
Abstract
Prostaglandins are lipid compounds that mediate many physiological effects. Prostaglandin E2 (PGE2) is the most abundant prostanoid in the human body, and synthesis of PGE2 is driven by cyclooxygenase enzymes including COX-2. Both elevated expression of COX-2 and increased PGE2 levels have been associated with many cancers including breast cancer. PGE2 exerts its effect by binding to the E series of prostaglandin receptors (EP) which are G protein-coupled receptors. Four EP receptor subtypes exist, EP1–4, and each is coupled to different intracellular signaling pathways. As downstream effectors of the COX-2 pathway, EP receptors have been shown to play a role in breast and other malignancies and in cancer metastasis. The role of each EP receptor in malignant behavior is complex and involves the interplay of EP receptor signaling on the tumor cell, on stromal cells, and on host immune effector cells. While preclinical and epidemiological data support the use of nonsteroidal anti-inflammatory drugs and selective COX-2 inhibitors (COXibs) for the prevention and treatment of malignancy, toxicities due to COXibs as well as less than promising results from clinical trials have laboratories seeking alternative targets. As knowledge concerning the role of EP receptors in cancer grows, so does the potential for exploiting EP receptors as therapeutic targets for the treatment or prevention of cancer and cancer metastasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Eicosanoids, which include prostaglandins and leukotrienes, are potent lipid mediators that have been connected to many pathological processes such as inflammation and cancer [1, 2]. Prostaglandin E2 (PGE2) is the most abundant prostanoid in the human body and exhibits the most versatile actions ranging from reproduction to neuronal, metabolic, and immune functions [1, 3]. Prostaglandin synthesis is driven by cyclooxygenases (COX) which exist in three isoforms: constitutively expressed COX-1, inducible COX-2 and COX-3, the latter is a splice variant of COX-1 [1]. COX-2 is normally absent from most cells; however, its expression can be induced by cytokines and growth factors, and it is involved in the regulation of inflammatory responses. Furthermore, COX-2 can be highly induced during tumor progression. Overexpression of COX-2 is detected in premalignant and malignant tissues and tumor cell lines including but not limited to breast, colon, biliary, skin, lung, and liver [4, 5]. PGE2 has been implicated in various tumorigenic processes as well along with the involvement of specific PGE2 receptors [1, 2, 6].
2 Eicosanoid biosynthesis pathway and cyclooxygenases
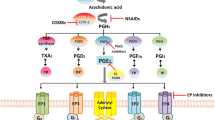
Eicosanoid biosynthesis begins with the mobilization of arachidonic acid (AA) from the plasma membrane by phospholipase A2 (PLA2) and, once free, COX enzymes convert AA to the precursor molecule prostaglandin H2 (PGH2). PGH2 can then be converted to one of five primary prostanoids prostaglandin D2, prostaglandin E2, prostaglandin F2α, prostaglandin I2, and thromboxane A2 through specific synthase molecules PGDS, PGES, PGFS, PGIS, and TXAS, respectively [2, 7, 8]. There are two classifications of PGES: cytosolic (cPGES) and microsomal or membrane-bound (mPGES). cPGES is predominantly coupled to COX-1, and mPGES is preferentially linked to COX-2 and exists in two isoforms, mPGES-1 and mPGES-2 [2, 7, 9]. The expression of mPGES-1 can be induced by proinflammatory signals, similar to COX-2, and mPGES-1 is the synthase that is primarily responsible for increasing the PGE2 levels during inflammation and tumorigenesis [9]. Once PGE2 is produced, it is exported into the extracellular microenvironment by a specific multidrug resistance-associated protein (MRP), MRP4, where PGE2 then exerts its biological effects in an autocrine or paracrine manner through binding to its cognate cell surface receptors, the E series of prostaglandin receptors (EP) (Fig. 1). After binding its receptor, PGE2 is metabolized in a two-step process in which the prostaglandin is transported into the cytoplasm through a passive mechanism or actively by prostaglandin transporter followed by inactivation by 15-hydroxyprostaglandin dehydrogenase [1, 7].

Eicosanoid biosynthesis and EP receptor signaling pathway. A phospholipids from the plasma membrane are mobilized and converted to arachidonic acid by phospholipase A2 (PLA 2 ). COX enzymes convert AA to prostaglandin H2 (PGH 2 ) precursor molecule which is then converted to prostaglandin E2 (PGE 2 ) by the synthase molecule PGES. Once produced PGE2 can exert its effects in one of two ways: (1) PGE2 can be exported into the extracellular microenvironment by multidrug resistance-associated protein four (MRP4) where PGE2 can bind to its cognate receptors, the E series of prostaglandin receptors (EP) on the plasma membrane of a tumor cell, stromal cell, or immune effector cell such as a T or natural killer (NK) cell and (2) after being synthesized by PGES, PGE2 can directly act on EP receptors located on the nuclear membrane. After binding its receptor, PGE2 can be transported back into the cytoplasm through a passive mechanism or actively through a prostaglandin transporter (PGT). PGE2 is inactivated by 15-hydroxyprostaglandin dehydrogenase (15-PGDH) and converted to 15-keto-PGE2. B EP receptors are G protein-coupled receptors of which four subtypes exist: EP1, EP2, EP3 and EP4. Each receptor is coupled to a different intracellular signaling pathway. The EP2 and EP4 receptors are linked to stimulation of cyclic AMP (cAMP) and protein kinase A (PKA) signaling through sequential activation of Gαs and adenylate cyclase (AC). EP4 can also activate phosphoinositide-3-kinase (PI3K) through Gαi. The EP1 receptor leads to elevation of intracellular calcium through Gαq. EP3 exists in multiple isoforms which are generated through alternative splicing and differing at their carboxy terminal tail. These isoforms are capable of eliciting different intracellular responses through multiple signal transduction pathways. The majority of the isoforms act to inhibit cAMP generation via Gαi. Ligand binding can also lead to an increase in IP3/intracellular calcium. Another isoform can act to stimulate adenylyl cyclase leading to an increase in cAMP. On immune effector cells, PGE2 acting through the EP receptors can modulate the function of various immune effector cells. PGE2 acting through its cognate receptor can inhibit NK cell activity and cytotoxic T-cell proliferation leading to a decrease in target cell lysis. C there is now growing evidence to support perinuclear and/or nuclear localization of functional EP receptors. To date EP1, EP2, EP3α, and EP4 have been shown to be colocalized at the nuclear membranes of a variety of cell types and tissues. Nuclear EP receptors could exert different effects from their plasma membrane counterparts; however, the signaling pathways for nuclear receptors have yet to be determined.
3 The role of COX-2 in breast cancer
Epidemiological data demonstrate a strong correlation between chronic inflammation and developing cancer. COX-2 can be rapidly induced by mitogens and proinflammatory cytokines, is an early response gene, and is an important component of the inflammatory response linked to carcinogenesis [4, 10]. COX-2 expression is not detectable in most healthy tissues while upregulation of COX-2 has been identified in many human cancers and precancerous lesions [11]. Initially recognized in the context of colorectal cancer, COX-2 has been shown, both experimentally and epidemiologically, to be involved in mammary carcinogenesis [11–13]. Experimentally COX-2 protein is present in rat mammary tumors induced by various carcinogens [12]. In addition, more than 85% of transgenic mice overexpressing COX-2 in mammary tissue, through the use of the mouse mammary tumor virus (MMTV), developed mammary tumors [14, 15]. This finding indicates that COX-2 overexpression alone is sufficient to cause breast carcinoma.
Clinical data also broadly support a protumorigenic role for COX enzymes in breast cancer. Elevated COX-2 protein levels have been detected immunohistochemically in approximately 40% of invasive breast carcinoma (as reviewed in [11]). Increased expression of COX-2 is more common in breast cancers with poor prognostic characteristics and is associated with an unfavorable outcome as well as worse survival independent of known prognostic factors [13, 16]. Specifically, overexpression of COX-2 was associated with large tumor size, high histological grade, negative hormone receptor status, high proliferation rate, ductal type histology, high p53 expression, HER-2 oncogene amplification, and axillary node involvement [13]. Progression-free survival may be better in patients with breast tumors that convert from COX-2 positive to COX-2 negative through treatment with neoadjuvant chemotherapy since reduction in COX-2 expression was mainly observed in clinical responders [17].
COX-2 has also been associated with breast cancer metastasis. Expression of COX-2 is correlated with the presence of lymph node metastases and distant metastasis [12, 18]. Likewise, COX-2 expression is positively correlated with tumorigenic and metastatic potential in a murine model of breast cancer [5, 19, 20]. Gene expression analyses and mouse model systems have also associated COX-2 with breast cancer metastasis to the lung, bone, and brain [14, 21, 22]. COX-2 overexpression increases motility and invasion of breast cancer cells [23]. Migration is a key functional activity of cancer cells and is associated with their metastatic potential. Silencing of COX-2 inhibits migration of human breast cancer cell line MDA-MB-231 in vitro and metastasis in vivo [24].
4 Nonsteroidal anti-inflammatory drugs and cancer
The involvement of COX-2 in tumorigenesis was revealed by complementary observations pertaining to colorectal cancer. Multiple epidemiological studies reported an inverse correlation between colon cancer incidence and regular use of nonsteroidal anti-inflammatory drugs (NSAIDs) including aspirin [12, 25–28]. NSAIDs are known to function by inhibiting cyclooxygenase enzyme activity; therefore, these observations suggested that aberrant prostaglandin biosynthesis may contribute to colorectal cancer. In addition, overexpression of COX-2 was detected in precancerous adenomas and colon carcinomas [12, 29–32]. These data suggest that COX-2 could be a useful target for chemoprevention providing impetus for clinical trials exploring the effect of NSAIDs, which inhibit COX-1 and COX-2, on colon cancer. The results from these clinical trials showed that COX inhibitors could decrease the size and number of polyps, but adverse side effects from the use of NSAIDs including peptic ulcer disease occurred [12].
Like colon cancer, experimental breast cancer can be suppressed by inhibiting COX activity with either NSAIDs or selective COX-2 inhibitors (COXibs) [11, 12, 20, 33]. The suppression of mammary tumor formation by COXibs applies to both chemically induced breast tumors, which tend to be hormone-dependent as well as hormone-independent models [11]. COX-2 expression is associated with breast cancer metastasis. Antagonism of COX-2 decreases breast cancer cell invasion and motility [34]. COX-1 or COX-2 inhibition decreases metastatic disease in a murine mammary carcinoma model system [20]. In a murine model of metastatic breast cancer, inhibition of COX-2 reduces the frequency of pulmonary metastases [12, 20, 35].
While the results from animal studies demonstrating the chemopreventative or chemotherapeutic efficacy of COXibs are promising, epidemiologic studies correlating breast cancer risk and NSAID use have been inconsistent. Several studies have reported reduced breast cancer incidence in association with NSAID use [11, 36–42]. Most recently, Holmes et al. reported that aspirin use was associated with a decreased risk of distant recurrence and breast cancer death [41]. Meta-analyses of either NSAID or aspirin use have found a 9% to 30% reduced risk of breast cancer incidence [41, 43–46]. Additionally, patients who take COX-2 inhibitors had a reduced risk of bone metastasis [47]. Conversely, multiple studies have shown no protective association between NSAID use and breast cancer incidence [48–51]. Various factors can contribute to the inconsistencies between these studies including differences in the quality and completeness of data on NSAID use.
Despite inconsistencies in the results of the epidemiological studies, there is evidence that COXibs could reduce the incidence of both familial and sporadic malignant disease, indicating that COX and prostaglandin signaling is a viable target; however, some clinical trials identified increased cardiovascular risk associated with COXib use [11, 52, 53]. The cardiovascular toxicity of COXibs has been attributed to their selective depression of prostacyclin (PGI2) levels which have a cardioprotective function [11, 54]. The cardiovascular toxicity of COXibs decreases the desirability of using this class of drugs in cancer prevention; therefore, alternative components of the COX signaling pathway that would offer a safer target need to be identified.
5 Functioning of G protein-coupled receptors in the cell
Prostaglandin cell surface receptors belong to the G protein-coupled receptor (GPCR) family. GPCRs have seven transmembrane-spanning α helices, an extracellular N terminus, an intracellular C terminus, and three interhelical loops on each side of the membrane. These receptors are coupled to heterotrimeric G proteins which initiate various intracellular signaling cascades in response to GPCR activation by extracellular stimuli [55]. Heterotrimeric G proteins are composed of three subunits, α, β, and γ and in their inactive state exist as a Gβγ monomer and a guanine diphosphate-bound Gα subunit. Heterotrimers are divided into four families based on α subunit sequence identity and signaling activity: Gαs, Gαi, Gαq/11, and Gα12/13. Following ligand activation, GPCRs catalyze the exchange of GDP for GTP on the Gα subunit, leading to decreased affinity of Gα for Gβγ. The resulting dissociation of the heterotrimer allows the GTP-bound Gα and free Gβγ to interact with several downstream effectors (Table 1) [55, 56].
6 The E series of prostaglandin receptors
PGE2 can bind any of four of the E series of prostaglandin EP receptor subtypes specified as EP1, EP2, EP3, and EP4 [2, 3, 7, 9, 57]. Each receptor has distinct biochemical properties, tissue and cellular localization and is coupled to different intracellular signaling pathways [3]. In addition to the different EP subtypes, isoforms generated through alternative mRNA splicing have been identified for two EP receptor subtypes, EP1 and EP3. The EP2 and EP4 receptors are linked to stimulation of cyclic AMP (cAMP) and protein kinase A (PKA) signaling through sequential activation of Gαs and adenylate cyclase [58–60]. However, functional coupling of the cAMP pathway seems to be more efficient for EP2 compared to EP4 [61]. EP4, unlike EP2, activates phosphoinositide-3-kinase (PI3K) through Gαi [61]. The coupling of EP4 to Gαi may in part explain the decrease in efficiency for the EP4 receptor to couple to the cAMP/PKA signaling pathway.
The EP1 receptor elicits the elevation of intracellular calcium through two distinct pathways coupling with Gαq, specifically Gq and/or G11, and receptor-activated Ca2+ channels (RACC) [62, 63]. Using CHO cells transfected with mouse EP1 cDNA, Katoh et al. demonstrated that PGE2 activation of the EP1 receptor elicits a large influx from extracellular calcium inducing PI hydrolysis and a very small Ca2+ mobilization from internal stores, most likely the endoplasmic reticulum, as a result of phospholipase C activation via Gq [62]. Further investigation of EP1-induced calcium mobilization by Tabata and colleagues identified transient receptor potential 5 as a possible candidate for the RACC coupled to EP1 and involved in Ca2+ influx [63]. In addition to coupling to Gq/11, utilizing human EP1 expressed in HEK cells, EP1 has been shown to couple to Gαi/o resulting in activation of PI3K [64]. A number of GPCRs that were traditionally considered to couple exclusively to Gq/11 have now been found to couple to Gi/o and to activate PI3K [65].
As mentioned previously, isoforms for two EP receptor subtypes, EP1 and EP3, have been identified. Okuda-Ashitaka, et al. identified a splice variant for the EP1 (EP1v) receptor from rat uterus cDNA [66]. This variant differs from EP1 from the middle of transmembrane segment VI to the carboxy terminus and results in a receptor with a transmembrane segment VII-like structure lacking an intracellular COOH terminal tail. The EP1v receptor retained ligand-binding capability but is no longer coupled to signal transduction systems. EP1v may affect the efficiency of EP1 and EP4 signal transduction since overexpression of the EP1v in CHO cells expressing EP1 or EP4 attenuates intracellular signaling mediated by EP1 and EP4 [66]. In addition to rat, EP1v has also been identified in murine cell lines including mast cell line MC/9 and mammary epithelial cell lines; however, functional characterization has yet to be carried out in a murine model system [67].
The EP3 receptors, due to the presence of multiple isoforms, are also capable of coupling to multiple G proteins. These isoforms, which are generated by alternative mRNA splicing, differ in their cytoplasmic carboxy terminal tail and signal transduction pathways [60, 68]. In humans, ten splice variants coding for eight different isoforms have been identified [68]. Previous studies have shown that alterations in the carboxyl tail impart differences in constitutive activity, G protein coupling and agonist-induced internalization [69–72]. EP3 receptor isoforms have been identified that couple to Gαi, Gαs, Gαq, and Gα12/13 [69, 73, 74]. Functionally, the majority of the isoforms (EP3-I, EP3-II, EP3-III, EP3-IV, EP3-e, and EP3-f) act to inhibit cAMP generation via Gαi. Ligand binding of EP3-I, EP3-II, and EP3-III could also increase IP3/intracellular calcium [68, 70, 75].
In the mouse, three EP3 receptor isoforms, EP3α, EP3β, and EP3γ have been identified and, similar to the human isoforms, are generated through alternative splicing, differ in their C-terminal tail, and couple to multiple G proteins [76–78]. The EP3 receptor signals are primarily involved in inhibition of adenylyl cyclase via Gi activation and Ca2+ mobilization through Gβγ from Gi [76, 78]. EP3γ is also coupled to Gs and stimulates adenylyl cyclase [78]. EP3β has the ability to superactivate adenylyl cyclase via the Gq/PLC/Ca2+ pathway in a lipid raft-dependent manner [79, 80]. In addition to affecting adenylate cyclase, all three isoforms are capable of Ca2+ mobilization mediated by Gβγ subunits from the Gi/o protein leading to activation of the PLCβ isoform [76, 81].
6.1 Subcellular localization of EP receptors
GPCRs are typically thought to work at the cell surface recognizing and responding to extracellular ligands at the plasma membrane; however, there is now growing evidence supporting perinuclear and/or nuclear localization of functional GPCRs, including the EP receptors (as reviewed in [82–84]). Enzymes essential for prostanoid biosynthesis and signaling are also located at the nucleus including PLA2, mPGES-1, COX-2, and G proteins which suggests that eicosanoids such as PGE2 can be produced directly at the nucleus and act through cognate receptors at the nuclear membrane [82]. Gobeil et al. has demonstrated that isolated nuclei with intact envelopes from porcine endothelial cells are capable of producing PGE2 which suggests a potential role for intracellular signaling for prostanoids [82, 85]. To date EP1, EP2, EP3α, and EP4 have been shown to be colocalized at the nuclear membranes of a variety of cell types and tissues [82, 84–87]. The plasma membrane and the nuclear receptors exhibit almost identical features based on: virtually indistinguishable kinetic profiles, immunoreactivity, molecular weight, and they arise from the same gene [82]. However, there is the possibility for differences in posttranslational modifications for the same receptor in different subcellular locations [82].
It has been suggested that plasma membrane GPCRs could exert different effects compared with GPCRs at the nucleus. In endothelial cells, there is a distinct signaling pathway and function for the EP3 receptor such that the plasma membrane receptor elicits immediate physiological actions (vasomotor effects with a decrease in cAMP); whereas, the nuclear EP3 receptor conveys gene regulation changes (induction of eNOS and nuclear calcium signals) without generation of second messengers such as cAMP or IP3 [82, 84, 85]. In breast cancer, nuclear EP1 expression has been identified via immunohistochemistry. In malignant cells, nuclear EP1 expression was correlated with good prognosis markers such as node-negative disease and PR expression and the absence of nuclear EP1 expression was correlated with worse survival [88, 89]. Conversely, in cholangiocarcinoma, nuclear EP1 was shown to play a role in the activation of signal transducer and activator of transcription-3 (stat 3) and induces tumor cell growth [90]. Considering the differences in the plasma versus nuclear membrane function of EP3 and the prognostic significance of nuclear EP1 and the potential role of nuclear EP1 in cholangiocarcinoma, nuclear EP receptors could play an important role in the function of normal and malignant cells.
7 PGE2 and the role of EP receptors in breast cancer
The relationship between elevated COX-2 expression and cancer was first suggested by reports of elevated prostaglandin levels in breast tumors especially from patients with metastatic disease [12, 91–93]. COX-2-derived prostanoids promote angiogenesis, induce invasion, and increase metastasis [13]. PGE2 is the principle COX-2 product in tumors and plays a predominant role in promoting cancer progression through its cognate receptors [57]. As mentioned previously, the cellular effects of PGE2 are mediated through four receptors, EP1, EP2, EP3, and EP4 that are coupled to different intracellular signaling pathways [57]. As downstream targets of the COX-2 pathway, experimental evidence has also indicated that EP receptors can play a role in cancer including breast cancer and breast cancer metastasis; however, the precise role of each EP receptor in malignant behavior has not been determined.
EP receptors play diverse roles in normal and malignant tissues, and each receptor may have a unique function in tumor behavior that can vary based on cell and tissue type and model system. In normal murine mammary gland, all four EP receptors are expressed at various points during gland development [94]. EP2 and EP4 receptors are induced during the proliferative phase of mammary gland development (pregnancy and lactation) and subsequently downregulated during the involution phase. EP3 is downregulated during the proliferative stage, and its expression is returned to high levels in the involuted mammary gland. In contrast, the EP1 receptor is expressed only in the involuted mammary gland [94]. The expression profile of the EP receptors changed in COX-2-induced mammary tumors. In these tumors, EP1, EP2 and EP4 receptors are strongly induced relative to normal mammary gland; whereas, the EP3 receptor is downregulated. The downregulation of the EP3 receptor would suggest that EP3 could have a protective role in mammary tumor development in this model system. Chang et al. suggested that EP2 and EP4 receptor subtypes are most likely to be involved in mammary tumor progression and angiogenesis since indomethacin, a nonselective COX-1 and COX-2 inhibitor, suppressed expression of EP2 and EP4 receptors [94]. In follow-up studies, Chang and colleagues focused on the role of EP2 specifically and determined that in COX-2-induced mammary tumors, EP2 was required for mammary epithelial hyperplasia and EP2 overexpression in mammary tumor cells mediates increased VEGF production via a cAMP/PKA-dependent pathway [95, 96]. Furthermore, EP2 plays a role in the oncogenic activities of transforming growth factor-β (TGF-β) during mammary tumorigenesis [97].
Estrogen plays a significant role in the development and progression of breast cancer. Cytochrome P450 (aromatase), encoded by the CYP19 gene, catalyzes the synthesis of estrogen from androgens [98]. Aromatase, located primarily in the adipose stromal cells of breast tumors, catalyzes estrogen biosynthesis and is fundamental to hormone-dependent growth of breast cancer [98, 99]. In a study conducted by Richards and Brueggemeier in normal nonneoplastic breast adipose stromal cells, PGE2 regulation of aromatase gene expression and activity involved EP1, EP2, and EP3 signaling pathways [99]. Stimulation of EP1 and EP2 with receptor agonists resulted in an increase in aromatase gene expression, and stimulation of EP2 resulted in an increase in aromatase activity most likely utilizing a PKA/cAMP mechanism. Stimulation of the adipose cells with the EP3 agonist sulprostone resulted in an inhibitory effect whereby EP3 activation blocked the PGE2-mediated increase in aromatase gene expression and activity. Zhao et al. also demonstrated upregulation of the aromatase gene through activation of EP1 and EP2 via the PKC and PKA pathways in human adipose tissue [100]. Conversely, Subbaramaiah and Dannenberg published results indicating that the EP2 and EP4 receptors act to regulate aromatase expression in human adipocytes and breast cancer cells through the cAMP protein kinase A pathway that resulted in an enhanced interaction between P-CREB, p300, and the aromatase promoter 1.3/II [98]. Reduction of the EP1 and EP3 receptors in adipocytes had no effect on PGE2-mediated increase in aromatase activity or expression. Neither EP1 or EP3 agonists were utilized in this study; therefore, the apparent discrepancy between these studies could be due to differences in the source and type of tissues used to study the effect of specific EP receptors on aromatase expression and activity [98, 99].
In another study, EP2 and EP4 have also been implicated in playing a role in increasing aromatase activity and expression in breast cancer cells after exposure to environmental toxicant o,p′-dichlorodiphenyltrichloroethane [101]. While some differences in experimental outcomes are present for EP1 and EP3, it would appear that there is a consensus that EP2 and EP4 receptors are involved in the PGE2-mediated increase in aromatase expression and activity in adipose stromal cells and breast cancer cells and could play a role in the development of hormone-dependent breast cancer. Recently, Subbaramaiah and Dannenberg reported a linkage between obesity, inflammation, and aromatase activity in the murine mammary gland [102]. Obesity leads to inflammation in the mammary gland resulting in increased levels of COX-2-derived PGE2, which could drive aromatase activity.
EP receptors have also been shown to play a role in inflammatory breast cancer. Inflammatory breast cancer (IBC) is the most aggressive form of locally advanced breast cancer and deviates from the phenotypic characteristics of either ductal or lobular breast tumors [103]. COX-2 is upregulated in primary IBC and metastatic lesions. Using human IBC tumor cell line SUM149, Robertson and colleagues demonstrate that stimulating the EP4 receptor with EP4 agonist PGE1 alcohol increased proliferation and invasion and conversely antagonizing the EP4 receptor, by pharmacologic or genetic means, inhibited proliferation and invasion [103]. Robertson then expanded on this work confirming that EP4 plays a role in regulating invasion of IBC cells, and, furthermore, the EP3 receptor was shown to regulate the ability of SUM149 cells to undergo vasculogenic mimicry [104]. Vasculogenic mimicry, a characteristic of tumor cells from aggressive tumors, occurs when tumor cells are able to form matrix rich capillary-like networks when placed in a three-dimensional culture in the absence of endothelial cells and fibroblasts [104]. EP3 stimulation with sulprostone inhibited the capacity of SUM149 cells to undergo vasculogenic mimicry, but antagonizing EP4 did not affect this response.
While activation of EP3 in inflammatory breast cancer and in adipose tissue appears to be beneficial, by inhibiting both the vasculogenic mimicry process and aromatase expression, respectively, there is limited additional information about the role of EP3 receptor in breast cancer [99, 104]. In CHO cells overexpressing the human EP3 receptor, stimulation with EP3 agonist M&B 28.767 can induce migration in a dose-dependent manner [105]. Much of the literature suggested that the EP3 receptor could play a role in angiogenesis. In murine mammary tumors from the MMTV-COX-2 transgenic mouse, EP2 may play a role in angiogenesis since EP3 was downregulated in the tumors [94]; however, in a sponge-induced granulation assay in ddy mice, EP3 agonist ONO-AEI-248 increased angiogenesis in a dose-dependent manner [106]. Furthermore, in EP3 knockout mice, using the same sponge model approach, angiogenesis was significantly reduced compared to wild-type mice. Tumor-associated angiogenesis and VEGF expression, induced in sarcoma-180 and Lewis lung carcinoma models, were reduced in EP3 knockout mice resulting in reduced tumor growth [106]. EP3-I isoform expressed in HEK-293 cells can induce the expression of vascular endothelial growth factor (VEGF) and VEGF receptor-1 mRNA via activation of PI3K and ERK [69, 107]. In Lewis lung carcinoma cells, EP3 receptor signaling was shown to regulate tumor-associated lymphangiogenesis through upregulation of VEGF-C and its receptor VEGFR-3 in tumor stromal tissues [108]. Whether EP3 plays a role in angiogenesis in breast cancer specifically has yet to be determined; however, Timoshenko et al. have demonstrated in several human breast cancer cell lines that antagonizing EP1 and EP4 resulted in inhibition of VEGF-C production [109].
Limited information is available about the potential role for EP1 in breast cancer. However, EP1 has been shown to play a role in other cancers. For example, EP1 has been implicated in UVB-induced inflammation and skin tumorigenesis [110] and signaling through the EP1 receptor can upregulate survivin expression in hepatocellular carcinoma [111]. Nuclear EP1 has been shown to play a growth-promoting role in cholangiocarcinoma [90]. Stimulation of EP1 can affect the transcription of aromatase in adipose cells of abdominal and breast origin and contribute to VEGF-C production in human breast cancer cell line MDA-MB-231 potentially playing a role in angiogenesis [99, 100, 109]. In a rat model of chemically induced breast cancer, an EP1 antagonist significantly inhibited breast cancer development [112]. In a murine mammary model of metastatic breast cancer, EP1 functioned as a suppressor of breast cancer metastasis [89]. Treatment of murine mammary metastatic cell lines with pharmacological antagonists against EP1 (SC19220) or EP1/EP2 (AH6809) or reducing EP1 gene expression with shRNA resulted in an increase in the number of lung tumor colonies. Manipulation of EP1 only affected the metastatic potential of the cells with no effect on the primary tumor. In the same study, Ma and colleagues investigated the relationship between EP1 expression and survival in invasive ductal carcinomas and determined that nuclear EP1 expression correlated with improved survival compared to women with no nuclear EP1 expression [89]. This result correlated with Thorat's study which stated that nuclear EP1 expression in primary human breast tumors was correlated with good prognostic markers of progesterone expression and lymph node-negative status [88, 89]. Based on the disparate results concerning EP1 and cancer, it is possible that EP1 could play a protumorigenic role in the primary tumor in some cancers but an antimetastatic role in breast cancer.
The role of EP4 receptor in cancer appears to be clearer in comparison to the other EP receptors. EP4 has been shown to be involved in multiple cancers including colon, gastric, prostate, and lung cancer [113–119]. In metastatic murine (C3L5) and human (MDA-MB-231) breast cancer cells, EP4 plays a role in mediating autocrine PGE2-mediated migration [120]. In a follow-up study, Timoshenko et al. provided an additional role for EP4 in cancer progression in C3L5 cells wherein activation of EP4 results in an increase of inducible nitric oxide synthase [121]. Tumor-derived nitric oxide has been shown to promote tumor growth and metastasis in a murine breast cancer model [122]. Additionally, EP4 is upregulated in castration-resistant hormone-naïve prostate cancer [117]. An EP4 antagonist inhibits growth in two xenograft models of castration-resistant disease.
In addition to migration, EP4 has been implicated in other aspects of metastasis. Osteolysis due to bone metastasis of breast cancer is linked to EP4 activation [123]. The stimulation of EP4 via an autocrine/paracrine mechanism results in an increase in RANKL in osteoblasts which leads to the induction of osteoclastogenesis and osteolysis in the bone. EP4 can act to enhance lymphatic invasion of breast cancer cells [124]. CCR7 is a chemokine receptor that plays an important role in the mediation of migration of leukocytes and dendritic cells toward lymphatic endothelial cells (LECs) that express the CCR7 ligand CCL21. Pan et al. demonstrated that in human breast cancer tissues, CCR7 expression in COX-2 overexpressing tumors was significantly correlated with lymph node metastasis. In addition, stimulation of the EP4 receptor in MDA-MB-231 cells lead to an increase in CCR7 expression through the PKA pathway which suggests that EP4 could play a role in lymphatic invasion in breast cancer. Since EP2 can activate the same pathway as EP4, EP2 often demonstrates functional similarities in regards to breast cancer. EP2 can also increase CCR7 expression in breast cancer cells leading to enhancement of migration towards LECs and promoting lymphatic invasion [124]. EP4, through activation of Erg-1 pathway through EGFR and ERK1/2, has also been connected to increasing transcription of inhibitors of DNA binding (Id-1) and cell invasion which can drive breast cancer metastasis [125].
In a murine model of metastatic breast cancer, antagonism of the EP4 receptor inhibited tumor metastasis in vivo [126]. Pretreatment of metastatic murine mammary tumor cell lines 410.4 and 66.1 with EP4 antagonist AH23848 or ONO-AE3-208 significantly inhibited the ability of these cells to colonize the lung. Additionally, EP4 antagonists also inhibited migration of 66.1 to PGE2 and modestly inhibited cell growth of 410.4 cells. The chemotactic response of 66.1 cells and proliferation of 410.4 cells was also inhibited by the EP1/EP2 antagonist AH6809. In a follow-up study, reduction of EP4 expression via shRNA led to a decrease in the ability of 66.1 cells to form lung colonies or to spontaneously metastasize from a primary tumor [127]. However, EP4 gene silencing did not inhibit local tumor growth which indicates that in this model system, the role of EP4 seems to be directed to processes unique to metastasis rather than to expansion of the primary tumor.
8 The role of EP receptors in the immune system
PGE2 has the ability to regulate the immune system. Within the immune system, PGE2 modulates the functions of different cell populations [i.e., natural killer (NK) cells, T lymphocytes, B lymphocytes, macrophages, dendritic cells (DC), and myeloid-derived suppressor cells (MDSC)]. For example, PGE2 inhibits the proliferation of CD4+ T cells via decreases in intracellular calcium release and activity of p59 protein tyrosine kinase [128–130]. PGE2 also has profound inhibitory effects on T cell apoptosis and decreases production of interferon gamma (IFNγ) and interleukin (IL)-2 [131, 132]. PGE2 affects the development and activity of B cells in a complex manner. PGE2 suppresses proliferation and induced apoptosis of immature B cells [133, 134]. On the other side, PGE2 does not induce cell death or inhibit proliferation in mature B cells but regulates their activity by enhancing Ig class switching [135]. In macrophages, PGE2 inhibits production of cytokines such as tumor necrosis factor α (TNFα) and IL-12, suppressing type-1 immune responses [136].
The concept that PGE2 inhibits NK cell function arises from both in vitro exposure of NK cells to PGE2 and in vivo administration of PGE2 inhibitors [137]. Initial work in the 1980s showed that prostaglandins inhibited NK cell activity and PGE2 had greater suppressive effects than other prostaglandins (PGF2α and PGD2) in both human and murine NK cells [138, 139]. Early studies observed the inability of NK cells to exert cytotoxic effects in patients with breast cancer or in animal models of breast cancer [140, 141]. The high levels of PGE2 produced by suppressors of NK function (i.e., macrophages, tumor cells) contribute to the inhibition of NK cells. With increasing tumor burden, host NK cell activity declines. Therapy with indomethacin, a dual COX-1 and COX-2 inhibitor, in tumor-bearing animals showed appreciable restoration of the NK cell activity. In one study, normal NK activity at maximum effector to target ratio was 26% specific cytotoxicity, but in tumor-bearing mice, NK cells were only able to lyse 6% of their targets; with indomethacin treatment, cytotoxicity was restored to 10–18% [138].
It is known that PGE2 downregulates IL2-activated lymphokine-activated killer (LAK) cell cytotoxicity through EP2 receptors [142, 143]. LAK cells (activated NK cells) are generated from adherent splenocytes cultured in IL2. In a study by Su et al. [142], LAK cells were treated with various EP receptor agonists and antagonists to identify which were involved in the immunosuppressive effects of PGE2. They show that an EP2 agonist significantly inhibits LAK cytotoxicity and that the inhibitory effects of PGE2 can be blocked using an EP2 antagonist. In contrast, neither an EP1/EP3 agonist nor EP1 or EP4 antagonists modulate LAK cell cytolytic activity. Thus, PGE2-mediated inhibition of LAK cytotoxic activity is through the EP2, but not the EP1, EP3, or EP4 receptors. We have recently investigated the role of individual EP receptors in regulating activities of endogenous, resident splenic NK cells [144]. Like PGE2, the EP4 agonist PGE1-OH blocked NK cell migration to a panel of chemokines. In contrast to the inhibitory actions of PGE2, the EP1/EP3 agonist sulprostone increased migration. Unlike the opposing effects of EP4 versus EP1/EP3 on migration, agonists of each EP receptor were uniformly inhibiting to NK-mediated cytotoxicity. The EP4 agonist PGE1-OH inhibited IFNγ production from NK cells. Agonists for EP1, EP2, and EP3 were not as effective at inhibiting IFNγ. Agonists of EP1, EP2, and EP4 all inhibited TNFα; EP4 agonists were the most potent. Thus, the EP4 receptor consistently contributed to loss of function. These results, taken together, support a mechanism whereby inhibiting PGE2 production or preventing signaling through the EP4 receptor may prevent suppression of NK functions that are critical to the control of breast cancer metastasis.
The EP4 and/or EP2 receptors have also been identified as managing inhibitory and suppressive properties of other immune cells (T cells, B cells, macrophages, etc.). CD4+ T cells have been divided into four distinct subtypes (Th1, Th2, Tfh, and Th17) based on the help they provide to other effector cells. Proliferation of Th1 T cells, also known as the cytotoxic T cells, is inhibited through EP2 [145]. T cells deficient in EP1 and EP3 were susceptible to PGE2 inhibition, while only at the highest concentration of PGE2 were T cells resistant to the inhibitory effects of PGE2 through EP4 receptor. This suggested that the EP2 and maybe the EP4 receptors mediated the suppressive effects of PGE2 on T cells. The generation of the Th17 subset of CD4+ T cells by TGF-β and IL-6 is suppressed by PGE2. The suppressive effects of PGE2 were mediated by both the EP2 and EP4 receptors [146]. PGE2 can exert opposing effects by enhancing Th17 cell expansion in human T cells. Using human peripheral blood T cells, Boniface et al. [147] reported that PGE2 drove the development of human Th17 cells in the presence of IL-23 and IL-1β through the EP2 and EP4 receptors. This shows that the combination of specific inflammatory cytokines during differentiation and activation determines the ultimate phenotype of Th17 cells. CD4+CD25+ T regulatory cells suppress potential antitumor responses. T regulatory cell-specific transcription factor, FOXp3, is induced by tumor-derived PGE2. Splenocytes treated with PGE2 had a 12-fold induction in FOXp3 gene expression which is associated with suppressed antitumor immune responses [148]. In vitro, the EP2/EP4 receptor agonist, 11-deoxy-PGE1 and the EP2 agonist, butaprost, induced FOXp3 expression in Treg cells by 25- and 16-fold, respectively. This was further confirmed using knockout mice where Treg cell FOXp3 expression was reduced in EP4−/− mice and ablated in EP2−/− mice.
PGE2 affects B cells differently depending on the stage of B cell development and activity. Secretion of IgG by splenic B cells was increased with the addition of PGE2 [135]. MHC class II molecules are antigen-presenting cells that are necessary for the endocytic and endogenous pathways, while CD23, FcεRII, represents maturing B cells and is important in the regulation of IgE levels. On quiescent B lymphocytes, EP2 or EP4 agonists inhibited the expression of class II major histocompatibility complex and CD23 [149]. This shows that PGE2 acts in an inhibitory manner on immature and developing cells. PGE2 promotes IgE switching in IgM-positive B cells. PGE2 promoted IL-4 and LPS stimulated B-lymphocyte Ig isotype switching from IgM to IgE mainly through the EP4 receptor [149]. Treatment of activated B lymphocytes with PGE2 increases numbers of IgE-expressing cells, which may lead to hypersensitivity and play a role in pathogenesis of allergy and asthma.
Macrophages are known to produce high levels of PGE2. Released PGE2 also acts on the macrophages themselves and exhibits inhibitory effects on early- and late-stage activation processes, producing a negative feedback. PGE2 exerts inhibitory actions on macrophages through the reduction of cytokine production. In C3H/HeJ macrophages it is reported that PGE2 inhibits TNFα and IL-12 through the EP4 receptor [136]. The interesting finding from this report was that stimulated macrophages undergo receptor switching from EP4 to EP2. This may be a mechanism for the macrophage to avoid EP4-mediated inhibition. It has been shown that chemokines can modulate the migratory capacity of macrophages in response to PGE2 [150]. In response to the CCR7 ligands, CCL19 and CCL21, the EP2 and EP4 receptors regulate the migratory response of macrophages. In a chemotaxis assay, MONO-MAC-1 cells migrated efficiently toward both CCL19 and CCL21. This response was further enhanced in the presence of the EP2 agonist, butaprost, EP2/EP4 agonist, 11-deoxy-PGE1, and the EP1/EP3/EP4 agonist, 17-PT-PGE2. In contrast, the EP1/EP3 agonist, sulprostone, was unable to induce MONO-MAC-1 cells to migrate. Therefore, the EP2 and EP4 receptors are required for the increased migratory capacity of macrophages to CCL19 and CCL21 in the presence of PGE2. Survivin, an inhibitor of apoptosis is increased in dendritic cells induced by PGE2. Monocyte-DC cells treated with the EP2 agonist, butaprost, induced a significant increase in survivin expression. The EP1/EP3 agonist, sulprostone, was unable to cause survivin induction in monocyte-DC cells. Therefore, the EP2 receptor has been implicated in regulating PGE2-mediated induction of dendritic cell survivin.
Tumor-derived PGE2 is known to have direct inhibitory effects on immune cell functions. Monocytes from patients with breast cancer contribute to increased levels of PGE2 produced by peripheral blood mononuclear cells in culture, and this correlates with a decrease in NK cell activity [140]. We show that NK functions (lysis, migration, cytokine production) are compromised in tumor-bearing mice and that tumor produced PGE2 interferes with the function of NK cells ([144], unpublished data). PGE2 inhibits the potential of NK cells to migrate, exert cytotoxic effects, and secrete IFNγ. This ability of PGE2 to inhibit NK cells from tumor-bearing mice is through EP2 and EP4 receptors. In contrast to the inhibitory effects of PGE2 on cytotoxicity, and IFNγ production, TNFα secretion was induced in NK cells from tumor-bearing mice. PGE2 is uniformly inhibitory to the production of TNFα by NK cells from normal mice. Taken together these data show that NK functions are depressed in tumor-bearing hosts and this suppression is mediated by tumor-derived PGE2 acting on EP2 and EP4 receptors.
In the human leukemic T-cell line, Jurkat, it is postulated that an EP receptor linked to adenylate cyclase, but pharmacologically distinct from all EP receptors described, is present on the cells [151]. These findings may represent the EP receptor profile changes that can occur during tumorigenesis ([144], unpublished results) or the lack of responsiveness to selective agonists present at the time. Fedyk et al. [152] showed that EP1, EP3β, and EP4 were expressed at the same level in B cells from normal splenocytes as in B cell lymphoma and myeloma cell lines. This was irrespective of the fact that PGE2 only exerts inhibitory effects on immature and developing B cells and not mature B cells. They also showed that LPS upregulated EP4 receptor expression in the mature B cell lymphoma line, however the polyclonal activator was unable to affect EP1 and EP3β receptor expression. This shows that the EP4 receptor may play a role in PGE2 mediated inhibition of B cells. Myeloid-derived suppressor cells (MDSCs) are induced in cancer patients and animals and are potent suppressors of immunity. PGE2 has been shown to promote 4T1 mammary carcinoma by inducing the accumulation of MDSC [153]. In EP2−/− mice injected with 4T1 mammary carcinoma cells, the number of MDSCs was reduced and tumor growth was inhibited. This indicated that PGE2 mediates MDSC accumulation through the EP2 receptor.
9 Summary
Elevated cyclooxygenase activity is common in many tumors. Preclinical and epidemiologic data support the use of NSAIDs and selective COXibs for the prevention and treatment of malignancy. Clinical trials with COXibs have been less successful, and significant toxicities have been associated with the use of COXibs. Many laboratories have investigated the possibility of targeting other aspects of the COX pathway. We have attempted to summarize the growing literature regarding the role of the prostaglandin E receptor family in malignant behavior. Our studies and those of many other laboratories have shown that EP receptors are expressed in malignant cells, and there is an emerging body of literature that some if not all EP receptors may be therapeutic targets. Each of the four EP receptors has distinct binding characteristics and is coupled to different intracellular signaling pathways, so it is not surprising that some receptors contribute to tumor progression and others may be protective. These different roles are tissue and cell dependent as well. Additional layers of complexity are contributed by the multiple subcellular locations of the same receptor. While EP receptors are classical plasma membrane-expressed G protein-coupled receptors, emerging data indicate that EP receptors are expressed at other subcellular locations where they may have distinct functions. Expression of EP receptors in the stroma and by host immune effector cells also plays a role in tumor behavior. EP receptors expressed by stromal cells drive aromatase expression in breast cancer. EP receptors expressed on host immune cells mediate the marked immune suppression that characterizes tumor progression. As the reagents to study these receptors improve and as additional selective EP agonists and antagonists become available, our knowledge regarding the role of each EP receptor in cancer will grow so that we can more effectively exploit these as therapeutic targets.
References
Legler, D. F., Bruckner, M., Uetz-von Allmen, E., & Krause, P. (2010). Prostaglandin E2 at new glance: novel insights in functional diversity offer therapeutic chances. The International Journal of Biochemistry & Cell Biology, 42(2), 198–201.
Wang, M.-T., Honn, K. V., & Nie, D. (2007). Cyclooxygenases, prostanoids, and tumor progression. Cancer and Metastasis Reviews, 26(3–4), 525–534.
Sugimoto, Y., & Narumiya, S. (2007). Prostaglandin E receptors. Journal of Biological Chemistry, 282(16), 11613–11617.
Dannenberg, A. J., & Subbaramaiah, K. (2003). Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell, 4(6), 431–436.
Kundu, N., Yang, Q., Dorsey, R., & Fulton, A. M. (2001). Increased cyclooxygenase-2 (cox-2) expression and activity in a murine model of metastatic breast cancer. International Journal of Cancer, 93(5), 681–686.
Greenhough, A., Smartt, H. J. M., Moore, A. E., Roberts, H. R., Williams, A. C., Paraskeva, C., et al. (2009). The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis, 30(3), 377–386.
Menter, D. G., Schilsky, R. L., & DuBois, R. N. (2010). Cyclooxygenase-2 and cancer treatment: understanding the risk should be worth the reward. Clinical Cancer Research, 16(5), 1384–1390.
Wang, D., & Dubois, R. N. (2010). Eicosanoids and cancer. Nature Reviews. Cancer, 10(3), 181–193.
Nakanishi, M., Gokhale, V., Meuillet, E. J., & Rosenberg, D. W. (2010). mPGES-1 as a target for cancer suppression: a comprehensive invited review “Phospholipase A2 and lipid mediators”. Biochimie, 92(6), 660–664.
Schetter, A. J., Heegaard, N. H. H., & Harris, C. C. (2010). Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis, 31(1), 37–49.
Howe, L. R. (2007). Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Research, 9(4), 210.
Howe, L. R., & Dannenberg, A. J. (2003). COX-2 inhibitors for the prevention of breast cancer. Journal of Mammary Gland Biology and Neoplasia, 8(1), 31–43.
Ristimäki, A., Sivula, A., Lundin, J., Lundin, M., Salminen, T., Haglund, C., et al. (2002). Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Research, 62(3), 632–635.
Singh, B., Berry, J. A., Shoher, A., Ayers, G. D., Wei, C., & Lucci, A. (2007). COX-2 involvement in breast cancer metastasis to bone. Oncogene, 26(26), 3789–3796.
Liu, C. H., Chang, S. H., Narko, K., Trifan, O. C., Wu, M. T., Smith, E., et al. (2001). Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. Journal of Biological Chemistry, 276(21), 18563–18569.
Zerkowski, M. P., Camp, R. L., Burtness, B. A., Rimm, D. L., & Chung, G. G. (2007). Quantitative analysis of breast cancer tissue microarrays shows high cox-2 expression is associated with poor outcome. Cancer Investigation, 25(1), 19–26.
Chuah, B. Y. S., Putti, T., Salto-Tellez, M., Charlton, A., Iau, P., Buhari, S. A., et al. (2011). Serial changes in the expression of breast cancer-related proteins in response to neoadjuvant chemotherapy. Annals of Oncology, 22(8), 1748–1754.
Ranger, G. S., Thomas, V., Jewell, A., & Mokbel, K. (2004). Elevated cyclooxygenase-2 expression correlates with distant metastases in breast cancer. Anticancer Research, 24(4), 2349–2351.
Ma, X., Yang, Q., Wilson, K. T., Kundu, N., Meltzer, S. J., & Fulton, A. M. (2004). Promoter methylation regulates cyclooxygenase expression in breast cancer. Breast Cancer Research, 6(4), R316–321.
Kundu, N., & Fulton, A. M. (2002). Selective cyclooxygenase (COX)-1 or COX-2 inhibitors control metastatic disease in a murine model of breast cancer. Cancer Research, 62(8), 2343–2346.
Minn, A. J., Gupta, G. P., Siegel, P. M., Bos, P. D., Shu, W., Giri, D. D., et al. (2005). Genes that mediate breast cancer metastasis to lung. Nature, 436(7050), 518–524.
Bos, P. D., Zhang, X. H.-F., Nadal, C., Shu, W., Gomis, R. R., Nguyen, D. X., et al. (2009). Genes that mediate breast cancer metastasis to the brain. Nature, 459(7249), 1005–1009.
Singh, B., Berry, J. A., Shoher, A., Ramakrishnan, V., & Lucci, A. (2005). COX-2 overexpression increases motility and invasion of breast cancer cells. International Journal of Oncology, 26(5), 1393–1399.
Stasinopoulos, I., O’Brien, D. R., Wildes, F., Glunde, K., & Bhujwalla, Z. M. (2007). Silencing of cyclooxygenase-2 inhibits metastasis and delays tumor onset of poorly differentiated metastatic breast cancer cells. Molecular Cancer Research, 5(5), 435–442.
Greenberg, E. R., Baron, J. A., Freeman, D. H., Jr., Mandel, J. S., & Haile, R. (1993). Reduced risk of large-bowel adenomas among aspirin users. The Polyp Prevention Study Group. Journal of the National Cancer Institute, 85(11), 912–916.
Logan, R. F., Little, J., Hawtin, P. G., & Hardcastle, J. D. (1993). Effect of aspirin and non-steroidal anti-inflammatory drugs on colorectal adenomas: case–control study of subjects participating in the Nottingham faecal occult blood screening programme. BMJ, 307(6899), 285–289.
Reeves, M. J., Newcomb, P. A., Trentham-Dietz, A., Storer, B. E., & Remington, P. L. (1996). Nonsteroidal anti-inflammatory drug use and protection against colorectal cancer in women. Cancer Epidemiology, Biomarkers & Prevention, 5(12), 955–960.
Thun, M. J., Namboodiri, M. M., & Heath, C. W., Jr. (1991). Aspirin use and reduced risk of fatal colon cancer. The New England Journal of Medicine, 325(23), 1593–1596.
Eberhart, C. E., Coffey, R. J., Radhika, A., Giardiello, F. M., Ferrenbach, S., & DuBois, R. N. (1994). Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology, 107(4), 1183–1188.
Kargman, S. L., O’Neill, G. P., Vickers, P. J., Evans, J. F., Mancini, J. A., & Jothy, S. (1995). Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Research, 55(12), 2556–2559.
Sano, H., Kawahito, Y., Wilder, R. L., Hashiramoto, A., Mukai, S., Asai, K., et al. (1995). Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Research, 55(17), 3785–3789.
Kutchera, W., Jones, D. A., Matsunami, N., Groden, J., McIntyre, T. M., Zimmerman, G. A., et al. (1996). Prostaglandin H synthase 2 is expressed abnormally in human colon cancer: evidence for a transcriptional effect. Proceedings of the National Academy of Sciences of the United States of America, 93(10), 4816–4820.
Howe, L. R., Subbaramaiah, K., Brown, A. M., & Dannenberg, A. J. (2001). Cyclooxygenase-2: a target for the prevention and treatment of breast cancer. Endocrine-Related Cancer, 8(2), 97–114.
Larkins, T. L., Nowell, M., Singh, S., & Sanford, G. L. (2006). Inhibition of cyclooxygenase-2 decreases breast cancer cell motility, invasion and matrix metalloproteinase expression. BMC Cancer, 6, 181.
Connolly, E. M., Harmey, J. H., O’Grady, T., Foley, D., Roche-Nagle, G., Kay, E., et al. (2002). Cyclo-oxygenase inhibition reduces tumour growth and metastasis in an orthotopic model of breast cancer. British Journal of Cancer, 87(2), 231–237.
Friedman, G. D., & Ury, H. K. (1980). Initial screening for carcinogenicity of commonly used drugs. Journal of the National Cancer Institute, 65(4), 723–733.
Harris, R. E., Chlebowski, R. T., Jackson, R. D., Frid, D. J., Ascenseo, J. L., Anderson, G., et al. (2003). Breast cancer and nonsteroidal anti-inflammatory drugs: prospective results from the Women’s Health Initiative. Cancer Research, 63(18), 6096–6101.
Harris, R. E., Namboodiri, K. K., & Farrar, W. B. (1996). Nonsteroidal antiinflammatory drugs and breast cancer. Epidemiology, 7(2), 203–205.
Harris, R. E., Beebe-Donk, J., & Alshafie, G. A. (2006). Reduction in the risk of human breast cancer by selective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer, 6, 27.
Rahme, E., Ghosn, J., Dasgupta, K., Rajan, R., & Hudson, M. (2005). Association between frequent use of nonsteroidal anti-inflammatory drugs and breast cancer. BMC Cancer, 5, 159.
Holmes, M. D., Chen, W. Y., Li, L., Hertzmark, E., Spiegelman, D., & Hankinson, S. E. (2010). Aspirin intake and survival after breast cancer. Journal of Clinical Oncology, 28(9), 1467–1472.
Ready, A., Velicer, C. M., McTiernan, A., & White, E. (2008). NSAID use and breast cancer risk in the VITAL cohort. Breast Cancer Research and Treatment, 109(3), 533–543.
Khuder, S. A., & Mutgi, A. B. (2001). Breast cancer and NSAID use: a meta-analysis. British Journal of Cancer, 84(9), 1188–1192.
Bosetti, C., Gallus, S., & La Vecchia, C. (2006). Aspirin and cancer risk: an updated quantitative review to 2005. Cancer Causes & Control, 17(7), 871–888.
Mangiapane, S., Blettner, M., & Schlattmann, P. (2008). Aspirin use and breast cancer risk: a meta-analysis and meta-regression of observational studies from 2001 to 2005. Pharmacoepidemiology and Drug Safety, 17(2), 115–124.
Takkouche, B., Regueira-Méndez, C., & Etminan, M. (2008). Breast cancer and use of nonsteroidal anti-inflammatory drugs: a meta-analysis. Journal of the National Cancer Institute, 100(20), 1439–1447.
Valsecchi, M. E., Pomerantz, S. C., Jaslow, R., & Tester, W. (2009). Reduced risk of bone metastasis for patients with breast cancer who use COX-2 inhibitors. Clinical Breast Cancer, 9(4), 225–230.
Gill, J. K., Maskarinec, G., Wilkens, L. R., Pike, M. C., Henderson, B. E., & Kolonel, L. N. (2007). Nonsteroidal antiinflammatory drugs and breast cancer risk: the multiethnic cohort. American Journal of Epidemiology, 166(10), 1150–1158.
Gierach, G. L., Lacey, J. V., Jr., Schatzkin, A., Leitzmann, M. F., Richesson, D., Hollenbeck, A. R., et al. (2008). Nonsteroidal anti-inflammatory drugs and breast cancer risk in the National Institutes of Health-AARP Diet and Health Study. Breast Cancer Research, 10(2), R38.
Zhang, S. M., Cook, N. R., Manson, J. E., Lee, I.-M., & Buring, J. E. (2008). Low-dose aspirin and breast cancer risk: results by tumour characteristics from a randomised trial. British Journal of Cancer, 98(5), 989–991.
Jacobs, E. J., Thun, M. J., Bain, E. B., Rodriguez, C., Henley, S. J., & Calle, E. E. (2007). A large cohort study of long-term daily use of adult-strength aspirin and cancer incidence. Journal of the National Cancer Institute, 99(8), 608–615.
Bresalier, R. S., Sandler, R. S., Quan, H., Bolognese, J. A., Oxenius, B., Horgan, K., et al. (2005). Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. The New England Journal of Medicine, 352(11), 1092–1102.
Solomon, S. D., McMurray, J. J. V., Pfeffer, M. A., Wittes, J., Fowler, R., Finn, P., et al. (2005). Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. The New England Journal of Medicine, 352(11), 1071–1080.
Grosser, T., Fries, S., & FitzGerald, G. A. (2006). Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. The Journal of Clinical Investigation, 116(1), 4–15.
Oldham, W. M., & Hamm, H. E. (2008). Heterotrimeric G protein activation by G-protein-coupled receptors. Nature Reviews Molecular Cell Biology, 9(1), 60–71.
Lappano, R., & Maggiolini, M. (2011). G protein-coupled receptors: novel targets for drug discovery in cancer. Nature Reviews. Drug Discovery, 10(1), 47–60.
Fulton, A. M., Ma, X., & Kundu, N. (2006). Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Research, 66(20), 9794–9797.
Ichikawa, A., Sugimoto, Y., & Tanaka, S. (2010). Molecular biology of histidine decarboxylase and prostaglandin receptors. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences, 86(8), 848–866.
Regan, J. W. (2003). EP2 and EP4 prostanoid receptor signaling. Life Sciences, 74(2–3), 143–153.
Dey, I., Lejeune, M., & Chadee, K. (2006). Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. British Journal of Pharmacology, 149(6), 611–623.
Fujino, H., & Regan, J. W. (2006). EP(4) prostanoid receptor coupling to a pertussis toxin-sensitive inhibitory G protein. Molecular Pharmacology, 69(1), 5–10.
Katoh, H., Watabe, A., Sugimoto, Y., Ichikawa, A., & Negishi, M. (1995). Characterization of the signal transduction of prostaglandin E receptor EP1 subtype in cDNA-transfected Chinese hamster ovary cells. Biochimica et Biophysica Acta, 1244(1), 41–48.
Tabata, H., Tanaka, S., Sugimoto, Y., Kanki, H., Kaneko, S., & Ichikawa, A. (2002). Possible coupling of prostaglandin E receptor EP(1) to TRP5 expressed in Xenopus laevis oocytes. Biochemical and Biophysical Research Communications, 298(3), 398–402.
Ji, R., Chou, C.-L., Xu, W., Chen, X.-B., Woodward, D. F., & Regan, J. W. (2010). EP1 prostanoid receptor coupling to G i/o up-regulates the expression of hypoxia-inducible factor-1 alpha through activation of a phosphoinositide-3 kinase signaling pathway. Molecular Pharmacology, 77(6), 1025–1036.
Voss, B., McLaughlin, J. N., Holinstat, M., Zent, R., & Hamm, H. E. (2007). PAR1, but not PAR4, activates human platelets through a Gi/o/phosphoinositide-3 kinase signaling axis. Molecular Pharmacology, 71(5), 1399–1406.
Okuda-Ashitaka, E., Sakamoto, K., Ezashi, T., Miwa, K., Ito, S., & Hayaishi, O. (1996). Suppression of prostaglandin E receptor signaling by the variant form of EP1 subtype. Journal of Biological Chemistry, 271(49), 31255–31261.
Gomi, K., Zhu, F. G., & Marshall, J. S. (2000). Prostaglandin E2 selectively enhances the IgE-mediated production of IL-6 and granulocyte-macrophage colony-stimulating factor by mast cells through an EP1/EP3-dependent mechanism. Journal of Immunology, 165(11), 6545–6552.
Kotelevets, L., Foudi, N., Louedec, L., Couvelard, A., Chastre, E., & Norel, X. (2007). A new mRNA splice variant coding for the human EP3-I receptor isoform. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 77(3–4), 195–201.
Israel, D. D., & Regan, J. W. (2009). EP(3) prostanoid receptor isoforms utilize distinct mechanisms to regulate ERK 1/2 activation. Biochimica et Biophysica Acta, 1791(4), 238–245.
Kotani, M., Tanaka, I., Ogawa, Y., Usui, T., Mori, K., Ichikawa, A., et al. (1995). Molecular cloning and expression of multiple isoforms of human prostaglandin E receptor EP3 subtype generated by alternative messenger RNA splicing: multiple second messenger systems and tissue-specific distributions. Molecular Pharmacology, 48(5), 869–879.
Bilson, H. A., Mitchell, D. L., & Ashby, B. (2004). Human prostaglandin EP3 receptor isoforms show different agonist-induced internalization patterns. FEBS Letters, 572(1–3), 271–275.
Jin, J., Mao, G. F., & Ashby, B. (1997). Constitutive activity of human prostaglandin E receptor EP3 isoforms. British Journal of Pharmacology, 121(2), 317–323.
Breyer, R. M., Bagdassarian, C. K., Myers, S. A., & Breyer, M. D. (2001). Prostanoid receptors: subtypes and signaling. Annual Review of Pharmacology and Toxicology, 41, 661–690.
An, S., Yang, J., So, S. W., Zeng, L., & Goetzl, E. J. (1994). Isoforms of the EP3 subtype of human prostaglandin E2 receptor transduce both intracellular calcium and cAMP signals. Biochemistry, 33(48), 14496–14502.
Schmid, A., Thierauch, K. H., Schleuning, W. D., & Dinter, H. (1995). Splice variants of the human EP3 receptor for prostaglandin E2. European Journal of Biochemistry, 228(1), 23–30.
Hatae, N., Sugimoto, Y., & Ichikawa, A. (2002). Prostaglandin receptors: advances in the study of EP3 receptor signaling. Journal of Biochemistry, 131(6), 781–784.
Sugimoto, Y., Negishi, M., Hayashi, Y., Namba, T., Honda, A., Watabe, A., et al. (1993). Two isoforms of the EP3 receptor with different carboxyl-terminal domains. Identical ligand binding properties and different coupling properties with Gi proteins. Journal of Biological Chemistry, 268(4), 2712–2718.
Irie, A., Sugimoto, Y., Namba, T., Harazono, A., Honda, A., Watabe, A., et al. (1993). Third isoform of the prostaglandin-E-receptor EP3 subtype with different C-terminal tail coupling to both stimulation and inhibition of adenylate cyclase. European Journal of Biochemistry, 217(1), 313–318.
Yamaoka, K., Yano, A., Kuroiwa, K., Morimoto, K., Inazumi, T., Hatae, N., et al. (2009). Prostaglandin EP3 receptor superactivates adenylyl cyclase via the Gq/PLC/Ca2+ pathway in a lipid raft-dependent manner. Biochemical and Biophysical Research Communications, 389(4), 678–682.
Hatae, N., Yamaoka, K., Sugimoto, Y., Negishi, M., & Ichikawa, A. (2002). Augmentation of receptor-mediated adenylyl cyclase activity by Gi-coupled prostaglandin receptor subtype EP3 in a Gbetagamma subunit-independent manner. Biochemical and Biophysical Research Communications, 290(1), 162–168.
Irie, A., Segi, E., Sugimoto, Y., Ichikawa, A., & Negishi, M. (1994). Mouse prostaglandin E receptor EP3 subtype mediates calcium signals via Gi in cDNA-transfected Chinese hamster ovary cells. Biochemical and Biophysical Research Communications, 204(1), 303–309.
Zhu, T., Gobeil, F., Vazquez-Tello, A., Leduc, M., Rihakova, L., Bossolasco, M., et al. (2006). Intracrine signaling through lipid mediators and their cognate nuclear G-protein-coupled receptors: a paradigm based on PGE2, PAF, and LPA1 receptors. Canadian Journal of Physiology and Pharmacology, 84(3–4), 377–391.
Gobeil, F., Fortier, A., Zhu, T., Bossolasco, M., Leduc, M., Grandbois, M., et al. (2006). G-protein-coupled receptors signalling at the cell nucleus: an emerging paradigm. Canadian Journal of Physiology and Pharmacology, 84(3–4), 287–297.
Gobeil, F., Jr., Vazquez-Tello, A., Marrache, A. M., Bhattacharya, M., Checchin, D., Bkaily, G., et al. (2003). Nuclear prostaglandin signaling system: biogenesis and actions via heptahelical receptors. Canadian Journal of Physiology and Pharmacology, 81(2), 196–204.
Gobeil, F., Jr., Dumont, I., Marrache, A. M., Vazquez-Tello, A., Bernier, S. G., Abran, D., et al. (2002). Regulation of eNOS expression in brain endothelial cells by perinuclear EP(3) receptors. Circulation Research, 90(6), 682–689.
Bhattacharya, M., Peri, K., Ribeiro-da-Silva, A., Almazan, G., Shichi, H., Hou, X., et al. (1999). Localization of functional prostaglandin E2 receptors EP3 and EP4 in the nuclear envelope. Journal of Biological Chemistry, 274(22), 15719–15724.
Bhattacharya, M., Peri, K. G., Almazan, G., Ribeiro-da-Silva, A., Shichi, H., Durocher, Y., et al. (1998). Nuclear localization of prostaglandin E2 receptors. Proceedings of the National Academy of Sciences of the United States of America, 95(26), 15792–15797.
Thorat, M. A., Morimiya, A., Mehrotra, S., Konger, R., & Badve, S. S. (2008). Prostanoid receptor EP1 expression in breast cancer. Modern Pathology, 21(1), 15–21.
Ma, X., Kundu, N., Ioffe, O. B., Goloubeva, O., Konger, R., Baquet, C., et al. (2010). Prostaglandin E receptor EP1 suppresses breast cancer metastasis and is linked to survival differences and cancer disparities. Molecular Cancer Research, 8(10), 1310–1318.
Han, C., Demetris, A. J., Stolz, D. B., Xu, L., Lim, K., & Wu, T. (2006). Modulation of Stat3 activation by the cytosolic phospholipase A2alpha and cyclooxygenase-2-controlled prostaglandin E2 signaling pathway. Journal of Biological Chemistry, 281(34), 24831–24846.
Rolland, P. H., Martin, P. M., Jacquemier, J., Rolland, A. M., & Toga, M. (1980). Prostaglandin in human breast cancer: evidence suggesting that an elevated prostaglandin production is a marker of high metastatic potential for neoplastic cells. Journal of the National Cancer Institute, 64(5), 1061–1070.
Bennett, A., Charlier, E. M., McDonald, A. M., Simpson, J. S., Stamford, I. F., & Zebro, T. (1977). Prostaglandins and breast cancer. Lancet, 2(8039), 624–626.
Tan, W. C., Privett, O. S., & Goldyne, M. E. (1974). Studies of prostaglandins in rat mammary tumors induced by 7,12-dimethylbenz(a)anthracene. Cancer Research, 34(12), 3229–3231.
Chang, S.-H., Liu, C. H., Conway, R., Han, D. K., Nithipatikom, K., Trifan, O. C., et al. (2004). Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proceedings of the National Academy of Sciences of the United States of America, 101(2), 591–596.
Chang, S.-H., Ai, Y., Breyer, R. M., Lane, T. F., & Hla, T. (2005). The prostaglandin E2 receptor EP2 is required for cyclooxygenase 2-mediated mammary hyperplasia. Cancer Research, 65(11), 4496–4499.
Chang, S.-H., Liu, C. H., Wu, M.-T., & Hla, T. (2005). Regulation of vascular endothelial cell growth factor expression in mouse mammary tumor cells by the EP2 subtype of the prostaglandin E2 receptor. Prostaglandins & Other Lipid Mediators, 76(1–4), 48–58.
Tian, M., & Schiemann, W. P. (2010). PGE2 receptor EP2 mediates the antagonistic effect of COX-2 on TGF-beta signaling during mammary tumorigenesis. The FASEB Journal, 24(4), 1105–1116.
Subbaramaiah, K., Hudis, C., Chang, S. H., Hla, T., & Dannenberg, A. J. (2008). EP2 and EP4 receptors regulate aromatase expression in human adipocytes and breast cancer cells. Evidence of a BRCA1 and p300 exchange. Journal of Biological Chemistry, 283(6), 3433–3444.
Richards, J. A., & Brueggemeier, R. W. (2003). Prostaglandin E2 regulates aromatase activity and expression in human adipose stromal cells via two distinct receptor subtypes. Journal of Clinical Endocrinology and Metabolism, 88(6), 2810–2816.
Zhao, Y., Agarwal, V. R., Mendelson, C. R., & Simpson, E. R. (1996). Estrogen biosynthesis proximal to a breast tumor is stimulated by PGE2 via cyclic AMP, leading to activation of promoter II of the CYP19 (aromatase) gene. Endocrinology, 137(12), 5739–5742.
Han, E. H., Kim, H. G., Hwang, Y. P., Choi, J. H., Im, J. H., Park, B., et al. (2010). The role of cyclooxygenase-2-dependent signaling via cyclic AMP response element activation on aromatase up-regulation by o,p′-DDT in human breast cancer cells. Toxicology Letters, 198(3), 331–341.
Subbaramaiah, K., Howe, L. R., Bhardwaj, P., Du, B., Gravaghi, C., Yantiss, R. K., et al. (2011). Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prevention Research (Philadelphia, Pa.), 4(3), 329–346.
Robertson, F. M., Simeone, A.-M., Mazumdar, A., Shah, A. H., McMurray, J. S., Ghosh, S., et al. (2008). Molecular and pharmacological blockade of the EP4 receptor selectively inhibits both proliferation and invasion of human inflammatory breast cancer cells. Journal of Experimental Therapeutics and Oncology, 7(4), 299–312.
Robertson, F. M., Simeone, A.-M., Lucci, A., McMurray, J. S., Ghosh, S., & Cristofanilli, M. (2010). Differential regulation of the aggressive phenotype of inflammatory breast cancer cells by prostanoid receptors EP3 and EP4. Cancer, 116(11 Suppl), 2806–2814.
Blindt, R., Bosserhoff, A.-K., vom Dahl, J., Hanrath, P., Schrör, K., Hohlfeld, T., et al. (2002). Activation of IP and EP(3) receptors alters cAMP-dependent cell migration. European Journal of Pharmacology, 444(1–2), 31–37.
Amano, H., Hayashi, I., Endo, H., Kitasato, H., Yamashina, S., Maruyama, T., et al. (2003). Host prostaglandin E(2)-EP3 signaling regulates tumor-associated angiogenesis and tumor growth. The Journal of Experimental Medicine, 197(2), 221–232.
Taniguchi, T., Fujino, H., Israel, D. D., Regan, J. W., & Murayama, T. (2008). Human EP3(I) prostanoid receptor induces VEGF and VEGF receptor-1 mRNA expression. Biochemical and Biophysical Research Communications, 377(4), 1173–1178.
Kubo, H., Hosono, K., Suzuki, T., Ogawa, Y., Kato, H., Kamata, H., et al. (2010). Host prostaglandin EP3 receptor signaling relevant to tumor-associated lymphangiogenesis. Biomedicine and Pharmacotherapy, 64(2), 101–106.
Timoshenko, A. V., Chakraborty, C., Wagner, G. F., & Lala, P. K. (2006). COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. British Journal of Cancer, 94(8), 1154–1163.
Tober, K. L., Wilgus, T. A., Kusewitt, D. F., Thomas-Ahner, J. M., Maruyama, T., & Oberyszyn, T. M. (2006). Importance of the EP(1) receptor in cutaneous UVB-induced inflammation and tumor development. The Journal of Investigative Dermatology, 126(1), 205–211.
Bai, X.-M., Jiang, H., Ding, J.-X., Peng, T., Ma, J., Wang, Y.-H., et al. (2010). Prostaglandin E2 upregulates survivin expression via the EP1 receptor in hepatocellular carcinoma cells. Life Sciences, 86(5–6), 214–223.
Kawamori, T., Uchiya, N., Nakatsugi, S., Watanabe, K., Ohuchida, S., Yamamoto, H., et al. (2001). Chemopreventive effects of ONO-8711, a selective prostaglandin E receptor EP(1) antagonist, on breast cancer development. Carcinogenesis, 22(12), 2001–2004.
Chandramouli, A., Mercado-Pimentel, M. E., Hutchinson, A., Gibadulinová, A., Olson, E. R., Dickinson, S., et al. (2010). The induction of S100p expression by the Prostaglandin E2 (PGE2)/EP4 receptor signaling pathway in colon cancer cells. Cancer Biology & Therapy, 10(10), 1056–1066.
Cherukuri, D. P., Chen, X. B. O., Goulet, A.-C., Young, R. N., Han, Y., Heimark, R. L., et al. (2007). The EP4 receptor antagonist, L-161,982, blocks prostaglandin E2-induced signal transduction and cell proliferation in HCA-7 colon cancer cells. Experimental Cell Research, 313(14), 2969–2979.
Yang, L., Huang, Y., Porta, R., Yanagisawa, K., Gonzalez, A., Segi, E., et al. (2006). Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Research, 66(19), 9665–9672.
Oshima, H., Popivanova, B. K., Oguma, K., Kong, D., Ishikawa, T. O., & Oshima, M. (2011). Activation of epidermal growth factor receptor signaling by the prostaglandin E(2) receptor EP4 pathway during gastric tumorigenesis. Cancer Science, 102(4), 713–719.
Terada, N., Shimizu, Y., Kamba, T., Inoue, T., Maeno, A., Kobayashi, T., et al. (2010). Identification of EP4 as a potential target for the treatment of castration-resistant prostate cancer using a novel xenograft model. Cancer Research, 70(4), 1606–1615.
Zheng, Y., Ritzenthaler, J. D., Sun, X., Roman, J., & Han, S. (2009). Prostaglandin E2 stimulates human lung carcinoma cell growth through induction of integrin-linked kinase: the involvement of EP4 and Sp1. Cancer Research, 69(3), 896–904.
Kim, J. I., Lakshmikanthan, V., Frilot, N., & Daaka, Y. (2010). Prostaglandin E2 promotes lung cancer cell migration via EP4-βArrestin1-c-Src signalsome. Molecular Cancer Research, 8(4), 569–577.
Timoshenko, A., Guoxiong, X., CHakrabarti, S., Lala, P., & Chakraborty, C. (2003). Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells. Experimental Cell Research, 289, 265–274.
Timoshenko, A. V., Lala, P. K., & Chakraborty, C. (2004). PGE2-mediated upregulation of iNOS in murine breast cancer cells through the activation of EP4 receptors. International Journal of Cancer, 108(3), 384–389.
Jadeski, L. C., Chakraborty, C., & Lala, P. K. (2002). Role of nitric oxide in tumour progression with special reference to a murine breast cancer model. Canadian Journal of Physiology and Pharmacology, 80(2), 125–135.
Ohshiba, T., Miyaura, C., & Ito, A. (2003). Role of prostaglandin E produced by osteoblasts in osteolysis due to bone metastasis. Biochemical and Biophysical Research Communications, 300(4), 957–964.
Pan, M.-R., Hou, M.-F., Chang, H.-C., & Hung, W.-C. (2008). Cyclooxygenase-2 up-regulates CCR7 via EP2/EP4 receptor signaling pathways to enhance lymphatic invasion of breast cancer cells. Journal of Biological Chemistry, 283(17), 11155–11163.
Subbaramaiah, K., Benezra, R., Hudis, C., & Dannenberg, A. J. (2008). Cyclooxygenase-2-derived prostaglandin E2 stimulates Id-1 transcription. Journal of Biological Chemistry, 283(49), 33955–33968.
Ma, X., Kundu, N., Rifat, S., Walser, T., & Fulton, A. M. (2006). Prostaglandin E receptor EP4 antagonism inhibits breast cancer metastasis. Cancer Research, 66(6), 2923–2927.
Kundu, N., Ma, X., Holt, D., Goloubeva, O., Ostrand-Rosenberg, S., & Fulton, A. M. (2009). Antagonism of the prostaglandin E receptor EP4 inhibits metastasis and enhances NK function. Breast Cancer Research and Treatment, 117(2), 235–242.
Choudhry, M., Ahmed, Z., & Sayeed, M. (1999). PGE(2)-mediated inhibition of T-cell p59 (fyn) is independent of cAMP. American Journal of Physiology, 277(2Pt1), C301–C309.
Harris, S., Padilla, J., Koumas, L., Ray, D., & Phipps, R. (2002). Prostaglandins as modulators of immunity. Trends in Immunology, 23(3), 144–150.
Choudhry, M., Hockberger, P., & Sayeed, M. (1999). PGE2 suppresses mitogen-induced Ca2+ mobilization in T cells. American Journal of Physiology, 277(6Pt2), R17410–R1748.
Porter, B., & Malek, T. (1999). Prostaglandin E2 inhibits T-cell activation-induced apoptosis and Fas-mediated cellular cytotoxicity by blockade of Fas-ligand induction. European Journal of Immunology, 29(7), 2360–2365.
Hilkens, C., Snijders, A., Snijdewint, F., Wierenga, E., & Kapsenberg, M. (1996). Modulation of T-cell cytokine secretion by accessory-cell-derived products. European Respiratory Journal Supplement, 22, 90s–94s.
Garrone, P., Galibert, L., Rousset, F., Fu, S., & Branchereau, J. (1994). Regulatory effects of prostaglandin E2 on the growth and differentiation of human B lymphocytes activated through their CD40 antigen. Journal of Immunology, 152(9), 4282–4290.
Brown, D., Warner, G., Ales-Marinez, S., Scott, D., & Phipps, R. (1992). Prostaglandin E2 induces apoptosis in immature normal and malignant B lymphocytes. Clinical Immunology and Immunopathology, 63(3), 221–229.
Roper, R., Brown, D., & Phipps, R. (1995). Prostaglandin E2 promotes B lymphocyte Ig isotype switching to IgE. The Journal of Immunology, 154, 162–170.
Ikegami, R., Sugimoto, Y., Segi, E., Katsuyama, M., Karahashi, H., Amano, F., et al. (2001). The expression of prostaglandin E2 receptors EP2 and EP4 and their different regulation by lipopolysaccharide in C3H/HeN peritoneal macrophages. The Journal of Immunology, 166(7), 4689–4696.
Voiculescu, C., Rosu, L., & Rogoz, S. (1988). Modulation of mouse spleen natural killer (NK) cell activity by beta-interferon, interleukin-1, and prostaglandins. Lymphology, 21, 144–151.
Brunda, M. J., Herberman, R. B., & Holden, H. T. (1980). Inhibition of murine natural killer cell activity by prostaglandin. The Journal of Immunology, 124(6), 2682–2687.
Bankhurst, A. (1982). The modulation of human natural killer cell activity by prostaglandins. Journal of Clinical & Laboratory Immunology, 7, 85–91.
Baxevanis, C., Reclos, G., Gritzapis, A., Dedousis, G., Missitzis, I., & Papamichail, M. (1993). Elevated prostaglandin E2 production by monocytes is responsible for the depressed levels of natural killer and lymphokine-activated killer cell function in patients with breast cancer. Cancer, 72(2), 491–501.
Fulton, A. (1988). Inhibition of experimental metastasis with indomethacin: role of macrophages and natural killer cells. Prostaglandins, 35(3), 413–425.
Su, Y., Huang, X., Raskovalova, T., Zacharia, L., Lokshin, A., Jackson, E., et al. (2008). Cooperation of adenosine and prostaglandin E2 (PGE2) in amplification of cAMP-PKA signaling and immunosuppression. Cancer Immunology and Immunotherapeutics, 57, 1611–1623.
Su, Y., Jackson, E., & Gorelik, E. (2011). Receptor desensitization and blockade of the suppressive effects of prostaglandin E2 and adenosine on the cytotoxic activity of human melanoma-infiltrating T lymphocytes. Cancer Immunology and Immunotherapy, 60(1), 111–122.
Holt, D., Ma, X., Kundu, N., Fulton, A., (2011). Prostaglandin E2 (PGE2) suppresses natural killer cell function through the PGE2 receptor EP4. Cancer Immunology and Immunotherapy. doi:10.1007/s00262-011-1064-9.
Nataraj, C., Thomas, D., Tilley, S., Nguyen, M., Mannon, R., Koller, B., et al. (2001). Receptors for prostaglandin E2 that regulate cellular immune responses in the mouse. The Journal of Clinical Investigation, 108(8), 1229–1235.
Yao, C., Sakata, D., Esaki, Y., Li, Y., Matsuoka, T., Kuroiwa, K., et al. (2009). Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nature Medicine, 15(6), 633–640.
Boniface, K., Bak-Jensen, K., Li, Y., Blumenschein, W., McGeachy, M., McClanahan, T., et al. (2009). Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. The Journal of Experimental Medicine, 206(3), 535–548.
Sharma, S., Yang, S., Zhu, L., Reckamp, K., Gardner, B., Baratelli, F., et al. (2005). Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+CD25+ T regulatory cell activities in lung cancer. Cancer Research, 65(12), 5211–5220.
Fedyk, E., & Phipps, R. (1996). Prostaglandin E2 receptors of the EP2 and EP4 subtype regulate activation and differentiation of mouse B lymphocytes to IgE-secreting cells. Proceedings of the National Academy of Sciences, 93, 10978–10983.
Panzer, U., & Uguccioni, M. (2004). Prostaglandin E2 modulates the functional responsiveness of human monocytes to chemokines. European Journal of Immunology, 34(12), 3682–3689.
De Vries, G., Guarino, P., McLaughlin, A., Chen, J., Andrews, S., & Woodward, D. (1995). An EP receptor with a novel pharmacological profile in the T-cell line Jurkat. British Journal of Pharmacology, 115, 1231–1234.
Fedyk, E., Ripper, J., Brown, D., & Phipps, R. (1996). A molecular analysis of PGE receptor (EP) expression on normal and transformed B lymphocytes: coexpression of EP1, EP2, EP3β, EP4. Molecular Immunology, 33(1), 33–45.
Sinha, P., Clements, V. K., Fulton, A. M., & Ostrand-Rosenberg, S. (2007). Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Research, 67(9), 4507–4513.
Acknowledgments
This work is supported by the United States Department of Health and Human Services, United States Department of Defense, United States Department of Veteran Affairs, and Susan G. Komen for the Cure.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reader, J., Holt, D. & Fulton, A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev 30, 449–463 (2011). https://doi.org/10.1007/s10555-011-9303-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-011-9303-2