor by increasing cell adhesion to extracellular matrix in prostate tumor cells. Journal of Urology, 169, 1157–1161.
Pan, Y. L., Zheng, S., Peng, J. P., et al. (2005). Effects of microencapsulated CHO cells modified with maspin gene on the motility and adhesiveness of breast carcinoma cells Bcap37. Zhonghua Zhong Liu Za Zhi, 27, 342–346.
Shi, H. Y., Zhang, W., Liang, R., et al. (2001). Blocking tumor growth, invasion, and metastasis by maspin in a syngeneic breast cancer model. Cancer Research, 61, 6945–6951.
Sheng, S., Carey, J., Seftor, E. A., et al. (1996). Maspin acts at the cell membrane to inhibit invasion and motility of mammary and prostatic cancer cells. Proceedings of the National Academy of Sciences of the United States of America, 93, 11669–11674.
Jiang, N., Meng, Y., Zhang, S., et al. (2002). Maspin sensitizes breast carcinoma cells to induced apoptosis. Oncogene, 21, 4089–4098.
Zhang, M., Volpert, O., Shi, Y. H., et al. (2000). Maspin is an angiogenesis inhibitor. Nature Medicine, 6, 196–199.
Seftor, R. E. B., Odero, V. A., Seftor, E. A., et al. (2002). Maspin suppresses breast cancer invasiveness by modulating integrin expression and function (pp. 84–95). Georgetown: Landes Bioscience, Maspin.
Denk, A. E., Bettstetter, M., Wild, P. J., et al. (2007). Loss of maspin expression contributes to a more invasive potential in malignant melanoma. Pigment Cell Research, 20, 112–119.
Law, R. H., Zhang, Q., McGowan, S., et al. (2006). An overview of the serpin superfamily. Genome Biology, 7, 216.
Huntington, J. A. (2011). Serpin structure, function and dysfunction. Journal of Thrombosis and Haemostasis, 9(Suppl 1), 26–34.
Hopkins, P. C., & Whisstock, J. (1994). Function of maspin. Science, 265, 1893–1894.
Pemberton, P. A., Wong, D. T., Gibson, H. L., et al. (1995). The tumor suppressor maspin does not undergo the stressed to relaxed transition or inhibit trypsin-like serine proteases. Evidence that maspin is not a protease inhibitory serpin. Journal of Biological Chemistry, 270, 15832–15837.
Al-Ayyoubi, M., Gettins, P. G., & Volz, K. (2004). Crystal structure of human maspin, a serpin with antitumor properties: reactive center loop of maspin is exposed but constrained. Journal of Biological Chemistry, 279, 55540–55544.
Bartuski, A. J., Kamachi, Y., Schick, C., et al. (1997). Cytoplasmic antiproteinase 2 (PI8) and bomapin (PI10) map to the serpin cluster at 18q21.3. Genomics, 43, 321–328.
Silverman, G. A., Bartuski, A. J., Cataltepe, S., et al. (1998). SCCA1 and SCCA2 are proteinase inhibitors that map to the serpin cluster at 18q21.3. Tumor Biology, 19, 480–487.
Nakashima, T., Pak, S. C., Silverman, G. A., et al. (2000). Genomic cloning, mapping, structure and promoter analysis of HEADPIN, a serpin which is down-regulated in head and neck cancer cells. Biochimica Et Biophysica Acta-Gene Structure and Expression, 1492, 441–446.
Scott, F. L., Eyre, H. J., Lioumi, M., et al. (1999). Human ovalbumin serpin evolution: phylogenic analysis, gene organization, and identification of new PI8-related genes suggest that two interchromosomal and several intrachromosomal duplications generated the gene clusters at 18q21–q23 and 6p25. Genomics, 62, 490–499.
Fitzpatrick, P. A., Wong, D. T., Barr, P. J., et al. (1996). Functional implications of the modeled structure of maspin. Protein Engineering, 9, 585–589.
Law, R. H., Irving, J. A., Buckle, A. M., et al. (2005). The high resolution crystal structure of the human tumor suppressor maspin reveals a novel conformational switch in the G-helix. Journal of Biological Chemistry, 280, 22356–22364.
Liu, T., Pemberton, P. A., & Robertson, A. D. (1999). Three-state unfolding and self-association of maspin, a tumor-suppressing serpin. Journal of Biological Chemistry, 274, 29628–29632.
Umekita, Y., Hiipakka, R. A., & Liao, S. (1997). Rat and human maspins: structures, metastatic suppressor activity and mutation in prostate cancer cells. Cancer Letters, 113, 87–93.
Zhang, M., Sheng, S., Maass, N., et al. (1997). mMaspin: the mouse homolog of a human tumor suppressor gene inhibits mammary tumor invasion and motility. Molecular Medicine, 3, 49–59.
Sheng, S., Pemberton, P. A., & Sager, R. (1994). Production, purification, and characterization of recombinant maspin proteins. Journal of Biological Chemistry, 269, 30988–30993.
Latha, K., Zhang, W., Cella, N., et al. (2005). Maspin mediates increased tumor cell apoptosis upon induction of the mitochondrial permeability transition. Molecular and Cellular Biology, 25, 1737–1748.
Sheng, S., Truong, B., Fredrickson, D., et al. (1998). Tissue-type plasminogen activator is a target of the tumor suppressor gene maspin. Proceedings of the National Academy of Sciences of the United States of America, 95, 499–504.
McGowen, R., Biliran, H., Jr., Sager, R., et al. (2000). The surface of prostate carcinoma DU145 cells mediates the inhibition of urokinase-type plasminogen activator by maspin. Cancer Research, 60, 4771–4778.
Biliran, H., Jr., & Sheng, S. (2001). Pleiotrophic inhibition of pericellular urokinase-type plasminogen activator system by endogenous tumor suppressive maspin. Cancer Research, 61, 8676–8682.
Ngamkitidechakul, C., Warejcka, D. J., Burke, J. M., et al. (2003). Sufficiency of the reactive site loop of maspin for induction of cell-matrix adhesion and inhibition of cell invasion. Conversion of ovalbumin to a maspin-like molecule. Journal of Biological Chemistry, 278, 31796–31806.
Ravenhill, L., Wagstaff, L., Edwards, D. R., et al. (2010). G-helix of maspin mediates effects on cell migration and adhesion. Journal of Biological Chemistry, 285, 36285–36292.
Zhang, M., Maass, N., Magit, D., et al. (1997). Transactivation through Ets and Ap1 transcription sites determines the expression of the tumor-suppressing gene maspin. Cell Growth & Differentiation, 8, 179–186.
Zhang, M., Magit, D., & Sager, R. (1997). Expression of maspin in prostate cells is regulated by a positive Ets element and a negative hormonal responsive element site recognized by androgen receptor. Proceedings of the National Academy of Sciences of the United States of America, 94, 5673–5678.
Zou, Z., Gao, C., Nagaich, A. K., et al. (2000). p53 regulates the expression of the tumor suppressor gene maspin. Journal of Biological Chemistry, 275, 6051–6054.
Zhang, W., & Zhang, M. (2002). Tissue microarray analysis of maspin expression and its reverse correlation with mutant p53 in various tumors. International Journal of Oncology, 20, 1145–1150.
Wang, S. E., Narasanna, A., Whitell, C. W., et al. (2007). Convergence of p53 and transforming growth factor beta (TGFbeta) signaling on activating expression of the tumor suppressor gene maspin in mammary epithelial cells. Journal of Biological Chemistry, 282, 5661–5669.
Eitel, J. A., Bijangi-Vishehsaraei, K., Saadatzadeh, M. R., et al. (2009). PTEN and p53 are required for hypoxia induced expression of maspin in glioblastoma cells. Cell Cycle, 8, 896–901.
Duan, H., Zhang, H. J., Yang, J. Q., et al. (2003). MnSOD up-regulates maspin tumor suppressor gene expression in human breast and prostate cancer cells. Antioxidants & Redox Signaling, 5, 677–688.
Maekawa, T., Sano, Y., Shinagawa, T., et al. (2008). ATF-2 controls transcription of Maspin and GADD45 alpha genes independently from p53 to suppress mammary tumors. Oncogene, 27, 1045–1054.
Kim, S., Han, J., Kim, J., et al. (2004). Maspin expression is transactivated by p63 and is critical for the modulation of lung cancer progression. Cancer Research, 64, 6900–6905.
Spiesbach, K., Tannapfel, A., Mossner, J., et al. (2005). TAp63gamma can substitute for p53 in inducing expression of the maspin tumor suppressor. International Journal of Cancer, 114, 555–562.
Domann, F. E., Rice, J. C., Hendrix, M. J., et al. (2000). Epigenetic silencing of maspin gene expression in human breast cancers. International Journal of Cancer, 85, 805–810.
Futscher, B. W., Oshiro, M. M., Wozniak, R. J., et al. (2002). Role for DNA methylation in the control of cell type specific maspin expression. Nature Genetics, 31, 175–179.
Maass, N., Biallek, M., Rosel, F., et al. (2002). Hypermethylation and histone deacetylation lead to silencing of the maspin gene in human breast cancer. Biochemical and Biophysical Research Communications, 297, 125–128.
Ohike, N., Maass, N., Mundhenke, C., et al. (2003). Clinicopathological significance and molecular regulation of maspin expression in ductal adenocarcinoma of the pancreas. Cancer Letters, 199, 193–200.
Tang, B., Peng, Z. H., Yu, P. W., et al. (2006). Transcription regulation of 5-Aza-2′-deoxycytidine on maspin gene demethylation in RKO human colorectal cell line. Zhonghua Wei Chang Wai Ke Za Zhi, 9, 260–263.
Murakami, J., Asaumi, J., Maki, Y., et al. (2004). Effects of demethylating agent 5-aza-2(′)-deoxycytidine and histone deacetylase inhibitor FR901228 on maspin gene expression in oral cancer cell lines. Oral Oncology, 40, 597–603.
Horswill, M. A., Narayan, M., Warejcka, D. J., et al. (2008). Epigenetic silencing of maspin expression occurs early in the conversion of keratocytes to fibroblasts. Experimental Eye Research, 86, 586–600.
Fujisawa, K., Maesawa, C., Sato, R., et al. (2005). Epigenetic status and aberrant expression of the maspin gene in human hepato-biliary tract carcinomas. Laboratory Investigation, 85, 214–224.
Dokras, A., Coffin, J., Field, L., et al. (2006). Epigenetic regulation of maspin expression in the human placenta. Molecular Human Reproduction, 12, 611–617.
Villares, G. J., Zigler, M., Dobroff, A. S., et al. (2011). Protease activated receptor-1 inhibits the Maspin tumor-suppressor gene to determine the melanoma metastatic phenotype. Proceedings of the National Academy of Sciences of the United States of America, 108, 626–631.
Futscher, B. W., O’Meara, M. M., Kim, C. J., et al. (2004). Aberrant methylation of the maspin promoter is an early event in human breast cancer. Neoplasia, 6, 380–389.
Boltze, C., Schneider-Stock, R., Quednow, C., et al. (2003). Silencing of the maspin gene by promoter hypermethylation in thyroid cancer. International Journal of Molecular Medicine, 12, 479–484.
Fitzgerald, M., Oshiro, M., Holtan, N., et al. (2003). Human pancreatic carcinoma cells activate maspin expression through loss of epigenetic control. Neoplasia, 5, 427–436.
Akiyama, Y., Maesawa, C., Ogasawara, S., et al. (2003). Cell-type-specific repression of the maspin gene is disrupted frequently by demethylation at the promoter region in gastric intestinal metaplasia and cancer cells. American Journal of Pathology, 163, 1911–1919.
Sugimoto, S., Maass, N., Takimoto, Y., et al. (2004). Expression and regulation of tumor suppressor gene maspin in human bladder cancer. Cancer Letters, 203, 209–215.
Yatabe, Y., Mitsudomi, T., & Takahashi, T. (2004). Maspin expression in normal lung and non-small-cell lung cancers: cellular property-associated expression under the control of promoter DNA methylation. Oncogene, 23, 4041–4049.
Wada, K., Maesawa, C., Akasaka, T., et al. (2004). Aberrant expression of the maspin gene associated with epigenetic modification in melanoma cells. The Journal of Investigative Dermatology, 122, 805–811.
Ogasawara, S., Maesawa, C., Yamamoto, M., et al. (2004). Disruption of cell-type-specific methylation at the Maspin gene promoter is frequently involved in undifferentiated thyroid cancers. Oncogene, 23, 1117–1124.
Rose, S. L., Fitzgerald, M. P., White, N. O., et al. (2006). Epigenetic regulation of maspin expression in human ovarian carcinoma cells. Gynecologic Oncology, 102, 319–324.
Wu, Y., Alvarez, M., Slamon, D. J., et al. (2010). Caspase 8 and maspin are downregulated in breast cancer cells due to CpG site promoter methylation. BMC Cancer, 10, 32.
Kwon, Y. J., Lee, S. J., Koh, J. S., et al. (2012). Genome-wide analysis of DNA methylation and the gene expression change in lung cancer. Journal of Thoracic Oncology, 7, 20–33.
Beltran, A., Parikh, S., Liu, Y., et al. (2007). Re-activation of a dormant tumor suppressor gene maspin by designed transcription factors. Oncogene, 26, 2791–2798.
Beltran, A. S., Sun, X., Lizardi, P. M., et al. (2008). Reprogramming epigenetic silencing: artificial transcription factors synergize with chromatin remodeling drugs to reactivate the tumor suppressor mammary serine protease inhibitor. Molecular Cancer Therapeutics, 7, 1080–1090.
Beltran, A. S., & Blancafort, P. (2011). Reactivation of MASPIN in non-small cell lung carcinoma (NSCLC) cells by artificial transcription factors (ATFs). Epigenetics, 6, 224–235.
Khalkhali-Ellis, Z., & Hendrix, M. J. (2003). Nitric oxide regulation of maspin expression in normal mammary epithelial and breast cancer cells. American Journal of Pathology, 162, 1411–1417.
Girish, G. V., Bhattacharya, G., & Sinha, A. K. (2006). The role of insulin dependent NO synthesis in the impaired production of maspin in human breast cancer. Journal of Cancer Research and Clinical Oncology, 132, 389–398.
Girish, G. V., Sinha, N., Chakraborty, K., et al. (2006). Restoration by aspirin of impaired plasma maspin level in human breast cancer. Acta Oncologica, 45, 184–187.
Bhattacharyya, M., Girish, G. V., Ghosh, R., et al. (2010). Acetyl salicylic acid (aspirin) improves synthesis of maspin and lowers incidence of metastasis in breast cancer patients [corrected]. Cancer Science, 101, 2105–2109.
Lim, S., Hung, A. C., & Porter, A. G. (2009). Focused PCR screen reveals p53 dependence of nitric oxide-induced apoptosis and up-regulation of maspin and plasminogen activator inhibitor-1 in tumor cells. Molecular Cancer Research, 7, 55–66.
Shao, Z. M., Radziszewski, W. J., & Barsky, S. H. (2000). Tamoxifen enhances myoepithelial cell suppression of human breast carcinoma progression in vitro by two different effector mechanisms. Cancer Letters, 157, 133–144.
Khalkhali-Ellis, Z., Christian, A. L., Kirschmann, D. A., et al. (2004). Regulating the tumor suppressor gene maspin in breast cancer cells: a potential mechanism for the anticancer properties of tamoxifen. Clinical Cancer Research, 10, 449–454.
Liu, Z., Shi, H. Y., Nawaz, Z., et al. (2004). Tamoxifen induces the expression of maspin through estrogen receptor-alpha. Cancer Letters, 209, 55–65.
Jiang, W. G., Hiscox, S., Horrobin, D. F., et al. (1997). Gamma linolenic acid regulates expression of maspin and the motility of cancer cells. Biochemical and Biophysical Research Communications, 237, 639–644.
Mueller, E., Sarraf, P., Tontonoz, P., et al. (1998). Terminal differentiation of human breast cancer through PPAR gamma. Molecular Cell, 1, 465–470.
Teoh, S. S., Whisstock, J. C., & Bird, P. I. (2010). Maspin (SERPINB5) is an obligate intracellular serpin. Journal of Biological Chemistry, 285, 10862–10869.
Odero-Marah, V. A., Khalkhali-Ellis, Z., Schneider, G. B., et al. (2002). Tyrosine phosphorylation of maspin in normal mammary epithelia and breast cancer cells. Biochemical and Biophysical Research Communications, 295, 800–805.
Narayan, M., Mirza, S. P., & Twining, S. S. (2011). Identification of phosphorylation sites on extracellular corneal epithelial cell maspin. Proteomics, 11, 1382–1390.
Nawata, S., Shi, H. Y., Sugino, N., et al. (2011). Evidence of post-translational modification of the tumor suppressor maspin under oxidative stress. International Journal of Molecular Medicine, 27, 249–254.
Zhang, M., Magit, D., Botteri, F., et al. (1999). Maspin plays an important role in mammary gland development. Developmental Biology, 215, 278–287.
Bailey, C. M., Khalkhali-Ellis, Z., Kondo, S., et al. (2005). Mammary serine protease inhibitor (Maspin) binds directly to interferon regulatory factor 6: identification of a novel serpin partnership. Journal of Biological Chemistry, 280, 34210–34217.
Bailey, C. M., Abbott, D. E., Margaryan, N. V., et al. (2008). Interferon regulatory factor 6 promotes cell cycle arrest and is regulated by the proteasome in a cell cycle-dependent manner. Molecular and Cellular Biology, 28, 2235–2243.
Bailey, C. M., Margaryan, N. V., Abbott, D. E., et al. (2009). Temporal and spatial expression patterns for the tumor suppressor Maspin and its binding partner interferon regulatory factor 6 during breast development. Development, Growth & Differentiation, 51, 473–481.
Gao, F., Shi, H. Y., Daughty, C., et al. (2004). Maspin plays an essential role in early embryonic development. Development, 131, 1479–1489.
Dokras, A., Gardner, L. M., Kirschmann, D. A., et al. (2002). The tumour suppressor gene maspin is differentially regulated in cytotrophoblasts during human placental development. Placenta, 23, 274–280.
Chim, S. S., Tong, Y. K., Chiu, R. W., et al. (2005). Detection of the placental epigenetic signature of the maspin gene in maternal plasma. Proceedings of the National Academy of Sciences of the United States of America, 102, 14753–14758.
Pemberton, P. A., Tipton, A. R., Pavloff, N., et al. (1997). Maspin is an intracellular serpin that partitions into secretory vesicles and is present at the cell surface. Journal of Histochemistry and Cytochemistry, 45, 1697–1706.
Katz, A. B., & Taichman, L. B. (1999). A partial catalog of proteins secreted by epidermal keratinocytes in culture. Journal of Investigative Dermatology, 112, 818–821.
Qin, L., & Zhang, M. (2010). Maspin regulates endothelial cell adhesion and migration through an integrin signaling pathway. Journal of Biological Chemistry, 285, 32360–32369.
Endsley, M. P., Hu, Y., Deng, Y., et al. (2011). Maspin, the molecular bridge between the plasminogen activator system and beta1 integrin that facilitates cell adhesion. Journal of Biological Chemistry, 286, 24599–24607.
Toillon, R. A., Lagadec, C., Page, A., et al. (2007). Proteomics demonstration that normal breast epithelial cells can induce apoptosis of breast cancer cells through insulin-like growth factor-binding protein-3 and maspin. Molecular & Cellular Proteomics, 6, 1239–1247.
Khalkhali-Ellis, Z., & Hendrix, M. J. (2007). Elucidating the function of secreted maspin: inhibiting cathepsin D-mediated matrix degradation. Cancer Research, 67, 3535–3539.
Yu, X., Harris, S. L., & Levine, A. J. (2006). The regulation of exosome secretion: a novel function of the p53 protein. Cancer Research, 66, 4795–4801.
Hojo, T., Akiyama, Y., Nagasaki, K., et al. (2001). Association of maspin expression with the malignancy grade and tumor vascularization in breast cancer tissues. Cancer Letters, 171, 103–110.
Sopel, M., Kasprzyk, I., & Berdowska, I. (2005). Maspin and c-erbB-2 expression in correlation with microvessel density in invasive ductal breast cancer. Folia Histochemica et Cytobiologica, 43, 109–116.
Sharma, G., Mirza, S., Parshad, R., et al. (2011). Clinical significance of Maspin promoter methylation and loss of its protein expression in invasive ductal breast carcinoma: correlation with VEGF-A and MTA1 expression. Tumour Biology, 32, 23–32.
Maass, N., Teffner, M., Rosel, F., et al. (2001). Decline in the expression of the serine proteinase inhibitor maspin is associated with tumour progression in ductal carcinomas of the breast. The Journal of Pathology, 195, 321–326.
Maass, N., Hojo, T., Rosel, F., et al. (2001). Down regulation of the tumor suppressor gene maspin in breast carcinoma is associated with a higher risk of distant metastasis. Clinical Biochemistry, 34, 303–307.
Machtens, S., Kuczyk, M., Serth, J., et al. (2000). P53 regulated maspin protein expression determines recurrence-free survival of patients with localised prostate cancer. Prostate Cancer and Prostatic Diseases, 3, S27.
Maass, N., Nagasaki, K., Ziebart, M., et al. (2002). Expression and regulation of tumor suppressor gene maspin in breast cancer. Clinical Breast Cancer, 3, 281–287.
Stark, A. M., Schem, C., Maass, N., et al. (2010). Expression of metastasis suppressor gene maspin is reduced in breast cancer brain metastases and correlates with the estrogen receptor status. Neurological Research, 32, 303–308.
Umekita, Y., Ohi, Y., Sagara, Y., et al. (2002). Expression of maspin predicts poor prognosis in breast-cancer patients. International Journal of Cancer, 100, 452–455.
Bieche, I., Girault, I., Sabourin, J. C., et al. (2003). Prognostic value of maspin mRNA expression in ER alpha-positive postmenopausal breast carcinomas. British Journal of Cancer, 88, 863–870.
Joensuu, K. M., Leidenius, M. H., Andersson, L. C., et al. (2009). High expression of maspin is associated with early tumor relapse in breast cancer. Human Pathology, 40, 1143–1151.
Umekita, Y., & Yoshida, H. (2003). Expression of maspin is up-regulated during the progression of mammary ductal carcinoma. Histopathology, 42, 541–545.
Lee, M. J., Suh, C. H., & Li, Z. H. (2006). Clinicopathological significance of maspin expression in breast cancer. Journal of Korean Medical Science, 21, 309–314.
Tsoli, E., Tsantoulis, P. K., Papalambros, A., et al. (2007). Simultaneous evaluation of maspin and CXCR4 in patients with breast cancer. Journal of Clinical Pathology, 60, 261–266.
Kim, D. H., Yoon, D. S., Dooley, W. C., et al. (2003). Association of maspin expression with the high histological grade and lymphocyte-rich stroma in early-stage breast cancer. Histopathology, 42, 37–42.
Boltze, C., Schneider-Stock, R., Meyer, F., et al. (2003). Maspin in thyroid cancer: its relationship with p53 and clinical outcome. Oncology Reports, 10, 1783–1787.
Ito, Y., Yoshida, H., Tomoda, C., et al. (2004). Maspin expression is directly associated with biological aggressiveness of thyroid carcinoma. Thyroid, 14, 13–18.
Boltze, C., Hoang-Vu, C., Schneider-Stock, R., et al. (2004). Role of the class II tumor suppressor gene maspin in thyroid carcinogenesis. Verhandlungen der Deutschen Gesellschaft für Pathologie, 88, 237–245.
Son, H. J., Sohn, T. S., Song, S. Y., et al. (2002). Maspin expression in human gastric adenocarcinoma. Pathology International, 52, 508–513.
Wang, M. C., Yang, Y. M., Li, X. H., et al. (2004). Maspin expression and its clinicopathological significance in tumorigenesis and progression of gastric cancer. World Journal of Gastroenterology, 10, 634–637.
Zheng, H. C., Wang, M. C., Li, J. Y., et al. (2004). Expression of maspin and kai1 and their clinicopathological significance in carcinogenesis and progression of gastric cancer. Chinese Medical Sciences Journal, 19, 193–198.
Kim, S. M., Cho, S. J., Jang, W. Y., et al. (2005). Expression of maspin is associated with the intestinal type of gastric adenocarcinoma. Cancer Research and Treatment, 37, 228–232.
Song, S. Y., Son, H. J., Kim, M. H., et al. (2007). Prognostic significance of maspin expression in human gastric adenocarcinoma. Hepato-Gastroenterology, 54, 973–976.
Ito, R., Nakayama, H., Yoshida, K., et al. (2004). Loss of maspin expression is associated with development and progression of gastric carcinoma with p53 abnormality. Oncology Reports, 12, 985–990.
Terashima, M., Maesawa, C., Oyama, K., et al. (2005). Gene expression profiles in human gastric cancer: expression of maspin correlates with lymph node metastasis. British Journal of Cancer, 92, 1130–1136.
Yu, M., Zheng, H., Tsuneyama, K., et al. (2007). Paradoxical expression of maspin in gastric carcinomas: correlation with carcinogenesis and progression. Human Pathology, 38, 1248–1255.
He, Y., Zhang, L., Li, Y. J., et al. (2007). Expression of maspin protein and correlation between maspin and P53 proteins in gastric carcinoma. Zhonghua Yi Xue Za Zhi, 87, 909–912.
Lee, D. Y., Park, C. S., Kim, H. S., et al. (2008). Maspin and p53 protein expression in gastric adenocarcinoma and its clinical applications. Applied Immunohistochemistry & Molecular Morphology, 16, 13–18.
Boltze, C. (2005). Loss of maspin is a helpful prognosticator in colorectal cancer: a tissue microarray analysis. Pathology Research and Practice, 200, 783–790.
Zheng, H., Tsuneyama, K., Cheng, C., et al. (2007). Maspin expression was involved in colorectal adenoma–adenocarcinoma sequence and liver metastasis of tumors. Anticancer Research, 27, 259–265.
Markl, B., Arnholdt, H. M., Jahnig, H., et al. (2010). Shift from cytoplasmic to nuclear maspin expression correlates with shorter overall survival in node-negative colorectal cancer. Human Pathology, 41, 1024–1033.
Song, S. Y., Lee, S. K., Kim, D. H., et al. (2002). Expression of maspin in colon cancers: its relationship with p53 expression and microvessel density. Digestive Diseases and Sciences, 47, 1831–1835.
Fung, C. L., Chan, C., Jankova, L., et al. (2010). Clinicopathological correlates and prognostic significance of maspin expression in 450 patients after potentially curative resection of node-positive colonic cancer. Histopathology, 56, 319–330.
Xia, W., Lau, Y. K., Hu, M. C., et al. (2000). High tumoral maspin expression is associated with improved survival of patients with oral squamous cell carcinoma. Oncogene, 19, 2398–2403.
Yasumatsu, R., Nakashima, T., Hirakawa, N., et al. (2001). Maspin expression in stage I and II oral tongue squamous cell carcinoma. Head & Neck, 23, 962–966.
Nakashima, M., Ohike, N., Nagasaki, K., et al. (2004). Prognostic significance of the maspin tumor suppressor gene in pulmonary adenocarcinoma. Journal of Cancer Research and Clinical Oncology, 130, 475–479.
Li, H. W., Leung, S. W., Cheung, A. N., et al. (2006). Expression of maspin in gestational trophoblastic disease. Gynecologic Oncology, 101, 76–81.
Vereecken, P., Reynaert, S., Lalmand, M. C., et al. (2006). Decreased immunoreactive maspin expression in intermediate thickness and thick primary melanoma lesions. The Journal of International Medical Research, 34, 52–57.
Surowiak, P., Materna, V., Drag-Zalesinska, M., et al. (2006). Maspin expression is characteristic for cisplatin-sensitive ovarian cancer cells and for ovarian cancer cases of longer survival rates. International Journal of Gynecological Pathology, 25, 131–139.
Beecken, W. D., Engl, T., Engels, K., et al. (2006). Clinical relevance of maspin expression in bladder cancer. World Journal of Urology, 24, 338–344.
Blandamura, S., Giacomelli, L., Leo, G., et al. (2006). Nuclear maspin detection in renal cell tumours: possible diagnostic role and correlation with p53 status. Histopathology, 49, 274–282.
Nakagawa, M., Katakura, H., Adachi, M., et al. (2006). Maspin expression and its clinical significance in non-small cell lung cancer. Annals of Surgical Oncology, 13, 1517–1523.
Romani, A. A., Soliani, P., Desenzani, S., et al. (2006). The associated expression of Maspin and Bax proteins as a potential prognostic factor in intrahepatic cholangiocarcinoma. BMC Cancer, 6, 255.
Iezzi, G., Piattelli, A., Rubini, C., et al. (2007). Maspin expression in oral squamous cell carcinoma. The Journal of Craniofacial Surgery, 18, 1039–1043.
Schwarz, S., Ettl, T., Kleinsasser, N., et al. (2008). Loss of Maspin expression is a negative prognostic factor in common salivary gland tumors. Oral Oncology, 44, 563–570.
Kashima, K., Ohike, N., Mukai, S., et al. (2008). Expression of the tumor suppressor gene maspin and its significance in intraductal papillary mucinous neoplasms of the pancreas. Hepatobiliary & Pancreatic Diseases International, 7, 86–90.
Marioni, G., Koussis, H., Gaio, E., et al. (2009). MASPIN’s prognostic role in patients with advanced head and neck carcinoma treated with primary chemotherapy (carboplatin plus vinorelbine) and radiotherapy: preliminary evidence. Acta Oto-Laryngologica, 129, 786–792.
Takanami, I., Abiko, T., & Koizumi, S. (2008). Expression of maspin in non-small-cell lung cancer: correlation with clinical features. Clinical Lung Cancer, 9, 361–366.
Klasa-Mazurkiewicz, D., Narkiewicz, J., Milczek, T., et al. (2009). Maspin overexpression correlates with positive response to primary chemotherapy in ovarian cancer patients. Gynecologic Oncology, 113, 91–98.
Chua, R., Setzer, S., Govindarajan, B., et al. (2009). Maspin expression, angiogenesis, prognostic parameters, and outcome in malignant melanoma. Journal of the American Academy of Dermatology, 60, 758–766.
Ma, Y., & Peng, Z. L. (2009). Expression of maspin and its relation to tumor vascularization in epithelian ovarian cancer. Sichuan Da Xue Xue Bao. Yi Xue Ban, 40, 223–227.
Yoshizawa, K., Nozaki, S., Okamune, A., et al. (2009). Loss of maspin is a negative prognostic factor for invasion and metastasis in oral squamous cell carcinoma. Journal of Oral Pathology & Medicine, 38, 535–539.
Marioni, G., Staffieri, A., Bertolin, A., et al. (2010). Laryngeal carcinoma lymph node metastasis and disease-free survival correlate with MASPIN nuclear expression but not with EGFR expression: a series of 108 cases. European Archives of Oto-Rhino-Laryngology, 267, 1103–1110.
Marioni, G., Koussis, H., Scola, A., et al. (2010). Expression of MASPIN and angiogenin in nasopharyngeal carcinoma: novel preliminary clinico-pathological evidence. Acta Oto-Laryngologica, 130, 952–958.
Adim, S. B., Filiz, G., Kanat, O., et al. (2010). Maspin expression in gastrointestinal stromal tumors. World Journal of Surgical Oncology, 8, 22.
Yoshizawa, K., Nozaki, S., Kitahara, H., et al. (2011). Expression of urokinase-type plasminogen activator/urokinase-type plasminogen activator receptor and maspin in oral squamous cell carcinoma: association with mode of invasion and clinicopathological factors. Oncology Reports, 26, 1555–1560.
Ghazy, S. E., Helmy, I. M., & Baghdadi, H. M. (2011). Maspin and MCM2 immunoprofiling in salivary gland carcinomas. Diagnostic Pathology, 6, 89.
Smith, S. L., Watson, S. G., Ratschiller, D., et al. (2003). Maspin—the most commonly-expressed gene of the 18q21.3 serpin cluster in lung cancer—is strongly expressed in preneoplastic bronchial lesions. Oncogene, 22, 8677–8687.
Xu, C., Quddus, M. R., Sung, C. J., et al. (2005). Maspin expression in CIN 3, microinvasive squamous cell carcinoma, and invasive squamous cell carcinoma of the uterine cervix. Modern Pathology, 18, 1102–1106.
Nakashima, D., Uzawa, K., Kasamatsu, A., et al. (2006). Protein expression profiling identifies maspin and stathmin as potential biomarkers of adenoid cystic carcinoma of the salivary glands. International Journal of Cancer, 118, 704–713.
Cao, D., Wilentz, R. E., Abbruzzese, J. L., et al. (2005). Aberrant expression of maspin in idiopathic inflammatory bowel disease is associated with disease activity and neoplastic transformation. International Journal of Gastrointestinal Cancer, 36, 39–46.
Maesawa, C., Ogasawara, S., Yashima-Abo, A., et al. (2006). Aberrant maspin expression in gallbladder epithelium is associated with intestinal metaplasia in patients with cholelithiasis. Journal of Clinical Pathology, 59, 328–330.
Abd El-Wahed, M. M. (2005). Expression and subcellular localization of maspin in human ovarian epithelial neoplasms: correlation with clinicopathologic features. Journal of the Egyptian National Cancer Institute, 17, 173–183.
Cao, D., Zhang, Q., Wu, L. S., et al. (2007). Prognostic significance of maspin in pancreatic ductal adenocarcinoma: tissue microarray analysis of 223 surgically resected cases. Modern Pathology, 20, 570–578.
Nash, J. W., Bhardwaj, A., Wen, P., et al. (2007). Maspin is useful in the distinction of pancreatic adenocarcinoma from chronic pancreatitis: a tissue microarray based study. Applied Immunohistochemistry & Molecular Morphology, 15, 59–63.
Wang, D. L., Wang, Y. F., Shi, G. S., et al. (2007). Correlation of hTERT expression to maspin and bFGF expression and their significance in glioma. Ai Zheng, 26, 601–606.
Shams, T. M., Samaka, R. M., & Shams, M. E. (2006). Maspin protein expression: a special feature of papillary thyroid carcinoma. Journal of the Egyptian National Cancer Institute, 18, 274–280.
Tsuji, T., Umekita, Y., Ohi, Y., et al. (2007). Maspin expression is up-regulated during the progression of endometrioid endometrial carcinoma. Histopathology, 51, 871–874.
Sood, A. K., Fletcher, M. S., Gruman, L. M., et al. (2002). The paradoxical expression of maspin in ovarian carcinoma. Clinical Cancer Research, 8, 2924–2932.
Secord, A. A., Lee, P. S., Darcy, K. M., et al. (2006). Maspin expression in epithelial ovarian cancer and associations with poor prognosis: a Gynecologic Oncology Group study. Gynecologic Oncology, 101, 390–397.
Li, H. W., Leung, S. W., Chan, C. S., et al. (2007). Expression of maspin in endometrioid adenocarcinoma of endometrium. Oncology Reports, 17, 393–398.
Shui, H. H., Luo, L., Liang, S. Z., et al. (2008). The expression of Maspin protein in oral squamous cell carcinoma and its significance. Hua Xi Kou Qiang Yi Xue Za Zhi, 26(604–6), 610.
Kim, J., Jang, K. T., Kim, K. H., et al. (2010). Aberrant maspin expression is involved in early carcinogenesis of gallbladder cancer. Tumour Biology, 31, 471–476.
Bauerschlag, D. O., Habermann, M., Weimer, J., et al. (2010). Heterogeneous expression of serine protease inhibitor maspin in ovarian cancer. Anticancer Research, 30, 2739–2744.
Umekita, Y., Ohi, Y., Souda, M., et al. (2011). Maspin expression is frequent and correlates with basal markers in triple-negative breast cancer. Diagnostic Pathology, 6, 36.
Bal, N., Kocer, N. E., Ertorer, M. E., et al. (2008). Maspin, E-selectin, and P-selectin expressions in papillary thyroid carcinomas and their correlation with prognostic parameters. Pathology Research and Practice, 204, 743–750.
Woenckhaus, M., Bubendorf, L., Dalquen, P., et al. (2007). Nuclear and cytoplasmic Maspin expression in primary non-small cell lung cancer. Journal of Clinical Pathology, 60, 483–486.
Webber, B. A., Lawson, D., & Cohen, C. (2008). Maspin and Mutant p53 expression in malignant melanoma and carcinoma: use of tissue microarray. Applied Immunohistochemistry & Molecular Morphology, 16, 19–23.
Cho, J. H., Kim, H. S., Park, C. S., et al. (2007). Maspin expression in early oral tongue cancer and its relation to expression of mutant-type p53 and vascular endothelial growth factor (VEGF). Oral Oncology, 43, 272–277.
Goulet, B., Chan, G., Chambers, A. F., et al. (2011). An emerging role for the nuclear localization of maspin in the suppression of tumor progression and metastasis. Biochemistry and Cell Biology, 90, 22–38.
Mohsin, S. K., Zhang, M., Clark, G. M., et al. (2003). Maspin expression in invasive breast cancer: association with other prognostic factors. The Journal of Pathology, 199, 432–435.
Goulet, B., Kennette, W., Ablack, A., et al. (2011). Nuclear localization of maspin is essential for its inhibition of tumor growth and metastasis. Laboratory Investigation, 91, 1181–1187.
Marioni, G., Blandamura, S., Giacomelli, L., et al. (2005). Nuclear expression of maspin is associated with a lower recurrence rate and a longer disease-free interval after surgery for squamous cell carcinoma of the larynx. Histopathology, 46, 576–582.
Marioni, G., D’Alessandro, E., Giacomelli, L., et al. (2006). Maspin nuclear localization is related to reduced density of tumour-associated micro-vessels in laryngeal carcinoma. Anticancer Research, 26, 4927–4932.
Solomon, L. A., Munkarah, A. R., Schimp, V. L., et al. (2006). Maspin expression and localization impact on angiogenesis and prognosis in ovarian cancer. Gynecologic Oncology, 101, 385–389.
Sopel, M., Surowiak, P., & Berdowska, I. (2010). Nuclear maspin expression as a good prognostic factor in human epithelial ovarian carcinoma. Folia Morphologica (Warsz), 69, 204–212.
Lonardo, F., Li, X. H., Siddiq, F., et al. (2006). Maspin nuclear localization is linked to favorable morphological features in pulmonary adenocarcinoma. Lung Cancer, 51, 31–39.
Marioni, G., Giacomelli, L., D’Alessandro, E., et al. (2008). Nuclear localization of mammary serine protease inhibitor (MASPIN): is its impact on the prognosis in laryngeal carcinoma due to a proapoptotic effect? American Journal of Otolaryngology, 29, 156–162.
Marioni, G., Staffieri, C., Staffieri, A., et al. (2009). MASPIN tumour-suppressing activity in head and neck squamous cell carcinoma: emerging evidence and therapeutic perspectives. Acta Oto-Laryngologica, 129, 476–480.
Frey, A., Soubani, A. O., Adam, A. K., et al. (2009). Nuclear, compared with combined nuclear and cytoplasmic expression of maspin, is linked in lung adenocarcinoma to reduced VEGF-A levels and in stage I, improved survival. Histopathology, 54, 590–597.
Marioni, G., Blandamura, S., Lionello, M., et al. (2011). Nuclear MASPIN expression relates to a better prognosis in elderly patients with laryngeal carcinoma. Acta Oto-Laryngologica, 131, 1220–1225.
Blandamura, S., D’Alessandro, E., Giacomelli, L., et al. (2008). Expression of maspin in papillary Ta/T1 bladder neoplasms. Anticancer Research, 28, 471–478.
Zheng, H. C., Saito, H., Masuda, S., et al. (2008). Cytoplasmic and nuclear maspin expression in lung carcinomas: an immunohistochemical study using tissue microarrays. Applied Immunohistochemistry & Molecular Morphology, 16, 459–465.
Bettstetter, M., Woenckhaus, M., Wild, P. J., et al. (2005). Elevated nuclear maspin expression is associated with microsatellite instability and high tumour grade in colorectal cancer. The Journal of Pathology, 205, 606–614.
Dietmaier, W., Bettstetter, M., Wild, P. J., et al. (2006). Nuclear Maspin expression is associated with response to adjuvant 5-fluorouracil based chemotherapy in patients with stage III colon cancer. International Journal of Cancer, 118, 2247–2254.
Jang, H. L., Nam, E., Lee, K. H., et al. (2008). Maspin polymorphism associated with apoptosis susceptibility and in vivo tumorigenesis. International Journal of Molecular Medicine, 22, 333–338.
Yeom, S. Y., Jang, H. L., Lee, S. J., et al. (2010). Interaction of testisin with maspin and its impact on invasion and cell death resistance of cervical cancer cells. FEBS Letters, 584, 1469–1475.
Chambers, A. F. (2009). MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer Research, 69, 5292–5293.
Cai, Z., Zhou, Y., Lei, T., et al. (2009). Mammary serine protease inhibitor inhibits epithelial growth factor-induced epithelial–mesenchymal transition of esophageal carcinoma cells. Cancer, 115, 36–48.
Shi, H. Y., Stafford, L. J., Liu, Z., et al. (2007). Maspin controls mammary tumor cell migration through inhibiting Rac1 and Cdc42, but not the RhoA GTPase. Cell Motility and the Cytoskeleton, 64, 338–346.
Reis-Filho, J. S., Milanezi, F., Silva, P., et al. (2001). Maspin expression in myoepithelial tumors of the breast. Pathology Research and Practice, 197, 817–821.
Lele, S. M., Graves, K., & Gatalica, Z. (2000). Immunohistochemical detection of maspin is a useful adjunct in distinguishing radial sclerosing lesion from tubular carcinoma of the breast. Applied Immunohistochemistry & Molecular Morphology, 8, 32–36.
Sternlicht, M. D., & Barsky, S. H. (1997). The myoepithelial defense: a host defense against cancer. Medical Hypotheses, 48, 37–46.
Sternlicht, M. D., Kedeshian, P., Shao, Z. M., et al. (1997). The human myoepithelial cell is a natural tumor suppressor. Clinical Cancer Research, 3, 1949–1958.
Shao, Z. M., Nguyen, M., Alpaugh, M. L., et al. (1998). The human myoepithelial cell exerts antiproliferative effects on breast carcinoma cells characterized by p21(WAF1/CIP1) induction, G(2)/M arrest, and apoptosis. Experimental Cell Research, 241, 394–403.
Shao, Z., & Shen, Z. (1999). Human breast myoepithelial cell blocks estrogen receptor-negative breast carcinoma cell invasion. Zhonghua Yi Xue Za Zhi, 79, 214–216.
Martins, M. T., Altemani, A., Freitas, L., et al. (2005). Maspin expression in carcinoma ex pleomorphic adenoma. Journal of Clinical Pathology, 58, 1311–1314.
Campbell, I. D. & Humphries, M. J. (2011). Integrin structure, activation, and interactions. Cold Spring Harbor Perspectives in Biology, 3, a004994.
Seftor, R. E., Seftor, E. A., & Hendrix, M. J. (1999). Molecular role(s) for integrins in human melanoma invasion. Cancer Metastasis Reviews, 18, 359–375.
Seftor, R. E., Seftor, E. A., Sheng, S., et al. (1998). maspin suppresses the invasive phenotype of human breast carcinoma. Cancer Research, 58, 5681–5685.
Cella, N., Contreras, A., Latha, K., et al. (2006). Maspin is physically associated with [beta]1 integrin regulating cell adhesion in mammary epithelial cells. The FASEB Journal, 20, 1510–1512.
Bass, R., Wagstaff, L., Ravenhill, L., et al. (2009). Binding of extracellular maspin to beta1 integrins inhibits vascular smooth muscle cell migration. Journal of Biological Chemistry, 284, 27712–27720.
Tahmatzopoulos, A., Sheng, S., & Kyprianou, N. (2005). Maspin sensitizes prostate cancer cells to doxazosin-induced apoptosis. Oncogene, 24, 5375–5383.
Li, X., Chen, D., Yin, S., et al. (2007). Maspin augments proteasome inhibitor-induced apoptosis in prostate cancer cells. Journal of Cellular Physiology, 212, 298–306.
Liu, J., Yin, S., Reddy, N., et al. (2004). Bax mediates the apoptosis-sensitizing effect of maspin. Cancer Research, 64, 1703–1711.
Zhang, W., Shi, H. Y., & Zhang, M. (2005). Maspin overexpression modulates tumor cell apoptosis through the regulation of Bcl-2 family proteins. BMC Cancer, 5, 50.
Zheng, A. H., Yi, Y. F., & Zhou, W. W. (2008). Construction of a eukaryotic vector expressing MASPIN gene and its effect on cell apoptosis in gastric cancer cell line SGC7901. Ai Zheng, 27, 1161–1165.
Lee, S. J., Jang, H., & Park, C. (2012). Maspin increases Ku70 acetylation and Bax-mediated cell death in cancer. International Journal of Molecular Medicine, 29, 225–230.
Blandamura, S., D’Alessandro, E., Guzzardo, V., et al. (2007). Maspin expression in adenocarcinoma of the ampulla of Vater: relation with clinicopathological parameters and apoptosis. Anticancer Research, 27, 1059–1065.
Payne, C. M., Holubec, H., Crowley-Skillicorn, C., et al. (2011). Maspin is a deoxycholate-inducible, anti-apoptotic stress-response protein differentially expressed during colon carcinogenesis. Clinical and Experimental Gastroenterology, 4, 239–253.
Cher, M. L., Biliran, H. R., Jr., Bhagat, S., et al. (2003). Maspin expression inhibits osteolysis, tumor growth, and angiogenesis in a model of prostate cancer bone metastasis. Proceedings of the National Academy of Sciences of the United States of America, 100, 7847–7852.
Nickoloff, B. J., Lingen, M. W., Chang, B. D., et al. (2004). Tumor suppressor maspin is up-regulated during keratinocyte senescence, exerting a paracrine antiangiogenic activity. Cancer Research, 64, 2956–2961.
Friedrich, M. G., Toma, M. I., Petri, S., et al. (2004). Expression of Maspin in non-muscle invasive bladder carcinoma: correlation with tumor angiogenesis and prognosis. European Urology, 45, 737–743.
Bolat, F., Gumurdulu, D., Erkanli, S., et al. (2008). Maspin overexpression correlates with increased expression of vascular endothelial growth factors A, C, and D in human ovarian carcinoma. Pathology Research and Practice, 204, 379–387.
Li, Z., Shi, H. Y., & Zhang, M. (2005). Targeted expression of maspin in tumor vasculatures induces endothelial cell apoptosis. Oncogene, 24, 2008–2019.
Bailey, C. M., Khalkhali-Ellis, Z., Seftor, E. A., et al. (2006). Biological functions of maspin. Journal of Cellular Physiology, 209, 617–624.
Yin, S., Li, X., Meng, Y., et al. (2005). Tumor-suppressive maspin regulates cell response to oxidative stress by direct interaction with glutathione S-transferase. Journal of Biological Chemistry, 280, 34985–34996.
Li, X., Yin, S., Meng, Y., et al. (2006). Endogenous inhibition of histone deacetylase 1 by tumor-suppressive maspin. Cancer Research, 66, 9323–9329.
Lei, K. F., Liu, B. Y., Wang, Y. F., et al. (2011). SerpinB5 interacts with KHDRBS3 and FBXO32 in gastric cancer cells. Oncology Reports, 26, 1115–1120.
Lei, K. F., Wang, Y. F., Wang, Q. Q., et al. (2012). Interaction of SerpinB5 and MAFbx in gastric cancer cell and its action site. Zhonghua Wei Chang Wai Ke Za Zhi, 15, 169–173.
Staunton, J. E., Slonim, D. K., Coller, H. A., et al. (2001). Chemosensitivity prediction by transcriptional profiling. Proceedings of the National Academy of Sciences of the United States of America, 98, 10787–10792.
Nakatsu, N., Yoshida, Y., Yamazaki, K., et al. (2005). Chemosensitivity profile of cancer cell lines and identification of genes determining chemosensitivity by an integrated bioinformatical approach using cDNA arrays. Molecular Cancer Therapeutics, 4, 399–412.
Salter, K. H., Acharya, C. R., Walters, K. S., et al. (2008). An integrated approach to the prediction of chemotherapeutic response in patients with breast cancer. PLoS One, 3, e1908.
Nam, E., & Park, C. (2010). Maspin suppresses survival of lung cancer cells through modulation of Akt pathway. Cancer Research and Treatment, 42, 42–47.
Ben Shachar, B., Feldstein, O., Hacohen, D., et al. (2010). The tumor suppressor maspin mediates E2F1-induced sensitivity of cancer cells to chemotherapy. Molecular Cancer Research, 8, 363–372.
Khalkhali-Ellis, Z., Abbott, D. E., Bailey, C. M., et al. (2008). IFN-gamma regulation of vacuolar pH, cathepsin D processing and autophagy in mammary epithelial cells. Journal of Cellular Biochemistry, 105, 208–218.
Khalkhali-Ellis, Z. (2006). Maspin: the new frontier. Clinical Cancer Research, 12, 7279–7283.
Capony, F., Rougeot, C., Montcourrier, P., et al. (1989). Increased secretion, altered processing, and glycosylation of pro-cathepsin D in human mammary cancer cells. Cancer Research, 49, 3904–3909.
Vignon, F., Capony, F., Chambon, M., et al. (1986). Autocrine growth stimulation of the MCF 7 breast cancer cells by the estrogen-regulated 52 K protein. Endocrinology, 118, 1537–1545.
Harvat, B. L., & Jetten, A. M. (1996). Gamma-interferon induces an irreversible growth arrest in mid-G1 in mammary epithelial cells which correlates with a block in hyperphosphorylation of retinoblastoma. Cell Growth & Differentiation, 7, 289–300.
Narayan, M., & Twining, S. (2010). Focus on molecules: maspin. Experimental Eye Research, 90, 2–3.
Acknowledgments
This work was supported by a grant from the National Institutes of Health to M. J. C. Hendrix (NIH CA75681) and a trainee award from the Northwestern University Robert H. Lurie Comprehensive Cancer Center’s National Cancer Institute funded training program in Oncogenesis and Developmental Biology to T. M. Bodenstine (T32 CA080621).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bodenstine, T.M., Seftor, R.E.B., Khalkhali-Ellis, Z. et al. Maspin: molecular mechanisms and therapeutic implications. Cancer Metastasis Rev 31, 529–551 (2012). https://doi.org/10.1007/s10555-012-9361-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-012-9361-0