
Abstract
Somatostatin is an important regulator of endocrine and exocrine secretion, affecting the release of many hormones. The effects of somatostatin are mediated through its interaction with one of five somatostatin receptors. Gastroenteropancreatic neuroendocrine tumors (GEP-NETs) express multiple somatostatin receptors, making them excellent potential therapeutic targets. Many trials have shown that treatment with somatostatin analogs is associated with disease stabilization and prolonged survival. More recently, somatostatin analogs have been shown to have antiproliferative effects, thus broadening the scope of their uses. In this review, we update the current data on the treatment of GEP-NETs with somatostatin analogs, with particular emphasis on the results of the PROMID study. In addition, we discuss the current state of knowledge of novel therapies against GEP-NETs, including the use of somatostatin analogs with broader receptor binding profiles, chimeric somatostatin–dopamine molecules, combinations of somatostatin analogs with other active chemotherapy agents, and peptide receptor-targeted radionuclide therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The peptide hormone somatostatin, also known as somatotropin release-inhibiting factor, was first isolated and identified as a hypothalamic inhibitor of growth hormone secretion in 1973 [1]. It is a cyclopeptide consisting of 14 or 28 amino acids expressed in many tissues throughout the body including the central nervous system, hypothalamus, and gastrointestinal tract [1, 2]. Somatostatin is an important regulator of endocrine and exocrine secretion and affects the release of many hormones such as growth hormone (GH), glucagon, insulin, gastrin, secretin, and thyroid-stimulating hormone [2]. The neuroendocrine activity of somatostatin is reflected by its effective inhibition of GH release from the pituitary gland thereby indirectly also affecting the release of insulin-like growth factor-1 (IGF-1) from the liver and other peripheral organs, such as the heart and kidneys [3]. In the central nervous system, somatostatin acts as a neuromodulator and neurotransmitter [4].
The effects of somatostatin are mediated through interaction with five receptors (sst1–5) [5], belonging to a family of G-protein coupled receptors with seven transmembrane domains. These receptors are expressed in target tissues where they exert a large number of biological effects and have been cloned over the past years [6]. The sequence homology between receptor subtypes varies between 39% and 57%, with greatest sequence homology seen in the transmembrane domains [7]. All five somatostatin receptor subtypes modulate various intracellular signaling pathways such as adenylate cyclase, ion channels (K+, Ca2+), serine/threonine and tyrosine phosphatases as well as phospholipase A2 [7]. Most gastroenteropancreatic neuroendocrine tumors (GEP-NETs) overexpress mainly the somatostatin receptor sst2 [8], making it a good target for therapeutics.
Natural somatostatins (somatostatin-14, somatostatin-28) bind with high affinity to all five human somatostatin receptor subtypes [6, 7]. However, the clinical utility of native human somatostatin is limited by its short half-life of approximately 2 min due to rapid proteolytic degradation in plasma, thereby requiring continuous infusion regimens. In order to improve the pharmacokinetic profile, synthetic somatostatin analogs have been developed by shortening the polypeptide chain, while retaining the binding affinity to somatostatin receptors. A number of short synthetic somatostatin analogs with improved metabolic stability have been synthesized in the past, but octreotide (SMS-201-995) and lanreotide (BIM-23014) are the only two synthetic somatostatin analogs currently approved for clinical use [9, 10]. These analogs are used to treat disorders characterized by the excessive production of certain hormones such as GH, gastrin, secretin, glucagon, and insulin [11]. Moreover, these drugs have been proposed for the treatment of other pathological conditions, such as diabetic retinopathy, portal hypertension, pancreatic fistula, and Graves’ ophthalmopathy [12].
2 Classical somatostatin analogs
Since the introduction of somatostatin analogs, multiple phase II trials and retrospective series have demonstrated that treatment with somatostatin analogs is associated with prolonged survival and disease stabilization in a large proportion of patients with GEP-NETs.
A single-institution retrospective study of 146 patients with metastatic midgut NETs, 91% of whom had received long term octreotide treatment, demonstrated a 5-year survival rate of 75% compared to 19% in historical controls [13]. Among the first prospective studies documenting the antiproliferative effects of somatostatin analogs in GEP-NETs is a study conducted by the German Sandostatin Study Group. In this study, 103 patients with metastatic carcinoid and pancreatic endocrine tumors were treated with octreotide 200 μg thrice a day until evidence of tumor progression. Thirty-seven percent of patients with confirmed tumor progression experienced stabilization of tumor growth lasting for at least 3 months. In the subgroup with stable disease before octreotide, stable disease continued in 54% of patients over 12 months. No objective tumor responses were observed [14]. Another phase II clinical trial testing octreotide as an antiproliferative agent in 34 patients with progressive metastatic NETs demonstrated a disease stabilization rate of 50% lasting a median of 5 months [15].
The antiproliferative effect of 30 mg of intramuscular lanreotide given every 10 or 14 days was evaluated in a phase II trial that included 46 patients with carcinoid and pancreatic endocrine tumors. Two patients (4%) achieved an objective radiographic response, while 19 patients (41%) experienced stable disease for a mean of 9.5 months [16]. In another phase II study of lanreotide 30 mg in 55 patients with GEP-NETs (48 with carcinoid tumors, six with gastrinomas, and one with vasoactive intestinal peptide-secreting tumor), 7% of 31 assessable patients achieved a partial response and 81% experienced disease stability [17]. Table 1 summarizes the results of several trials on the antiproliferative effect of somatostatin analogs in patients with advanced GEP-NETs.
The antiproliferative and antitumoral activity of somatostatin and its analogs appears to be mediated through direct and indirect mechanisms. Direct mechanisms involve activation of somatostatin receptors on tumor cells leading to modulation of intracellular signaling transduction pathways [18]. Multiple in vitro studies using cell lines transfected with somatostatin receptors indicate that all receptor subtypes (sst1–5) may mediate inhibition of cell proliferation [19], whereas specific receptor subtypes (sst2 and 3) may mediate apoptosis [20]. These actions appear to be regulated primarily via the mitogen-activated protein kinase signaling pathway and through activation of phosphotyrosine phosphatases [21]. Somatostatin and its synthetic analogs also exert a number of indirect antitumor actions, including inhibition of the release of growth factors and hormones that drive tumor growth (such as IGF-1), antiangiogenic effects that reduce tumor blood flow through interaction with somatostatin receptors on endothelial cells, and immunomodulatory effects to stimulate the body’s natural antitumor mechanisms [18, 22].
NETs generally express multiple somatostatin receptors [23], all of which may mediate the antiproliferative effects of somatostatin analogs. Somatostatin-14 binds with high affinity to all five human somatostatin receptor subtypes (sst1–5) with the binding affinity for sst2 and sst5 being the highest. Many human tumors express more than one somatostatin receptor subtype, with sst2 being predominant. In contrast, the two short synthetic somatostatin analogs used in the clinic, octreotide and lanreotide, bind with high affinity to the sst2 subtype, with moderate affinity to sst5, and with little or no affinity to the sst1, 3, and 4 subtypes [24].
2.1 PROMID study: does octreotide have an anti-proliferative effect?
Although the studies described above provide initial evidence for the antitumor effects of somatostatin analogs, they have a number of features that prevent them from providing conclusive evidence. Uncontrolled studies have shown certain tumor regression in response to short-acting somatostatin analogs [25] and their combination with interferon alpha (INF-α) [26]. In subsequent trials, tumor stabilization was observed in up to 50% of patients, although complete regression could not be confirmed. Moreover, the combination of somatostatin analogs and INF-α did not show to improve survival [14–16, 27–30]. Limitations of these early studies investigating the antitumor activity of somatostatin analogs include lack of a randomized placebo control group, relatively small patient cohorts, and analysis of heterogeneous tumor populations. In addition, the majority of trials did not include treatment-naïve patients and did not focus on the evaluation of the antiproliferative effect of somatostatin analogs, and the observed effects on tumor growth may have reflected spontaneous phases of tumor growth or stabilization.
Long-acting somatostatin analogs are widely used for symptomatic, low-grade NETs such as carcinoid tumors. The survival impact of this therapy was never examined prospectively. As such, to demonstrate an antiproliferative effect of octreotide long-acting repeatable (LAR), a placebo-controlled, prospective, randomized study in patients with metastatic NET midgut tumors, the PROMID study, was initiated in 18 German academic centers [31]. This phase III trial is among the very few randomized trials performed in patients with this rare tumor type. To avoid confounding variables and to avoid a heterogeneous patient population, only patients with well-differentiated inoperable or metastatic midgut tumors were included. Patients included were randomly assigned to receive either octreotide LAR 30 mg intramuscularly or placebo every month until radiographic evidence of progression or death. The primary endpoint of this study was time to progression. Secondary end points included survival, response rate, quality of life, and safety.
Although the PROMID study planned to include 162 patients, enrollment stopped after an interim analysis with 85 patients. Most patients (74%) had evidence of somatostatin receptor expression as evidenced by radiotracer uptake of octreoscan. Nearly half of patients (39%) manifested carcinoid syndrome (flushing and/or diarrhea associated with elevation in urine of 5-hydroxyindole acetic acid). Median time to tumor progression was 14.3 months in the octreotide LAR 30 mg group vs 6.0 months in the placebo group (p < 0.001). Thus, antiproliferative efficacy was demonstrated with a reduction of 66% in the risk of tumor progression. This significantly greater time-to-tumor progression was seen in the overall study population, regardless of tumor functionality, chromogranin A level, or age. After 6 months, tumor progression was observed in 24% of patients on the octreotide LAR 30 mg arm vs 53% of patients receiving placebo (p < 0.01). Stable disease was observed in 67% of octreotide LAR recipients and in 37% of placebo recipients. Only one partial response was seen. No completed responses occurred. Occurrence of serious adverse events was balanced as it was observed in 11 patients in the octreotide LAR 30 mg arm and in ten patients in the placebo arm.
On multivariate analysis, the highest rates of disease stabilization were observed in patients with low hepatic tumor load (<10%) and resected primary tumor; however, both of these subgroups contained the majority of study patients. Even in patients with higher hepatic tumor burden (>10%), time to progression in the octreotide LAR arm was almost double the rate observed in the control arm. The low death rate in both treatment arms (seven patients in the octreotide LAR 30 mg arm and nine in the placebo arm) precluded any analysis of differences in survival.
It was concluded that octreotide LAR significantly lengthens median time to tumor progression compared with placebo in patients with functionally active and inactive metastatic NETs of the midgut. This study is likely to expand the use of somatostatin analogs to this subgroup of patients, although it should be noted that these results are based on a limited cohort of patients for whom no overall survival data are currently available.
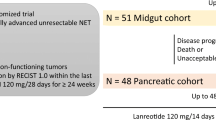
In addition to PROMID, the ongoing “Controlled study of Lanreotide Antiproliferative Response In NET” (CLARINET) trial has been designed to examine the efficacy of lanreotide (autogel 120 mg) in metastatic disease and/or locally advanced inoperable tumors non-functioning enteropancreatic tumors of unknown origin, tumors with a known primary location in the pancreas, mid-gut, or hindgut, or adequately controlled gastrinomas. The 96-week, double-blind, randomized, stratified comparative, placebo-controlled, parallel-group, multicenter phase III study aims to enroll 200 patients. The primary endpoint is time to either disease progression (as measured by Response Evaluation Criteria in Solid Tumors criteria) or death [32].
3 Novel somatostatin analogs
Somatostatin receptor subtypes can undergo heterodimerization with each other and with other receptor families, such as the dopamine receptor family, thereby enhancing their binding affinities and internalization [33]. Thus, novel somatostatin analogs that bind to multiple receptor subtypes as well as analogs capable of binding to different families of receptors may prove to be effective antisecretory and antiproliferative agents in patients refractory to octreotide or lanreotide.
Pasireotide, SOM230, is one such novel somatostatin analog that exhibits a very different binding profile to human somatostatin receptors. It binds avidly to four of the five somatostatin receptors (sst1, 2, 3, and 5). When compared with octreotide, pasireotide has a 40-, 30-, and 5-fold higher binding affinity for sst5, sst1, and sst3, respectively, and a lower affinity for sst2 [24, 34]. Interestingly, pasireotide demonstrates one of the highest binding affinities to sst5 ever reported for a somatostatin analog, which is two times higher than that observed for somatostatin-14 [24, 34].
In an in vitro study evaluating the use of octreotide and pasireotide on HEK293 cells expressing somatostatin receptor subtype sst2, treatment with octreotide resulted in an internalization of sst2 receptors at 30 min, whereas treatment with pasireotide did not lead to sst2 internalization. Such findings may suggest that a persistent and more durable efficacy could be obtained with pasireotide [35].
In an open-label phase II trial, the activity of subcutaneous pasireotide in patients with metastatic carcinoid tumors (predominantly of midgut origin) whose symptoms (flushing and diarrhea) were inadequately controlled with octreotide LAR was evaluated. Clinical response was based on the number of flushing episodes and bowel movements. Preliminary efficacy data from 44 patients indicated activity in this refractory population, with complete responses being achieved by 5% of patients at the 600 and 900 μg bid doses and partial responses by 20% of patients at 600–1,200 μg bid doses. Objective tumor response was observed in 11 patients, with nine showing stable disease and two showing progressive disease at 6 months. Patients in both treatment arms exhibited a similar safety profile [36]. Based on these encouraging results, clinical trials are being carried out to test the antiproliferative effects of pasireotide in NETs, including a phase III trial in patients with metastatic refractory or resistant carcinoid tumors (NCT00690430).
4 Chimeric somatostatin–dopamine molecules
Therapy using somatostatin analogs (either octreotide or lanreotide) in patients with functional GEP-NETs is not universally efficacious and the effects of treatment decline with time [23]. Although somatostatin analogs produce an effective long-term control of hormonal hypersecretion from GH-secreting pituitary adenomas, their effects on carcinoid tumors are often transient. Additionally, considerable differences exist between patients harboring islet cell and carcinoid tumors with respect to the development of tolerance. This may be related to a differential expression of somatostatin receptor subtypes among tumors [23].
The different mechanisms that are potentially involved in this adaptation or tolerance to continuous exposure to somatostatin or somatostatin analogs may be associated with processes such as receptor phosphorylation, G protein uncoupling, receptor internalization, and/or degradation. The majority of patients show desensitization of the inhibitory effect of octreotide on tumor-related hormone secretion within weeks or months. This effect may be initially reversed by increasing the dosage of octreotide, but eventually the drug becomes ineffective in most patients [37].
As stated above, receptor subtypes can undergo heterodimerization with each other, thereby enhancing binding affinity and internalization [38]. Thus, novel somatostatin analogs, such as pasireotide (SOM230) [34] or BIM-23244 [39] which binds to multiple receptor subtypes, as well as analogs capable of binding to different families of receptors, may prove to be effective antisecretory and antiproliferative agents in patients refractory to octreotide or lanreotide. Somatostatin receptors may also heterodimerize with other G protein-coupled receptors such as the dopamine D2 receptor [33], resulting in a novel receptor state with properties distinct from the individual receptors in terms of enhanced internalization, reduced agonist-induced desensitization, and functional activity.
Besides the development of novel somatostatin analogs with a broader receptor binding profile, there are several dopamine agonists, such as bromocriptine and cabergoline, which have been used clinically in combination with somatostatin analogs [40, 41]. These combinations reduce both GH and IGF-1 levels more effectively than treatment with either drug alone, especially in patients with GH/prolactin-secreting adenomas. This allows the possibility of less frequent drug administrations, lower doses of octreotide, and, consequently, lower treatment costs. However, in vitro studies have shown a lack of direct additivity at the cellular level between somatostatin analogs and D2 receptor agonists in suppressing GH release in cultured human adenoma cells. In cell culture studies, the dose-related inhibition of GH and prolactin secretion by the combination of somatostatin and dopamine analogs is similar to that achieved by the most potent of the individual compounds, predominantly the sst2-preferential analogs [42].
However, when chimeric compounds incorporating both dopaminergic and somatostatinergic pharmacological activities into a single molecule are used, the results are markedly different. A recently synthesized hybrid somatostatin–dopamine molecule, BIM-23A387, has been shown to retain high affinity binding to both sst2 and D2 receptors and has a remarkably enhanced potency in suppressing GH and prolactin release in primary cultures of human pituitary adenoma cells, compared with individual sst2- and D2-specific analogs, either alone or in combination [42]. Another chimeric molecule, BIM-23A760, with even greater efficacy in suppressing GH from primary GH-secreting adenomas has also been shown to inhibit cell proliferation in the non-small cell lung cancer cell line Calu-6, which expresses sst2, 5, and dopamine D2 receptors, with a higher potency and efficacy than individual sst2 and D2 receptor analogs [43]. A recent study that compared receptor expression in GH-secreting pituitary adenomas and in GEP-NETs indicates that GEP tumors have the appropriate sst2, sst5, and D2 receptor profile to potentially respond to somatostatin–dopamine chimeric molecules, such as BIM-23A760 [44]. Table 2 shows dopamine receptor D2 and somatostatin receptor (sst1–5) binding affinities of various dopamine and somatostatin analogs.
The enhanced activity of these chimeric molecules cannot be explained by a change in the affinity for either the sst2 or D2 receptor because the individual affinity of the chimeric molecule for each of these receptors is roughly equivalent to those of individual somatostatin and dopamine analogs that show a considerably lower efficacy in suppressing GH secretion [42]. The mechanism by which these chimeric molecules exert their potent action is presently unknown; however, a key hypothesis may be oligomerization of the dopamine and somatostatin receptors, thus creating a new receptor with distinct and enhanced functionality. The ligand-induced oligomerization of G protein-coupled receptors has now been demonstrated for several different receptors [45]. This oligomerization could be responsible for the enhanced biological activities observed with the somatostatin–dopamine chimeric molecules [42].
5 Combinations of somatostatin analogs with other active agents
Although somatostatin analogs will almost certainly continue to be a major treatment option for functioning NETs, in order to improve clinical outcomes, they will probably need to be combined with other targeted therapies and cytotoxics, such as IFN-α, and vascular endothelial growth factor (VEFG) and mammalian target of rapamycin (mTOR) inhibitors. The new multireceptor somatostatin analog pasireotide (SOM230) described above, as well as chimeric molecules, such as BIM-23A760 (a single compound with both somatostatinergic and dopaminergic activity), may also become part of the clinical management of GEP-NETs.
Somatostatin analogs and IFN-α have been used in combination to add the positive effect of somatostatin analogs on hypersecretory syndromes and to reduce the dose of IFN-α and thus the number of IFN-α-related side effects. This combination seems of benefit in patients in whom the usual octreotide treatment fails to achieve biochemical and symptomatic control [46]. This combination therapy has been shown to significantly lower the risk of progressive disease and increase median survival as compared with octreotide alone [47].
Carcinoid tumors are vascular and are known to express VEGF [48]. In a xenograft model of a human carcinoid, treatment with bevacizumab, an anti-VEGF monoclonal antibody, was found to inhibit tumor growth and metastases [49]. The activity of bevacizumab, or pegylated IFN-α-2b, as monotherapy, followed by a combination of the two agents in patients with advanced carcinoid tumors that previously were receiving octreotide, was examined in a randomized phase II study [50]. Bevacizumab therapy resulted in a higher objective response rate, reduction of tumor blood flow, and longer progression-free survival than pegylated IFN-α-2b treatment in patients with advanced carcinoid tumors.
The mTOR is a conserved serine/threonine kinase that regulates the cell cycle and metabolism in response to environmental factors. It mediates signaling transduction downstream of receptor tyrosine kinases. Evidence suggests that the mTOR pathway may be involved in the pathogenesis of NETs [51]. GEP-NETs are known to co-express IGF-1 and IGF-1 receptor [52]. It has been shown that IGF-1 activates mTOR and increases cell proliferation [52]. In contrast, mTOR inhibition suppresses tumor growth [52, 53]. Everolimus (RAD001) is an mTOR inhibitor that inhibits kinase activity. The rationale for combining octreotide and everolimusis is that upregulation of the upstream IGF-1 pathway is thought to be a potential resistance mechanism for RAD001, while octreotide has been shown to reduce serum IGF-1 levels in patients with solid tumors [54]. This combination therapy led to an overall median progression-free survival of 60 weeks and a 3-year survival rate of 78%. There were 22% of patients with partial responses, 70% with stable disease, and 8% progressed. The phase II trial RAD001 in Advanced Neuroendocrine Tumors-1 evaluated everolimus in patients with metastatic pancreatic NETs whose disease progressed while on prior cytotoxic chemotherapy [55]. Patients were enrolled into two strata based on whether they had previously received octreotide LAR therapy or not. Patients in stratum 1 (n = 115) received oral everolimus 10 mg/day alone, and patients in stratum 2 (n = 45) received oral everolimus 10 mg/day plus octreotide LAR intramuscularly every 28 days at its current dose. Most patients had been diagnosed for more than 2 years before study entry, and over 90% of patients in both strata had liver metastases. The overall response rate (confirmed by radiology) was 9.6% in stratum 1 and 4.4% in stratum 2. Stable disease was seen in 68% of patients in stratum 1 and 80% of patients in stratum 2. Median progression-free survival (by radiology) was 9.7 months in stratum 1 and 16.7 months in stratum 2, and median overall survival was 24.9 months in patients in stratum 1 and was not reached in patients in stratum 2. Treatment was generally well tolerated by patients in both strata. Based on these encouraging results, two subsequent studies are now ongoing.
6 Peptide receptor radionuclide therapy
Radiolabeled monoclonal antibodies have become very popular as potential “magic bullets” to be used to localize and treat human neoplasms at an early stage of development. However, real-life application of this simple principle has turned out to be much more difficult than expected, mainly because of the excessive molecular mass of the antibodies [56].
Only recently an alternative to radiolabeled antibodies appeared in the form of a small radiolabeled peptide, a somatostatin analog, which led to a major breakthrough in this field. On the basis that most human NETs express a high density of somatostatin receptors [6, 7], it has been possible to develop a method for localizing these tumors and their metastases by in vivo somatostatin receptor scintigraphy, using an intravenous injection of radiolabeled somatostatin analogs [57]. From a therapeutic point of view, recent pilot studies using high doses of somatostatin analogs radiolabeled with yttrium (90Y) have shown a reduction or at least a stabilization of the tumor growth [58]. The presence of somatostatin receptors in a higher density in NETs and their ability to form a receptor–ligand complex may allow internalization and accumulation of radiopharmaceuticals inside the tumor [59]. If isotopes emitting β-particles are used for peptide labeling, the radiation emitted from a radiolabeled peptide bound to a tumor cell may also kill neighboring cells because the path length of β-particles can extend over several cell diameters. The crossfire of β-particles can, in theory, destroy both somatostatin receptor-positive and somatostatin receptor-negative tumor cells.
Among radiolabeled somatostatin analogs, octreotide was first used as an iodinated (125I- or 123I-[Tyr3]octreotide) compound [57]. Linking chelators to this analog, such as diethylenetriaminopentaacetic acid (DTPA) or 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), creates molecules that accept certain types of metallic radioisotopes and greatly improve the biodistribution profile, with a shift from a gastrointestinal excretion pathway mainly to renal excretion [60]. Octreotide may be linked to these molecules plus several radionuclides, such as 90Y, gallium (67Ga), lutetium (177Lu), or indium (111In). 111In-DTPA-(D-Phe1)-octreotide (111In-pentetreotide) (OctreoScan®) has become the most widely used tracer for somatostatin receptor scintigraphy. It emits γ-rays and Auger electrons. The γ-rays are required for scintigraphy, whereas the Auger electrons may be used for radiotherapy. In radiotherapy, the most frequently used analog has been 90Y-DOTA-(D-Phe1, Tyr3)octreotide (90Y-DOTATOC), with 90Y as β-emitter [61].
Recently, the search for improved radiolabeled somatostatin analogs has intensified, and second-generation somatostatin radiopeptides are being investigated. Because sst2 appears to be the main somatostatin receptor subtype in many human tumors [8], improvement of sst2 affinity has been one goal of recent research. Apparently, minimal changes in DOTATOC, such as the replacement of one metal (In or Y) by another (Ga), resulting in the tracer 68Ga-DOTATOC, markedly improves sst2 binding affinity and in vivo tumor imaging [61]. Octreotate, which is an octreotide derivative lacking the alcohol moiety at threonine, shows great improvement in its biodistribution profile, affinity to sst2, and tumor scintigraphy quality [61]. The resulting 177Lu-(DOTA0, Tyr3)-octreotate (177Lu-DOTATATE) has a DOTA-(D-Phe1, Tyr3)-octreotide structure, but with a threonine instead of threoninol (octreotate instead of octreotide) [61]. Results of a phase I trial using 111In-pentetreotide showed that this radiolabeled somatostatin analog produced limited objective responses, probably due to the small particle range and short tissue penetration of Auger electrons emitted by the 111In isotope [62]. 68Ga-DOTATOC seems to be a very promising new positron emission tomography tracer for imaging somatostatin receptors, offering excellent imaging properties and a very high tumor-to-background ratio [63].
The latest research efforts in radiolabeled somatostatin analogs have focused on 177Lu-(DOTA0, Tyr3)-octreotate. This radionuclide has a half-life of 6.7 days and emits β and γ radiation, allowing imaging and dosimetry after therapy, with a shorter range of tissue penetration (2 mm) than 90Y. In a recent phase II clinical trial, the efficacy of 177Lu-(DOTA0, Tyr3)-octreotate was assessed in 310 patients with GEP-NETs [64]. Partial responses and complete responses were observed in 30% of patients, and median progression-free survival was 33.0 months. Tumor size reductions were achieved in 46% of patients. Upon comparing overall survival with other reference studies, 177Lu-(DOTA0, Tyr3)-octreotate achieved a survival benefit of 40–72 months from the time of diagnosis [64]. Treatment with 177Lu-(DOTA0, Tyr3)-octreotate produced few serious adverse events and therefore was regarded as safe. In addition, upon comparing the residence time in tumors between 177Lu-(DOTA0, Tyr3)-octreotide and 177Lu-(DOTA0, Tyr3)-octreotate in the same patients in a similar therapeutic setting, a factor of 2.1 in favor of 177Lu-(DOTA0, Tyr3)-octreotate was observed. Moreover, when compared with histological controls, a benefit in overall survival of several years from time of diagnosis is observed. Therefore, 77Lu-(DOTA0, Tyr3)-octreotate may be considered the analog of choice when performing peptide receptor radionuclide therapy with radiolabeled somatostatin analogs [64].
In another study, patients completed European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire C30 before therapy with 177Lu-(DOTA0, Tyr3)-octreotate and at a follow-up visit 6 weeks after the last cycle. After that period, the score increased significantly to a mean of 78.2, up from 69.0. 177Lu-(DOTA0, Tyr3)-octreotate therapy significantly improved the global health/quality of life and several function and symptom scales in patients with metastasized GEP-NETs, but especially in patients with proven tumor regression. Significant improvement was observed in the role, emotional, and social function scales. Symptom scores for fatigue, insomnia, and pain were significantly decreased [65].
Future directions aim toward the combination of drugs in the treatment of GEP-NETs, such as the combination of 177Lu-(DOTA0, Tyr3)-octreotate with capecitabine [66]. Treatment with both drugs is feasible and safe when considering the occurrence of acute and subacute side effects. Due to these encouraging results, a randomized, controlled clinical trial comparing the efficacy and safety of this combination with 177Lu-(DOTA0, Tyr3)-octreotate as single agent is currently ongoing.
7 Conclusions
Somatostatin analogs were initially developed as antisecretory agents used primarily to control hormonal syndromes associated with NETs. In recent years, data obtained from several studies have supported their role as antiproliferative agents, capable of stabilizing tumor growth in a large proportion of patients with GEP-NETs. The recently published PROMID study provides high-level evidence to validate the role of octreotide LAR as an antiproliferative agent in patients with metastatic NETs of the midgut. New somatostatin analogs, both single-receptor and multi-receptor, have to be investigated and developed in order to treat patients refractory to classical analogs such as octreotide and lanreotide; such is the case for pasireotide. Also, chimeric somatostatin–dopamine molecules as well as a combination of somatostatin analogs with other biological agents such as IFN-α, VEFG inhibitors, mTOR inhibitors, and dopamine agonists provide a new perspective in the treatment of GEP-NETs. Lastly, peptide receptor radionuclide therapy using somatostatin analogs has provided a method of locating and treating these particular tumors.
References
Brazeau, P., Vale, W., Burgus, R., et al. (1973). Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science, 179(68), 77–79.
Reichlin, S. (1983). Somatostatin. The New England Journal of Medicine, 309(24), 1495–1501.
Serri, O., Brazeau, P., Kachra, Z., et al. (1992). Octreotide inhibits insulin-like growth factor-I hepatic gene expression in the hypophysectomized rat: Evidence for a direct and indirect mechanism of action. Endocrinology, 130(4), 1816–1821.
Epelbaum, J. (1986). Somatostatin in the central nervous system: Physiology and pathological modifications. Progress in Neurobiology, 27(1), 63–100.
Maurer, R., & Reubi, J. C. (1985). Somatostatin receptors. JAMA, 253(18), 2741.
Bruns, C., Raulf, F., Hoyer, D., et al. (1996). Binding properties of somatostatin receptor subtypes. Metabolism, 45(8 Suppl 1), 17–20.
Hoyer, D., Lubbert, H., & Bruns, C. (1994). Molecular pharmacology of somatostatin receptors. Naunyn Schmiedebergs Arch Pharmacol, 350(5), 441–453.
Oberg, K. E., Reubi, J. C., Kwekkeboom, D. J., et al. (2010). Role of somatostatins in gastroenteropancreatic neuroendocrine tumor development and therapy. Gastroenterology, 139(3), 742–753. 753 e1.
Bauer, W., Briner, U., Doepfner, W., et al. (1982). SMS 201-995: A very potent and selective octapeptide analogue of somatostatin with prolonged action. Life Sciences, 31(11), 1133–1140.
Murphy, W. A., Lance, V. A., Moreau, S., et al. (1987). Inhibition of rat prostate tumor growth by an octapeptide analog of somatostatin. Life Sciences, 40(26), 2515–2522.
Karashima, T., Cai, R. Z., & Schally, A. V. (1987). Effects of highly potent octapeptide analogs of somatostatin on growth hormone, insulin and glucagon release. Life Sciences, 41(8), 1011–1019.
Mazziotti, G., Floriani, I., Bonadonna, S., et al. (2009). Effects of somatostatin analogs on glucose homeostasis: A metaanalysis of acromegaly studies. The Journal of Clinical Endocrinology and Metabolism, 94(5), 1500–1508.
Strosberg, J., Gardner, N., & Kvols, L. (2009). Survival and prognostic factor analysis of 146 metastatic neuroendocrine tumors of the mid-gut. Neuroendocrinology, 89(4), 471–476.
Arnold, R., Trautmann, M. E., Creutzfeldt, W., et al. (1996). Somatostatin analogue octreotide and inhibition of tumour growth in metastatic endocrine gastroenteropancreatic tumours. Gut, 38(3), 430–438.
Saltz, L., Trochanowski, B., Buckley, M., et al. (1993). Octreotide as an antineoplastic agent in the treatment of functional and nonfunctional neuroendocrine tumors. Cancer, 72(1), 244–248.
Ducreux, M., Ruszniewski, P., Chayvialle, J. A., et al. (2000). The antitumoral effect of the long-acting somatostatin analog lanreotide in neuroendocrine tumors. The American Journal of Gastroenterology, 95(11), 3276–3281.
Wymenga, A. N., Eriksson, B., Salmela, P. I., et al. (1999). Efficacy and safety of prolonged-release lanreotide in patients with gastrointestinal neuroendocrine tumors and hormone-related symptoms. Journal of Clinical Oncology, 17(4), 1111.
Susini, C., & Buscail, L. (2006). Rationale for the use of somatostatin analogs as antitumor agents. Annals of Oncology, 17(12), 1733–1742.
Weckbecker, G., Lewis, I., Albert, R., et al. (2003). Opportunities in somatostatin research: Biological, chemical and therapeutic aspects. Nature Reviews. Drug Discovery, 2(12), 999–1017.
Lattuada, D., Casnici, C., Venuto, A., et al. (2002). The apoptotic effect of somatostatin analogue SMS 201-995 on human lymphocytes. Journal of Neuroimmunology, 133(1–2), 211–216.
Florio, T. (2008). Somatostatin/somatostatin receptor signalling: Phosphotyrosine phosphatases. Molecular and Cellular Endocrinology, 286(1–2), 40–48.
Florio, T., Morini, M., Villa, V., et al. (2003). Somatostatin inhibits tumor angiogenesis and growth via somatostatin receptor-3-mediated regulation of endothelial nitric oxide synthase and mitogen-activated protein kinase activities. Endocrinology, 144(4), 1574–1584.
Hofland, L. J., & Lamberts, S. W. (2003). The pathophysiological consequences of somatostatin receptor internalization and resistance. Endocrine Reviews, 24(1), 28–47.
Bruns, C., Lewis, I., Briner, U., et al. (2002). SOM230: A novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. European Journal of Endocrinology, 146(5), 707–716.
Imtiaz, K. E., Monteith, P., & Khaleeli, A. (2000). Complete histological regression of metastatic carcinoid tumour after treatment with octreotide. Clinical Endocrinology (Oxford), 53(6), 755–758.
Joensuu, H., Katka, K., & Kujari, H. (1992). Dramatic response of a metastatic carcinoid tumour to a combination of interferon and octreotide. Acta Endocrinologica (Copenhagen), 126(2), 184–185.
Arnold, R., Rinke, A., Klose, K. J., et al. (2005). Octreotide versus octreotide plus interferon-alpha in endocrine gastroenteropancreatic tumors: A randomized trial. Clinical Gastroenterology and Hepatology, 3(8), 761–771.
Faiss, S., Pape, U. F., Bohmig, M., et al. (2003). Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors—the International Lanreotide and Interferon Alfa Study Group. Journal of Clinical Oncology, 21(14), 2689–2696.
Aparicio, T., Ducreux, M., Baudin, E., et al. (2001). Antitumour activity of somatostatin analogues in progressive metastatic neuroendocrine tumours. European Journal of Cancer, 37(8), 1014–1019.
di Bartolomeo, M., Bajetta, E., Buzzoni, R., et al. (1996). Clinical efficacy of octreotide in the treatment of metastatic neuroendocrine tumors. A study by the Italian trials in medical oncology group. Cancer, 77(2), 402–408.
Rinke, A., Müller, H.-H., Schade-Brittinger, C., et al. (2009). Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID study group. Journal of Clinical Oncology, 27(28), 4656–4663.
Blumberg J, Gómez-Panzani E, Latapie-Martínez S, et al. Somatuline® Autogel® 120 mg (lanreotide) evaluation of tumor progression-free survival in patients with non-functioning entero-pancreatic endocrine tumors: An ongoing, double-blind, randomized, placebo-controlled, multicenter study (the CLARINET Study). NANETS—2010 Neuroendocrine Tumor Symposium (abstr. 726).
Rocheville, M., Lange, D. C., Kumar, U., et al. (2000). Receptors for dopamine and somatostatin: Formation of hetero-oligomers with enhanced functional activity. Science, 288(5463), 154–157.
Schmid, H. A. (2008). Pasireotide (SOM230): Development, mechanism of action and potential applications. Molecular and Cellular Endocrinology, 286(1–2), 69–74.
Van Vugt, H. H., Schmid, H. A., Sailer, A. W. (2008) Ligand-dependent internalization of somatostatin receptors. In: Endocrine abstract. p. 654.
Kvols, L., Wiedenmann, B., Oberg, K., et al. (2006). Safety and efficacy of pasireotide (SOM230) in patients with metastatic carcinoid tumors refractory or resistant to octreotide LAR: Results of a phase II study. Journal of Clinical Oncology (Meeting Abstracts), 24(18_suppl), 4082-.
Lamberts, S. W., van der Lely, A. J., de Herder, W. W., et al. (1996). Octreotide. The New England Journal of Medicine, 334(4), 246–254.
Rocheville, M., Lange, D. C., Kumar, U., et al. (2000). Subtypes of the somatostatin receptor assemble as functional homo- and heterodimers. The Journal of Biological Chemistry, 275(11), 7862–7869.
Saveanu, A., Gunz, G., Dufour, H., et al. (2001). Bim-23244, a somatostatin receptor subtype 2- and 5-selective analog with enhanced efficacy in suppressing growth hormone (GH) from octreotide-resistant human GH-secreting adenomas. The Journal of Clinical Endocrinology and Metabolism, 86(1), 140–145.
Minniti, G., Jaffrain-Rea, M. L., Baldelli, R., et al. (1997). Acute effects of octreotide, cabergoline and a combination of both drugs on GH secretion in acromegalic patients. La Clinica Terapeutica, 148(12), 601–607.
Flogstad, A. K., Halse, J., Grass, P., et al. (1994). A comparison of octreotide, bromocriptine, or a combination of both drugs in acromegaly. The Journal of Clinical Endocrinology and Metabolism, 79(2), 461–465.
Saveanu, A., Lavaque, E., Gunz, G., et al. (2002). Demonstration of enhanced potency of a chimeric somatostatin–dopamine molecule, BIM-23A387, in suppressing growth hormone and prolactin secretion from human pituitary somatotroph adenoma cells. The Journal of Clinical Endocrinology and Metabolism, 87(12), 5545–5552.
Kidd, M., Modlin, I. M., Black, J. W., et al. (2007). A comparison of the effects of gastrin, somatostatin and dopamine receptor ligands on rat gastric enterochromaffin-like cell secretion and proliferation. Regulatory Peptides, 143(1–3), 109–117.
O’Toole, D., Saveanu, A., Couvelard, A., et al. (2006). The analysis of quantitative expression of somatostatin and dopamine receptors in gastro-entero-pancreatic tumours opens new therapeutic strategies. European Journal of Endocrinology, 155(6), 849–857.
Hebert, T. E., & Bouvier, M. (1998). Structural and functional aspects of G protein-coupled receptor oligomerization. Biochemistry and Cell Biology, 76(1), 1–11.
Tiensuu Janson, E. M., Ahlstrom, H., Andersson, T., et al. (1992). Octreotide and interferon alfa: A new combination for the treatment of malignant carcinoid tumours. European Journal of Cancer, 28A(10), 1647–1650.
Kolby, L., Persson, G., Franzen, S., et al. (2003). Randomized clinical trial of the effect of interferon alpha on survival in patients with disseminated midgut carcinoid tumours. The British Journal of Surgery, 90(6), 687–693.
Terris, B., Scoazec, J. Y., Rubbia, L., et al. (1998). Expression of vascular endothelial growth factor in digestive neuroendocrine tumours. Histopathology, 32(2), 133–138.
Konno, H., Arai, T., Tanaka, T., et al. (1998). Antitumor effect of a neutralizing antibody to vascular endothelial growth factor on liver metastasis of endocrine neoplasm. Japanese Journal of Cancer Research, 89(9), 933–939.
Yao, J. C., Phan, A., Hoff, P. M., et al. (2008). Targeting vascular endothelial growth factor in advanced carcinoid tumor: A random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. Journal of Clinical Oncology, 26(8), 1316–1323.
Yuan, R., Kay, A., Berg, W. J., et al. (2009). Targeting tumorigenesis: Development and use of mTOR inhibitors in cancer therapy. J Hematol Oncol, 2, 45.
von Wichert, G., Jehle, P. M., Hoeflich, A., et al. (2000). Insulin-like growth factor-I is an autocrine regulator of chromogranin A secretion and growth in human neuroendocrine tumor cells. Cancer Research, 60(16), 4573–4581.
Moreno, A., Akcakanat, A., Munsell, M. F., et al. (2008). Antitumor activity of rapamycin and octreotide as single agents or in combination in neuroendocrine tumors. Endocrine-Related Cancer, 15(1), 257–266.
Yao, J. C., Phan, A. T., Chang, D. Z., et al. (2008). Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: Results of a phase II study. Journal of Clinical Oncology, 26(26), 4311–4318.
Yao, J. C., Lombard-Bohas, C., Baudin, E., et al. (2010). Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: A phase II trial. Journal of Clinical Oncology, 28(1), 69–76.
Behr, T. M., Memtsoudis, S., Sharkey, R. M., et al. (1998). Experimental studies on the role of antibody fragments in cancer radio-immunotherapy: Influence of radiation dose and dose rate on toxicity and anti-tumor efficacy. International Journal of Cancer, 77(5), 787–795.
Krenning, E. P., Bakker, W. H., Breeman, W. A., et al. (1989). Localisation of endocrine-related tumours with radioiodinated analogue of somatostatin. Lancet, 1(8632), 242–244.
Waldherr, C., Pless, M., Maecke, H. R., et al. (2002). Tumor response and clinical benefit in neuroendocrine tumors after 7.4 GBq (90)Y-DOTATOC. Journal of Nuclear Medicine, 43(5), 610–616.
Appetecchia, M., & Baldelli, R. (2010). Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine tumours, current aspects and new perspectives. Journal of Experimental & Clinical Cancer Research, 29, 19.
Heppeler, A., Froidevaux, S., Eberle, A. N., et al. (2000). Receptor targeting for tumor localisation and therapy with radiopeptides. Current Medicinal Chemistry, 7(9), 971–994.
Reubi, J. C. (2003). Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocrine Reviews, 24(4), 389–427.
Valkema, R., De Jong, M., Bakker, W. H., et al. (2002). Phase I study of peptide receptor radionuclide therapy with [In-DTPA]octreotide: The Rotterdam experience. Seminars in Nuclear Medicine, 32(2), 110–122.
Henze, M., Schuhmacher, J., Hipp, P., et al. (2001). PET imaging of somatostatin receptors using [68GA]DOTA-D-Phe1-Tyr3-octreotide: First results in patients with meningiomas. Journal of Nuclear Medicine, 42(7), 1053–1056.
Kwekkeboom, D. J., de Herder, W. W., Kam, B. L., et al. (2008). Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0, Tyr3]octreotate: Toxicity, efficacy, and survival. Journal of Clinical Oncology, 26(13), 2124–2130.
Teunissen, J. J., Kwekkeboom, D. J., & Krenning, E. P. (2004). Quality of life in patients with gastroenteropancreatic tumors treated with [177Lu-DOTA0, Tyr3]octreotate. Journal of Clinical Oncology, 22(13), 2724–2729.
van Essen, M., Krenning, E. P., Kam, B. L., et al. (2008). Report on short-term side effects of treatments with 177Lu-octreotate in combination with capecitabine in seven patients with gastroenteropancreatic neuroendocrine tumours. European Journal of Nuclear Medicine and Molecular Imaging, 35(4), 743–748.
Arnold, R., Benning, R., Neuhaus, C., et al. (1993). Gastroenteropancreatic endocrine tumours: Effect of Sandostatin® on tumour growth. Digestion, 54(Suppl. 1), 72.
Eriksson, B., Renstrup, J., Imam, H., et al. (1997). High-dose treatment with lanreotide of patients with advanced neuroendocrine gastrointestinal tumors: Clinical and biological effects. Annals of Oncology, 8(10), 1041–1044.
Ricci, S., Antonuzzo, A., Galli, L., et al. (2000). Long-acting depot lanreotide in the treatment of patients with advanced neuroendocrine tumors. American Journal of Clinical Oncology, 23(4), 412–415.
Acknowledgments
The authors acknowledge Dr. Fernando Sánchez-Barbero from HealthCo SL (Madrid, Spain) for his assistance in the preparation of this manuscript and Pfizer Spain for the financial support of medical writing services.
Conflicts of interest
The authors declare that they do not have any conflict of interest that may inappropriately influence this work. Michael Culler is an employee of IPSEN.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Culler, M.D., Öberg, K., Arnold, R. et al. Somatostatin analogs for the treatment of neuroendocrine tumors. Cancer Metastasis Rev 30 (Suppl 1), 9–17 (2011). https://doi.org/10.1007/s10555-011-9293-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-011-9293-0