
Abstract
According to a “canonical” view, reactive oxygen species (ROS) positively contribute, in different ways, to carcinogenesis and to malignant progression of tumor cells: they drive genomic damage and genetic instability, transduce, as signaling intermediates, mitogenic and survival inputs by growth factor receptors and adhesion molecules, promote cell motility and shape the tumor microenvironment by inducing inflammation/repair and angiogenesis. Chemopreventive and tumor-inhibitory effects of endogenous, diet-derived or supplemented antioxidants largely support this notion. However, emerging lines of evidence indicates that tumor cells also need to defend themselves from oxidative damage in order to survive and successfully spread at distance. This “heresy” has recently received important impulse from studies on the role of antioxidant capacity in cancer stem cells self-renewal and resistance to therapy; additionally, the transforming activity of some oncogenes has been unexpectedly linked to their capacity to maintain elevated intracellular levels of reduced glutathione (GSH), the principal redox buffer. These studies underline the importance of cellular antioxidant capacity in metastasis, as the result of a complex cell program involving enhanced motility and a profound change in energy metabolism. The glycolytic switch (Warburg effect) observed in malignant tissues is triggered by mitochondrial oxidative damage and/or activation of redox-sensitive transcription factors, and results in an increase of cell resistance to oxidants. On the other hand, cytoskeleton rearrangement underlying cell motile and tumor-aggressive behavior use ROS as intermediates and are therefore facilitated by oxidative stress. Along this line of speculation, we suggest that metastasis represents an integrated strategy for cancer cells to avoid oxidative damage and escape excess ROS in the primary tumor site, explaning why redox signaling pathways are often up-regulated in malignancy and metastasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Metastasis represents the final, most devastating stage of malignancy and the leading cause of death by cancer. Death occurs because of failure of colonized organs, para-neoplastic syndromes, and side effect of systemic anticancer therapy. In spite of significant progress in cancer prevention and early detection, a significant number of malignancies already presents distant metastases or is at high risk for these complications at the time of diagnosis [1].
Our molecular knowledge of the metastatic process is still very limited, but systematic gene expression studies comparing primary tumors with their metastases have recently shed new light on the cellular and biochemical cascades that are activated during tumor spread at distance. These studies have helped to identify large classes of genes linked to different stages of tumor dissemination, as those involved in metastasis “initiation” at the primary tumor site, “progression” to distant localization sites, and “virulent” colonization of the latter [2]. Important implications of these new findings have been the identification of genetic “signatures” potentially capable of predicting, in a primary cancer lesion, the tendency to form metastasis in different organs [3]. Moreover, prompted by these indications, detailed biochemical studies have provided new information on the complex interaction between metastatic cells and the components (stromal vascular and inflammatory cells) of their ectopic microenvironment, leading to a new molecular definition of the old Paget’s seed and soil theory [4].
Metastasis has been traditionally interpreted, in a darwinistic perspective, as a process whereby genetic instability in the primary tumor fuels cell heterogeneity, allowing few metastatic clones to eventually emerge and be positively selected to disseminate cancer at distance. It has also been suggested, based on genetic disparity between primary and metastatic tumors, that metastases may arise from cells leaving the primary tumor lesion at an early stage of its local progression, and thus develop in a parallel rather than sequential fashion with respect to the lesion of origin [5]. Either way, it is becoming increasingly clear that only a minority of malignant cells undertakes the metastatic route, and, of those, an even smaller fraction succeeds in this task. Evidence also exists that these cells may share some properties with somatic or embryonic stem cells [6, 7], which has led to the hypothesis that metastases are initiated by cell clones endowed of unique self-renewal and tumor propagating capacity, i.e., by cancer stem cells (CSC).
While molecular strategies employed by metastatic cells to leave the primary tumor, intravasate, disseminate at distance and grow into new colonies are being intensively and successfully investigated, less clear is what actually prompts some tumor cells to leave the primary tumor, or, in other words, what kind of environmental pressure operates on cells bound to a metastatic fate. We know, however, that a tumor can be a very hostile environment, due to shortage of oxygen and nutrients, inflammation and immune system attacks, and that both hypoxia and inflammation promote metastasis [8, 9]. Thus, metastasis may ultimately represent an escape strategy from cell death.
1.1 A tale of 3Ms: motility and metabolism in metastasis
In order to complete their journey to a secondary site, metastatic cells need to overcome several physical obstacles and to cross distances that are remarkable if considered on a cell’s scale of magnitude. The typical sequence of events whereby an epithelial cell (e.g., breast or colon carcinoma cell) eventually colonizes a distant anatomical site and establishes a full blown metastasis, includes, as initial steps: (1) loss of cell polarity and detachment from surrounding cells in the primary tissue, (2) increased interaction with the extracellular matrix (ECM) through integrins, and (3) migration through the ECM towards blood and lymphatic vessels [1]. This process is often indicated as epithelial to mesenchymal transition (EMT) and has been extensively investigated as an early event of carcinoma metastasis. Of note, EMT also occurs during normal embryonic development as precursor cells migrate along directions dictated by morphogenetic gradients [10, 11]. At a molecular level, EMT is accompanied by loss of cell-to-cell adhesion through down-regulation of E-cadherin expression, and by an increased cell capacity to interact with ECM proteins through integrins, degrade them through matrix metalloproteases (MMPs), and move forward via a complex cascade of cytoskeleton rearrangement brought about by the coordinated action of small Rho family GTPases. Among those, Rac-1 and Cdc42 promote membrane protrusion into lamellopodia and filopodia and the establishment of focal contacts at the leading edge of the cell, while Rho, via the Rho Kinase (ROCK), induces the formation of cytoplasmic stress fibers, stabilization of focal contacts into mature focal adhesion and cell body contraction through activation of the acto-myosin cytoskeleton by phosphorylation of myosin light chain [12, 13]. Cell detachment at the trailing edge, coupled to cytoskeleton contraction, is in turn necessary to allow cell to move forward [14]. Importantly, these continuous attachment–detachment cycles also involve the localized and highly coordinated action of tyrosine kinases (such as Src and focal adhesion kinase, FAK, [15]) and tyrosine phophatases, like phosphatase and tensin homologue deleted on chromosome 10 (PTEN) [16] and SH2 domain-containing protein tyrosine phosphatase (SHP-2) [17].
An alternative strategy of migration for malignant cells is amoeboid invasion, whereby cells lose their polarity, detach from the ECM and move through the path of least resistance, using cortical actin contraction for their propulsion [18]. This alternative motility style, used by lymphocytes and several types of cancer cells to invade ECM, requires the activation of the small GTPase Rho [19]. Of note, many of the molecular players involved in early invasion events have been mechanistically linked to metastasis in experimental and clinical settings; among those, GTPases or their activators/inhibitors (Tiam-1, Rho-C), [20–22] and receptor tyrosine kinase (RTK) upstream of Rho GTPases as hepatocyte growth factor receptor (HGF-R)/c-Met [23]or Trk-A [24].
A second critical step in the formation of hematogenous metastases is cell survival in the bloodstream. Normal and most cancer cells undergo apoptosis (anoikis) when detached from their matrices, and metastatic cells must develop specific molecular strategies in order to survive or even proliferate in an anchorage-independent fashion before they can arrest in the metastatic site and extravasate. Evidence exists that c-Src or some activated RTKs (for instance through Erb-B2 or TrkA) can effectively prevent anoikis by constitutive, ligand-independent activation of integrin signaling [22, 25, 26].
Finally, the full accomplishment of the metastatic programs requires that cells that have reached a distant site grow into secondary tumor masses. This involves in turn their capacity to proliferate, stimulate angiogenesis, and cross-talk with the component of the new microenvironment including parenchymal, stromal, and inflammatory cells ([2, 3] and references therein). A balance between cell proliferation and apoptosis, lack of neoangiogenesis and cell entrance in a quiescent, non-proliferating state have all been invoked as mechanisms underlying the phenomenon of metastasis “dormancy”, whereby successfully disseminated tumor cells (DTC) transiently or irreversibly fail to develop into full blown secondary tumors[2, 27].
A great deal of information has recently become available on the crucial role of the microenvironment in tissue colonization by metastatic cells. This knowledge has helped to understand some aspect of metastasis tropism for specific organs/tissues like bone, liver or lung. Relevant aspects of this interaction involve not only angiogenesis, but also promotion tumor cell proliferation/survival by stromal growth factors and cyto/chemokines. Inflammatory signals appear to significantly contribute to the establishment of metastatic lesions [28], and precursors of inflammatory cells may even precede cancer cells in the site of metastasis, preparing the soil for their engraftment, the so called “pre-metastatic niche” [29]. Importantly, stromal interactions also contribute to invasion at the primary tumor sites, where newly formed, leaky blood vessels facilitate cancer cell intravasation, and growth factors and cytokines produced by stromal cells like tumor-associated macrophages stimulate, in trans, EMT [30, 31].
Gain of metastatic capacity by malignant cells involves a profound genetic reprogramming. This is in part the product of genetic instability fuelling cell heterogeneity within the primary tumor site, in part also the result of multiple and complex environmental epigenetic effects. However, recurrence of specific genetic profiles in metastatic tumors (“metastatic signatures”), also suggests the frequent involvement of key, “nodal” transcriptional regulators, that, once activated during tumorigenesis, are capable of orchestrating transcriptional responses eventually leading to metastatic growth. Emerging lines of evidence indicates in the hypoxia-induced factor (HIF) one of these nodes (Fig. 1).

HIF as the metastasis’ compass. The indicated major aspects of cell metastatic phenotype result, at least in part, from a HIF-dependent genetic program. The figure refers collectively to both HIF-1 and HIF-2. Molecular details are given in the text. HIF regulation of cellular motility and antioxidant defenses are particularly relevant to this article
HIFs are master regulators of normal cell response to hypoxia; rapidly degraded in the presence of oxygen due to prolyl hydroxylation and subsequent ubiquitination and proteasomal degradation (two events brought about by, respectively, the oxygen-dependent prolyl hydroxylases (PHDs) family and the HIF-specific ubiquitin ligase Von Hippel Lindau (VHL)) [32], HIF accumulates in hypoxic cells and trans-activates genes relevant to cell and body adaptation to hypoxia, including those encoding for erythropoietin, angiogenic factors, glucose transporters and glycolytic enzymes like hexokinase and lactate dehydrogenase [33]. Besides stimulating anaerobic glycolysis and glucose uptake, HIF inhibits mitochondrial respiration through the up-regulation of pyruvate dehydrogenase kinase [34] and a change in subunit composition of the cytochrome oxidase complex [35].
Hypoxia frequently occurs in rapidly growing, poorly vascularized tumor masses [36], and promotes cell invasiveness and metastasis in large part through the activation of HIF; additionally, normoxic accumulation of HIF can be triggered by oncogenic signaling through the mitogen activated protein kinases (MAPK) and phosphatydyl-inositol-3-phosphate (PI3K)/ mammalian target of rapamycin (mTOR) phosphorylation cascades. HIF-dependent angiogenesis directly links hypoxia to metastasis initiation at the primary tumor site, since bloodstream is a major route of cancer dissemination and newly formed vessels are leaky and ideally shaped to allow malignant cell intravasation. Effects of HIF on cell invasion, however, are assorted. It is becoming clear, in fact, that increased cell motility and EMT are directly linked to HIF transcriptional activity via the up-regulation of “motogenic” factors like the c-Met/HGF-R [37] and the matrix modifying enzyme lysyl oxidase [38]. Finally, activation of these cell scattering/invasion pathways by HIF may represent an escape, oxygen-seeking strategy to avoid hypoxic death. In parallel, increasing evidence indicates that HIF is largely responsible for the “glycolytic switch” observed in the majority of malignancies, and known since decades as the Warburg Effect [39–41]. Besides providing cancer cells with abundant biosynthetic intermediates necessary for robust proliferation and reducing the risk of mitochondria-initiated apoptosis, preference for glycolytic versus oxidative metabolism, even in the presence of oxygen, leads to accumulation of lactate and to acidification of the extracellular space by HIF-induced carbonic anhydrase. Cancer cells selected by acid resistance may gain a significant selective advantage against surrounding normal cells, leading to enhanced invasiveness [42]. Furthermore, low pH has been demonstrated to favor matrix degradation by MMPs and cathepsins, and to increase the expression of angiogenic factors such as vascular endothelial growth factor-A (VEGF-A) and interleukin-8 (IL-8) [43]. While consequences of HIF activation on angiogenesis and on cellular metabolism and motility are consistent with experimental and epidemiological evidence for the involvement of this factor in metastasis, recent insights in the role of HIF and hypoxia in the maintenance of normal and cancer stem cells has shed new light on this potential linkage.
A growing body of evidence suggests that cancer tissues, like their normal counterparts, are hierarchically organized, in such a way that a small subset of undifferentiated cells endowed of nearly unlimited self-renewal capacity can generate all the other tumor cell populations, including multipotent progenitors, amplifying, and terminally differentiated cells. Notably, small numbers of these cells, when isolated from a tumor mass and transferred to immunodeficient mice, can fully reproduce the tumor of origin in all its cellular components. These cells, identified based on their functional properties and on expression of surface markers (like CD44 or CD133), have been termed CSC or “tumor propagating cells” [44].
Recently described also in several classes of solid tumors, CSC has been originally discovered in hematological malignancies as the malignant counterpart of hemopoietic stem cells (HSC). Interestingly, bone marrow, where HSCs reside, is a relatively hypoxic tissue, and exposure of bone marrow cells to low oxygen concentration in vitro has been shown to increase the success rate of engraftment in reconstitution experiments [45]. Likewise, low pO2 may represent a general feature of stem cell “niches” also in other developing or adult tissues.
Hypoxia plays a critical role in embryogenesis ([46] and references therein). Low oxygen promotes self-renewal and inhibits terminal differentiation of human embryonic cells in vitro [47]; in keeping with this finding, glycolytic enzymes phospho-glycerate mutase and glucose-phosphate isomerase positively regulate cellular life span, and are highly expressed both in immortalized mouse fibroblasts and in embryonic cells [48]. Along parallel lines of evidence, leukemia stem cells are remarkably resistant to hypoxia due to their mainly glycolytic energy metabolism, and can be strongly enriched upon culture exposure to low oxygen tension [49]. Moreover, hypoxia enhances tumor stemness, and a highly tumorigenic cell subset is localized in the hypoxic zones of solid tumors in vivo [50]. Thus, hypoxia and its metabolic consequences [51] seem to regulate important aspects of stem cell biology, both in normal and malignant contexts.
It is also widely accepted that cancer stem cells are involved in tumor metastatic spread. Not only, in fact, metastases are intuitively initiated by single malignant cells able to seed the primary tumor at distance (a definition close to that of cancer stem cells), but some features of stem cells, including a mesenchymal-like, motile phenotype closely resemble those of highly invasive, metastatic cells (see above). Collectively, these lines of evidence suggest that linkage between hypoxia and metastasis may be, at least in part, mediated by the effects of low oxygen on maintenance, differentiation and migration of CSCs. With this respect, while not all effects of hypoxia are dependent on HIF, it is of note that this molecule has been convincingly shown to regulate multiple cell pathways (i.e., those involving, Notch, Wnt, and Sox-4) related to stem cell identity and self-renewal (reviewed in Ref. [47]).
Hence, pleiotropic effects of HIF on angiogenesis, cell migration/invasion and energy metabolism clearly identify HIFs as nodal signaling components at the intersection of stem cell biology and cancer dissemination, and by extension as major molecular determinants of metastasis.
The emerging theme of redox signaling
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are constantly generated inside cells by dedicated enzyme complexes (NADPH oxidase and nitric oxide synthases (NOS), respectively) or as by-products of oxidation–reduction reactions including those underlying mitochondrial respiration [52, 53] (Table 1). While some of these intermediates are useful against pathogens in the context of innate immunity, most are harmful to cells because irreversibly damage proteins, lipids and nucleic acids. Nevertheless, species like hydrogen peroxide, superoxide anion and nitric oxide, endowed of mild reactivity, have been convincingly demonstrated to participate as signaling molecules in cellular physiology and in cell stress responses. In order to do so, they need to modify their molecular targets in a reversible fashion, and their intracellular concentration has to be finely tunable and responsive to a wide array of environmental cues [52].
Signal transduction by oxidant species (“redox signaling”) has been an emerging theme in biomedicine over the last decade, major areas of investigations being its basic biochemical mechanisms, the sources of oxidants and their regulation, the targets of oxidant species and their roles in cell homeostasis, stress responses, and cellular pathophysiology.
Oxidants operate as signaling molecules mainly through the oxidation of iron–sulfur centers or the formation of cysteine oxidative adducts (nitrosocysteine, SNO, glutathionylcysteine, SSG) or disulfide bonds (S–S) on their protein targets ([52] and references therein) (Fig. 2). Interestingly, different cysteine-based oxidative modifications may occur on the same residue of the same protein, with different biochemical and functional effects [54].

Established mechanisms and molecular targets of redox signaling. Reversible cysteine modification with formation of disulfide bonds, mixed disulfides and S-nitrosocysteine represent the most relevant signaling mechanisms, and occur in several classes of signaling-related proteins. Reversibility of these modifications is guaranteed by enzymatic reactions involving thioredoxins (Trx), glutaredoxins (Grx), peroxiredoxins (Prx), sulfiredoxins (Srx), and nitrosoglutathione reductase (GSNO-R). Redox cycling of heme–iron occurs in several electron transfer reactions; in some instances this modification may directly impinge on signaling functions; oxidation/inactivation of PHDs by hydrogen peroxide [84], and redox regulation of the cytosolic aconitase balance [237], represent remarkable examples. Effect of iron oxidation in these cases can be reverted by ascorbate [86, 89]
While generation of NO is largely due to NOSes, ligand-dependent generation of superoxide anion and, via superoxide dismutation, of hydrogen peroxide, is brought about by membrane oxidases of the NOX/DUOX family, whose prototypical member is the phagocytic NADPH oxidase (Phox/NOX2) [55]. Notably, the formation of both NO and hydrogen peroxide (that derives from dismutation of superoxide) involves electron transfer from NADPH, which couples redox signaling to the metabolic status of the cell.
Superoxide generating membrane oxidases are multicomponent enzymes made of membrane-bound catalytic (gp91phox, p22phox) and cytosolic regulatory (p47phox/NOX1, p67phox/NOXA1) subunits, whose assembly involves phosphorylation events, lipid signaling and, at least for NOX1 and NOX2, the activation of the small GTPase Rac1 (or Rac2 in neutrophils). Unlike the first discovered phagocytic NADPH oxidase (NOX2), NOXes are present in nearly all the cell types in a constitutive or inducible fashion, and, similar to NOSes, serve signaling roles, downstream of cytokine and growth factor receptors [56–58], G-protein coupled receptors (GPCRs) [59] and adhesion molecules [60].
Of note, besides NOXes, also mitochondria have been identified as sources of ROS involved in signaling and in cell homeostasis [52, 61], in particular in response to changes in nutrient availability [62] and hypoxia [63]. While mitochondrial ROS generally operate in the context of stress cascades eventually linked to cell death [64], lines of evidence exist for a deliberate and finely regulated formation of oxidants in the organelle [65–67].
Acting in concert with ROS and RNS generating enzymes, enzymatic antioxidants or scavengers like peroxiredoxins, catalase, and glutathione peroxidases (for hydrogen peroxide), superoxide dismutases (for superoxide) and nitrosoglutathione reductase (for GSNO) contribute to regulate the intracellular tone of oxidant intermediates [52] (Table 2). Additionally, cysteine oxidative modifications on target proteins, including disulfide formation, S-glutathionylation and S-nitrosylation are reversed by thioredoxins, glutaredoxins, and nitrosoglutathione reductase, respectively, using NADPH or GSH as electron donors. These enzymatic systems guarantee reversibility to redox cascades and add further layers of regulation to oxidant dependent signal transduction [68, 69].
Reversible cysteine oxidation plays a pivotal role in redox signaling cascades, major molecular targets being protein tyrosine phosphatase or lipid phosphatases (like PTEN), proteases, signaling adaptors and transcription factors (Fig. 2). Among protein tyrosine phosphatases, protein tyrosine phosphatase 1B (PTP1B), SHP-2, PTEN and low molecular weight-PTP (LMW-PTP) are transiently inactivated by hydrogen peroxide in the context of growth factor, cytokine or integrin signaling, as a necessary step for the propagation of tyrosine phosphorylation cascades ([70] and references therein). Conversely, a density-dependent decrease of NOX-Rac1 activity and ROS production in response to RTK engagement mediates growth inhibition by cell–cell contact in fibroblasts [71]. While invariably inhibitory for PTPs, cysteine oxidation may be either inhibitory for proteases of the caspase family, underlying anti-apoptotic signaling by S-NO [72], or activatory for MMPs, through the disruption of the inhibitory interaction between the pro-domain and the catalytic site [73].
Other than by directly targeting enzyme activities, cysteine oxidation can modulate signal transduction by impinging on protein–protein interactions. Two elegant examples of this kind of regulation are the oxidation-dependent dissociation of thioredoxin from the apoptosis stimulating kinase-1 (ASK-1), leading to activation of the kinase and of its downstream target Jun and stress kinases [74], and the nuclear translocation of nuclear respiratory factor-2 (NRF-2) following disruption of its interaction with the adaptor KEAP-1, upon oxidation of the latter on cysteines 273 and 288. Once in the nucleus, NRF-2 activates antioxidant response element (ARE)-dependent gene transcription, up-regulating several antioxidant and detoxifying enzymes to maintain cell homeostasis in response to oxidative and chemical stress [75, 76].
Finally, reversible cysteine oxidation of transcription factors affects DNA binding and transcriptional activity of several nuclear factors including activating-protein-1 (AP-1), nuclear factor-κB (NF-κB), p53, HIF [76]. In most cases, oxidation interferes with DNA binding and inhibits transcriptional activity that is reactivated upon cysteine reduction assisted by the redox factor-1 (REF-1) [77]. The activity of these and other factors is modulated by redox cascades also in an indirect fashion, i.e., through the regulation of their phosphorylation, acetylation, hydroxylation. Particularly intriguing is the redox regulation of the forkhead box class-O (FOXO) transcription factors, involved in crucial cellular processes including cell cycle regulation, apoptosis and resistance to oxidative stress. FoxO activity is inhibited by hydrogen peroxide by two distinct mechanisms: one involves FoxO association with the hystone acetylase p300 via the formation of an intermolecular disulfide bond, followed by protein acetylation and impaired transcriptional activity [78]. Importantly, acetylation-dependent inhibition can be reversed by the NAD+ dependent deacetylase sirt-1 [79]. A second, indirect mechanism requires ROS-dependent phosphorylation of FOXO by Akt, leading to nuclear exclusion of the factor [80, 81]. Interestingly, the two mechanisms appear to be largely independent from each other [79].
Compelling lines of evidence indicate that also HIF is subdued to multiple mechanism of redox regulation. S-nitrosylation of HIF at Cys-800 has been shown to increase factor stability and transcriptional activity, likely by promoting HIF binding to p300 [82]. Interestingly, also Cys-388 and 393 on p300 are necessary for this interaction [83], but the possibility that a disulfide bond may form between the two molecules has not been investigated. On the other hand, hydrogen peroxide indirectly promotes HIF stabilization by oxidizing catalytic Fe++ of PHDs and inhibiting their activity [84]. This second mechanism may be implicated in physiological stabilization of HIF under mild (1–3%), but not deep, hypoxia, a condition reportedly accompanied by mitochondrial production of ROS [63, 85]. Vitamin C has been shown to decrease HIF-1 levels by preventing the oxidation of the catalytic ferrous ion [86]. Furthermore, PHD inhibition by ROS may contribute to normoxic accumulation of HIF in cancer cells [87, 88]. In keeping, it has been recently reported in three different tumorigenic mouse models that antioxidants as N-acetyl cysteine (NAC) and vitamin C exert their antitumoral effect mainly acting on HIF-1α level [89].
Cysteine-based modifications of chromatin remodeling factors like histone acetylases/deacetylases is emerging as an additional mechanism of redox-based transcriptional regulation. For instance, in neurotrophin-stimulated neurons, nitrosylation of histone deacetylase-2 induces its release from chromatin, which increases acetylation of histones surrounding neurotrophin-dependent gene promoters and promotes transcription by the cAMP-responsive element binding (CREB) factor [90]. This elegant mechanism adds a new perspective to the already established notion that gene transcription by CREB is subdued to redox control [91, 92]. Of note, redox regulation of deacetylase activity may also occur via tyrosine nitration, in the context of inflammation [93].
ROS and cell dynamics
Roles played by oxidant species and redox cascades in cell–matrix interaction, cytoskeleton rearrangements and cell motility have been in the focus of intense research over the last decade. Initial observations were driven by the question of whether the long established effects of the small GTPase Rac-1 on cytoskeleton dynamics and its capacity to elicit ligand-controlled generation of ROS by membrane-bound oxidases may represent related biochemical events. Along this line of investigation, Moldovan and colleagues showed that actin reorganization induced by Rac-1 in human endothelial cells is accompanied by and dependent from the formation of ROS [94]. This in spite of a previous report indicating superoxide production by Rac-1 to be dispensable for cytoskeleton rearrangement in quiescent fibroblasts, although required for mitogenesis [95]. From a different angle, Zena Werb’s group demonstrated that cell shape changes, dictated by interference with integrin-ECM binding, lead to NF-kB activation and expression of collagenase through Rac-1 and the generation of ROS [96]; surprisingly, in this model, mitochondria, instead of NOXes, were identified as the major source of oxidant species triggered by Rac-1 upon cell detachment [97]. These observations, together with other studies addressing cell responses to mechanical stretch or shear stress [98], clearly indicate that the relation between cytoskeleton changes and production of ROS is biunivocal, i.e., ROS regulates cytoskeleton dynamics and the other way around.
Redox signaling by adhesion molecules has been investigated in detail. Chiarugi et al. initially reported that integrin engagement activates Rac-1 and ROS in adhering fibroblasts, and that this transient oxidative event is crucial to cell adhesion and spreading onto fibronectin [60]. Additionally, analysis of molecular events downstream of Rac-1 and NOX-driven hydrogen peroxide revealed that the transient and reversible inhibition of LMW-PTP is instrumental for activation of FAK in response to integrin engagement. A deeper analysis of ROS sources involved in integrin signaling confirmed engagement of both mitochondria and membrane-bound oxidases (NOXes and 5-lipoxygenase), although with different kinetics: mitochondrial ROS are involved in the regulation of the early contact with ECM through focal contacts, while membrane oxidases drive the spreading and actin dynamics of a moving cell [99].
The oxidative cascade involving Rac-1, ROS and LMW-PTP has been correlated by Nimnual and Bar-Sagi with the antagonistic cross-talk between Rac-1 and the related small GTPase RhoA [100]. This coordinated and opposed activity of Rac-1 and RhoA is crucial to cellular dynamics, the former promoting membrane protrusion, cell polarity and spreading, the second cytoplasm contractility and tail retraction [12]. In neurons, for instance, Rac-1 elicits neurite outgrowth and branching, while RhoA induces axon retraction in response to repulsive signals mediated by plexins and Ephrin receptors [101]; in epithelial cells Rac-1 strengthens, and RhoA loosens cell–cell interaction allowing epithelial-to-mesenchymal transition [102]. Rac-1 activity dominates at the highly motile leading edge of the cell, while RhoA is more active at the trailing edge during oriented migration [12]. Finally, Rac-1 and Rho A appear to orchestrate two different and mutually exclusive motility styles in invading malignant cells. Indeed, Rac-1 is a key molecular motor of polarized mesenchymal motility style, while RhoA-mediated cytoskeleton contractility has been involved in amoeboid motility, a successful non-oriented motility method used by lymphocytes and several cancer cells to invade ECM [12, 18].
In the attempt to clarify the mechanism whereby Rac-1 regulates RhoA signaling, Nimnual found that Rac-1-driven ROS, by inactivating LMW-PTP, causes hyperphosphorylation of the PTP substrate p190Rho-GTPase activating protein (GAP), thereby increasing its inhibitory activity on RhoA. In a specular fashion, activation of the repulsive EphA2 receptor in prostate carcinoma cells is accompanied by reduced Rac-1 activity and attenuated generation of ROS, which leads to LMW-PTP activation, p190RhoGAP dephosphorylation and to an increase of Rho signaling [103]. In keeping, Rac-1 activation and ROS generation have been correlated with collagenase-mediated motility [97] (a key feature of mesenchymal motility), while RhoA activation and attenuated generation of ROS has been associated with amoeboid motility [103, 104]. Finally, activated RhoA is able to inhibit Rac-1 through the chimerin2-ARHGAP2 [19], a GTPase activating protein for Rac-1, thereby completing the circuitry of Rac1-RhoA antagonism. Collectively, the above lines of evidence indicate that the intricate cross-talk between Rho family GTPase that underlies dynamic cell responses is in large part redox-regulated (Fig. 3).

Rac and Rho small GTPases reciprocal regulation in cell dynamics. ROS act as a balance for Rac-1/RhoA antagonism. Indeed Rac-1, which drives oriented mesenchymal motility, leading edge protrusion and lamellipodia formation, is a key molecular player of regulated intracellular ROS sources as NADPH oxidase, lipoxygenase, cycloxygenase, and mitochondria. Hence, oxidation/inactivation of the PTP which normally activate the Rho regulator p190Rho-GTPase, leads to RhoA down-regulation [100]. Conversely, low ROS intracellular content lead to RhoA activation, through PTP activation and p190RhoGAP dephosphorylation/inactivation (Buricchi, ref 104). Activated RhoA is able to inhibit Rac-1 through the ARHGAP2 (also named chimerin-2) [19]. Rho activation is responsible for amoeboid motility, a non-oriented movement which enables the cell to squeeze between gaps of ECM instead of proteolytically degrade it
Of note, redox switches relevant to cytoskeleton reorganization are not restricted to LMW-PTP or other phosphatases. The tyrosine kinase Src, a major effector in integrin signaling, is transiently oxidized and activated upon cell adhesion to the substrate [105]. Besides c-Src, activation via oxidation is mandatory also for the oncoprotein v-Src, as revealed by abolishment of xenograft growth and anchorage-independent growth by antioxidants. Moreover, cytoskeleton components can be directly modulated by oxidation; a proteomic screen for cysteine glutathionylated proteins in T cells exposed to oxidant agents diamide or hydrogen peroxide identified several cytoskeleton components (actin, vimentin, myosin, tropomyosin, cofilin, profilin) [106], and adhesion-triggered glutathionylation of actin at cysteine 374 is necessary for actinomyosin complex disassembly and for F-actin accumulation and cell spreading [107].
In line with the emerging role of ROS in mediating cell cytoskeleton reorganization in response to extracellular cues is the identification of molecule interacting with Cas-ligand (MICAL) as a protein involved in neurite repulsion by the semaphorin/plexin ligand/receptor system in neurons [108, 109]. Initially described as a cytoskeleton-associated multidomain protein expressed in several epithelial tissues [110], MICAL is also structurally related to FAD-dependent monoxygenases, and has been shown to generate hydrogen peroxide from molecular oxygen and NADPH, both in vitro and in intact cells [111]. A genetic screen for proteins functioning in plexin-mediated axonal repulsion in C. elegans identified MICAL as a component of this cascade, and interference with its redox activity has been shown to block neurite collapse in response to semaphorin, thereby phenocopying MICAL deletion [108]. MICALs also interacts with Rab GTPases and may be involved in vesicle trafficking between endoplasmic reticulum and the Golgi compartment [112], as well as in the resolution of intercellular junctions during epithelial to mesenchymal transition; accordingly, MICAL-L2 and Rab-13 have been found to co-localize at lamellipodia with F-actin during cell scattering [113]. Whether also these functions of MICAL rely on its oxidase activity is still to be determined.
Two other molecules involved in cell–ECM interaction and cell motility, the HGF receptor c-Met and lysyl oxidase, appear to exploit redox signaling cascades. The tyrosine kinase receptor c-Met triggers scattering and invasive behaviors and activates epithelial–mesenchymal transition during development and in malignant growth [23]. c-Met has been shown to activate Rac-1 and to elicit a rise in intracellular hydrogen peroxide in melanoma and in lung carcinoma cells [114, 115]. Although not directly demonstrated, the involvement of NOXes in this cascade is likely, since multiple members of the NOX gene family are highly expressed in cancer cells [55]. Importantly, Rac-dependent oxidants in B16 melanoma correlate with c-Met expression, and are essential for metastasis formation in vivo (see below).
Lysyl oxidase is a nearly ubiquitous secreted and membrane-associated enzyme that cross-links ECM proteins (collagens and elastin) increasing matrix stiffness and tensile forces. By doing so, lysyl oxidase generates hydrogen peroxide, which appears to contribute to cell adhesion and motility/migration via a redox mechanism [116]. In particular, lysyl oxidase-derived peroxide activates the Src-FAK axis in breast carcinoma cells, likely contributing to the invasiveness and poor prognosis of breast cancers that overexpress this enzyme.
The diffusive and reactive nature of oxidant species raises the question of how these intermediates can regulate highly localized molecular reactions as those underlying the above described complex cellular dynamics. NOXes provide an excellent example for subcellular compartmentalization of redox signals: assembly of active oxidases requires the contribution of membrane-bound catalytic subunits with cytosolic organizers, in turn subdued to multiple levels of regulatory interactions. These interactions provide the scaffold for NOX activity to be spatially restricted [117] (Fig. 4). Wu et al. have demonstrated that p47phox interaction with the orphan adaptor TRAF1 recruits the former at nascent focal contacts within lamellopodia [118]; mechanistic studies have revealed that ROS so generated are actually instrumental to membrane ruffling and endothelial cell migration, and that this happens through the transient inactivation of the tyrosine phosphatase PTP-PEST. Interestingly, PTP-PEST inhibition further reinforces Rac-1 activation and ROS formation, thereby creating a feed-forward loop [118]. Along similar lines, Diaz et al. have identified the Src tyrosine kinase substrates Tks4 and Tks5 as molecular organizers that specifically activate Nox1, Nox3, and Nox 4 at invadopodia, dynamic actin-rich protrusions of cancer cell membrane involved in pericellular proteolysis and in invasive behavior [119, 120]. Interestingly, down-regulation of Tks5, structurally homolog to p47phox, decreases intracellular ROS in Src-transformed 3T3 cells, and in turn ROS blockade decreases the number of invadopodia, directly involving NOXes and ROS in Src-induced cell invasiveness. Also, in these studies, ROS effects have been found at least in part mediated by the inhibition of PTP-PEST, which in turn leads to increased tyrosine phosphorylation of Tsks and further activation of NOXs.

Localized generation of ROS at invadopodia (and lamellipodia). Activation of NOX is spatially restricted to specialized membrane areas by molecular adaptors Tks, in turn regulated by upstream signals [119, 120]. Phosphorylation of Tks triggers NOX- and ROS-dependent formation of invadopodia in Src-transformed cells. The PEST-PTP has been identified as a main target of ROS in this circuitry. Inhibition of PEST further amplifies Tks phosphorylation, and the same effect may also be elicited by oxidative activation of c-Src [107]. The presence of redox-sensitive components upstream of NOXes establishes feed-forward activation loops and allows that NOX activity be also sensitive to extracellular or mitochondrial ROS
Feed-forward loops are a constant finding in redox signaling. In the examples above, c-Src, that triggers the localized activation of NOXes, may be itself activated by ROS [105]; likewise, the phosphatase PTP-PEST operates both upstream and downstream of the Rac-1/p47Phox/ROS cascade as a negative regulator of motility at endothelial cell lammellipodia [118]. These loops are likely to amplify redox signaling cascades and set thresholds for their “explosion” under the appropriate stimuli, but also create the conditions for these cascades to integrate redox changes initiated in different cell compartments, or even triggered in the extracellular space by neighboring cells (Fig. 4).
Lipid rafts and caveolae, specialized membrane domains enriched in cholesterol and glycosphingolipids that act as signaling platforms for GPCRs and RTKs, may also contribute to the spatial compartmentalization of redox signals involved in cell dynamics. In keeping with this emerging concept, nitric oxide synthase and NOXes concentrate in these areas, and several extracellular ligands known to trigger intracellular redox cascades induce lipid raft clustering [121]. Integrin signaling also leads to changes in membrane order and fluidity, and focal adhesions co-stain with highly ordered rafts [122]. Therefore, it appears conceivable that lipid raft redox signaling plays a role in cell–matrix interactions and in the intricate signaling network regulating cell adhesion and motility.
In addition to compartmentalization of oxidant sources, spatio-temporal restriction of redox cascades involved in cell dynamics requires also an elevated reductive potential, to allow a rapid reset of redox switches, as well as strong redox buffers (such as glutathione) to limit indiscriminate diffusion and maintain a polarized distribution of oxidant intermediates. Glutathione and the thioredoxin system are well suited to this function and guarantee that the intracellular environment is, with the remarkable exception of the endoplasmic reticulum lumen, substantially reductive. Both systems require NADPH (via glutathione reductase and thioredoxin reductase, respectively), mainly generated via the pentose phosphate pathway, as the electron donor to be regenerated [52], and, intriguingly, NADPH also provides reducing equivalents that fuel NOX, lipoxygenase and cycloxygenase activity [70]. Thus, oxidases and reductases appear to work coordinately under the control of glucose metabolism near the cell border where most motile activities take place. In keeping with this, in part, speculative view is the evidence that inactivation of SH2-domain protein tyrosine phosphatase (SHP-2), a cytosolic phosphatase regulated by cysteine oxidation [123], reduces cell motility [17], and that inhibition of glutathione synthesis impairs cell spreading [107]; moreover, glucose lowers the threshold for vascular smooth muscle cells chemotaxis towards PDGF [124], while inhibition of glucose-6-phosphate dehydrogenase, the first enzyme of the pentose phosphate pathway that generates NADPH, inhibits endothelial cell migration in response to VEGF [125].
Why oxidants are the cause of cancer
Compelling experimental, clinical, and epidemiological evidence indicates that ROS and RNS are cause of cancer. The carcinogenic activity of oxidants strongly depends from their mutagenic potential (initiation), their effects on intracellular signaling pathways controlling cell proliferation and survival (promotion), their impact on cell motility and invasiveness, and their established role in stromal reactions key to cancer development and dissemination, like inflammation/repair and angiogensis (tumor progression). The chemopreventive and tumor-inhibitory properties of several natural, dietary, or synthetic antioxidants [126], and the strong linkage between antioxidant enzymes and genetic predisposition to cancer, both in humans [127, 128] and animals [129], are strong arguments in support of this general and widespread notion.
While radical reactions have been long known to participate in the mutagenic effects of ionizing/exciting radiations and chemical carcinogens, endogenous oxygen and nitrogen reactive species also account for a large fraction of “background” DNA damage in mammalian cells, with the hydroxyl radical (OH·) and peroxinitrite (ONOO− ) being the best recognized candidates for formation of 8-oxo-guanine and single/double strand breaks [130]. Oxidative cell stress may also damage DNA indirectly through lipid peroxidation products (namely malondialdehyde, and 4-hydroxy-nonenal) endowed of the capacity to form adducts with DNA purines and pyrimidines. Additionally, some oxidants, like NO, may attack and inactivate DNA repair enzymes, thereby increasing genomic instability. Of course unrepaired DNA damages can lead to cell death rather than to initiation of carcinogenesis [131]. The cell fate will be dictated by the integrity of DNA damage responses and cell cycle checkpoints, the function of the apoptotic machinery, the presence of survival cues from the surrounding microenvironment.
Tumor promoting actions of oxidants involve proliferation and survival of carcinogen-initiated cells [132]. The presence at nodal points of signal transduction cascades initiated by growth factors, adhesion molecules cytokines and hormones, of molecular components (i.e., phosphatases/kinase, proteases, transcription factors, multidomain adaptors) subdued to redox regulation sets the stage for tumor promotion by oxygen and nitrogen species. Oxidants can be generated in excess as “physiological” signaling intermediates downstream of deregulated oncogenes (for instance, by oncogenic Ras [133] or overexpressed/mutated tyrosine kinase receptors [114]); alternatively pathologic pro-oxidant states (e.g., due to inflammation or tissue aging) may hijack and alter redox signaling cascades, leading to uncontrolled cell proliferation or defective cell death. In the latter case, oxidant species may be chemically different from those normally involved in redox signaling and lead (for instance through protein carbonylation, tyrosine nitration or cysteine hyperoxidation) to irreversible modifications of redox switches. This may often be the case for oxidant species of extracellular origin, like those generated by phagocytes during inflammatory reactions.
Redox signaling and the hallmarks of cancer
Evidence exists for a contribution by ROS and RNS signaling to all the seven “hallmarks” (i.e., self-sufficiency for growth signals, insensitivity to anti-growth signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, invasion and metastasis) listed by Hanahan and Weinberg as fundamental to malignancy [134] (Fig. 5). Cancer cell lines are long known to produce more oxygen radicals then normal cells in vitro [135]. Early conclusions placing the source of these oxidant species in flavin-dependent oxidases sensitive to flavin inhibitor diphenyleneiodonium (DPI) but not to electron transport blockers, have been more recently corroborated by the finding of elevated expression levels of NOXes in malignant cells [136]. Cell transformation by oncogenic Ras is accompanied by, and dependent on, increased generation of superoxide, and NOX1, that is transcriptionally up-regulated by K-Ras, is functionally required for these oxidative responses and for the establishment of Ras-transformed phenotypes, including anchorage-independent growth, morphological changes, and production of tumors in athymic mice [137, 138]. Interestingly, Ras is itself redox-regulated, and can be activated by NO (and likely also by hydrogen peroxide) via oxidation of Cys131 [139]. NOX-dependent overproduction of oxidants also occurs in fibroblasts transformed by the Src tyrosine kinase [119], while overexpression of the epidermal growth factor receptor (EGF-R) raises the level of ROS in NIH-3T3 cells, partially inhibited by the NOX inhibitor DPI [140].

ROS and/or RNS play important and multiple roles in each of the six hallmarks of cancers. Based on the model by Hanahan and Weinberg [134], cellular and molecular mechanism for which a role for ROS/RNS has been demonstrated are indicated for each hallmark, in the same color (see text for details). 1. GROWTH EVEN IN THE ABSENCE OF NORMAL “GO” SIGNALS: Most normal cells wait for an external message before dividing. Conversely, cancer cells often counterfeit their individual proliferative messages. 2. GROWTH DESPITE “STOP” COMMANDS: As the tumor enlarges, it squeezes adjacent tissues and therefore receive messages that would normally stop cell division. Malignant cells ignore these commands. 3. ABILITY TO INVADE TISSUES AND SPREAD TO OTHER ORGANS: Cancers usually lead to death only after they overcome their confines to the particular organ in which they arose. Cancer cells need to escape the primary tumor, invade matrix of different organs, find a suitable metastatic niche and then grow in this secondary site. 4. EFFECTIVE IMMORTALITY: Healthy cells can divide no more than 70 times, but malignant cells need more than 70 cycles to make tumors. Hence, tumors need to enforce the reproductive limit of cells. 5. ABILITY TO STIMULATE BLOOD VESSEL DE-NOVO ASSEMBLY: Tumors need oxygen and nutrients to survive and secrete factors recruiting new branches that run throughout the growing mass. 6. EVASION FROM ENDOGENOUS AUTODESTRUCT MECHANISMS: In healthy cells, several conditions (including genetic damage or lack of ECM adhesion) activates a suicide program, but tumor cells bypass these mechanisms, thereby surviving to death messages
Besides being generated as a consequence of oncogenic transformation, oxidant species can be increased in initiated or tumor cells by chemical promoters like phorbol esters, inflammatory cytokines or complete carcinogens like cigarette smoke. Irrespective of their origin, these species may reduce cell dependence on growth factors by lowering the activation thresholds of the cognate RTKs, or by transactivating receptors in a ligand-independent fashion [141]. Since RTKs couple to multiple downstream signaling cascade, many growth-related signaling events reportedly triggered by oxidants, such as activation of MAPKs or induction of early responsive genes Jun, Fos, and Myc could ultimately reflect, at least in part, the upstream activation of tyrosine kinase-dependent signaling. Exogenous oxidants may also target specific steps downstream of RTK signaling, remarkable examples being the direct regulation of Ras by NO [139], and the activation of PKCδ by hydrogen peroxide, the latter likely mediated by the tyrosine kinase Abl [142].
Growth inhibitory signals that normally restrain cell proliferative capacity may also be subverted by indiscriminate generation of ROS or exposure to pathologic pro-oxidant states. Normal adherent cells stop proliferating, even in the presence of mitogenic stimuli, upon reaching confluence (“contact inhibition”) or when detached from their substrate (“anchorage dependence”). These inhibitory responses are typically lost following oncogenic transformation, in parallel with increased generation of ROS. Contact inhibition in non-transformed fibroblasts is associated with reduced activity of Rac-1 and by low level of intracellular hydrogen peroxide that activates PTPs and impairs RTK signaling [71]. Interestingly, exogenous hydrogen peroxide can partially restore proliferation of contact-inhibited cells. Along similar lines, fibroblasts plated on polylysine (a substrate that does not activate integrins) display impaired generation of hydrogen peroxide in response to platelet derived growth factor (PDGF), and have blunted growth capacity [60]. Overexpression of constitutively active Rac-1 or oncogenic Ras in these cells restores anchorage-independent generation of ROS and, in parallel, allows colony formation in soft agar [143]. In both these examples, growth deregulation by oxidant species occurs at the early steps of growth factor signaling and involves redox regulation of SHP-2 and LMW-PTP, respectively.
At a downstream level, multiple antiproliferative cues, including those listed above, converge on the transcriptional up-regulation of the cyclin-dependent kinase inhibitor p27, a molecule dictating cell arrest at the G1/S boundary [144]. p27 is transcriptionally induced by FOXO-3a, whose phosphorylation by Akt in response to growth factors prevents nuclear translocation and gene trans-activation [145]. Phosphorylation and nuclear exclusion of FOXO-3a can also be elicited by hydrogen peroxide through a pathway involving Akt and the longevity related protein p66shc [81]. Although the p66Shc/FOXO-3a cascade has been primarily linked to cell and body aging, this circuit may also have a role in tumor progression, especially in cell that have already acquired some resistance to oxidant-induced cell death. Of note, FOXO-3a can also be inactivated by oxidant-induced association with p300 (see above); it is likely that the intensity of oxidative stress dictates these different responses in distinct pathophysiological conditions or cellular contexts.
Replicative senescence limits proliferation of initiated cells and may occur even before critical telomere shortening as a consequence of DNA damage or oncogenic signaling [146], and is triggered by a p53 and p16 dependent cascade [147]. Tumor development requires that cells gain unlimited replicative capacity. It is generally accepted that oxidative stress promotes cell senescence, and the finding that hypoxia delays senescence of human and rodent cells and decreases DNA damage supports the role of ROS in limiting cell lifespan [148, 149]; however, an alternative explanation has been proposed whereby hypoxic response, through HIF, induces the expression and activity of telomerase, thus favoring cell immortalization. Paradoxically, ROS triggered by low oxygen appear to have a positive role in this phenomenon [150]. While the importance of oxidant species in cell senescence and in adaptation to hypoxia will be discussed below, it is of note that telomerase expression has been found to be inhibited in ovarian cancer cells by vitamin E, a molecule endowed of strong antioxidant capacity [151].
Although indiscriminate oxidative stress has been extensively linked to apoptosis of both normal and cancer cells, several examples exist of the involvement of oxidants in survival signaling. Oxidative inactivation of the protein/lipid phosphatase PTEN [152] following engagement of growth factor receptors by the cognate ligands (PDGF, EGF, and insulin) increases PI3K signaling to Akt, a proto-oncogenic kinase playing pivotal roles in multiple cancer related cell functions, including cell cycle progression, motility and protection from apoptosis. Survival signaling by Akt in the context of growth factor and cytokine signaling is largely mediated by the inflammatory transcription factor NF-κB [153, 154]. Interestingly, NF-κB has been long known as a factor subdued to redox regulation; while direct oxidation within the DNA binding domain inhibits its transcriptional activity, oxidants indirectly activate NF-κB-dependent gene transcription through the phosphorylation-mediated degradation of the NF-κB inhibitor IkB. Early reports of NF-κB activation by exogenous hydrogen peroxide in Jurkat T cells [155] have been more recently confirmed by the finding that in cervical carcinoma HeLa cells ROS generated by NADPH oxidase are necessary for NF-κB activation in response to tumor necrosis factor-α (TNF-α) and interleukin 1 [156], although a role for PTEN and Akt in this response has not been demonstrated. Additionally, oncogenic Ras activates NF-κB through superoxide in rat kidney epithelial cells [157], and Rac-1, a component of NOX, is necessary for NF-κB activation and prevention of apoptosis in several Ras-transformed cell lines [158]. Finally, during hypoxia, the tyrosine kinase Src undergoes redox activation and mediates phosphorylation of IκB and activation of an NFκB-dependent pro-survival program [159]. Given the relevance of NF-κB in tumor cell survival in the context of oncogenic transformation [160, 161], these observations further underscore the importance of ROS-dependent anti-apoptotic signaling in tumor promotion and progression.
Besides activation of Akt and NF-κB, other survival mechanisms sustained by oxidant species have been described in tumor cells. In undifferentiated pheo-chromocytoma cells PC12, cell death by serum deprivation is prevented by nerve growth factor through a Rac-1 and ROS-dependent cascade leading to activation of the CREB transcription factor, and to transcriptional up-regulation of the mitochondrial scavenger Mn-superoxide dismutase (SOD) [91]. A similar protective cascade has been described in human neuroblastoma cells, whereby exogenous NO or overexpression of NOS activate CREB through guanylate cyclase and cGMP to prevent apoptosis by growth factor starvation [162]. Importantly, NO has recently emerged as a major regulator of apoptosis through the S-nitrosylation of critical protein targets [163]. Among these, nitrosylation of caspases and thioredoxin clearly favor cell survival, while their denitrosylation heralds the onset of apoptosis. Inhibitory modification of caspases by cysteine glutathiolation has also been described [164]. Although these mechanisms have been little investigated in the context of carcinogenesis, they may contribute to the tumor promoting effect of oxidant species, for instance at sites of chronic inflammation.
Loss of contact with an appropriate ECM rapidly triggers programmed cell death in normal cells, a phenomenon known as anoikis (i.e. “homelessness”). Malignant cells need to escape anoikis when detach from neighboring cells, degrade ECM and undertake metastatic dissemination through blood or lymphatic vessels. Accordingly, proto-oncogenes like c-Src or activated RTKs (TrkB, EGF-R) can provide protection from anoikis by releasing constitutive adhesion-related signals even in the absence of proper integrin–matrix interaction. Giannoni et al. have recently identified a novel ROS-mediated circuit allowing anchorage-independent survival of prostate cancer cells; in these malignant cells, high levels of intracellular ROS activate c-Src via cysteine oxidation, leading to trans-phosphorylation and trans-activation of the EGF-R, and to prevention of anoikis. Conversely, elimination of ROS restores death and inhibits cell growth in semisolid medium, a strong correlate of metastatic capacity [165, 166]. Although in this model oxidative signals are generated inside cells (in a 5-lipoxygenase dependent fashion), a similar mechanism could also operate in the context of environmental oxidative stress, like that sustained by inflammation, cigarette smoke or hypoxia, thus helping the initial invasive steps of malignancy.
Angiogenesis and invasion: setting the stage for metastasis
Sustained neoangiogenesis (the process of new blood vessel formation) is necessary for primary tumor growth and provides anatomic substrate for cancer cell intravasation and hematogenous dissemination at distance. Oxidant species and ROS in particular have emerged as critical signaling molecules operating at multiple levels of both normal and tumor angiogenesis [167], and by extension as potential targets of novel antiangiogenic intervention. During new blood vessel formation, endothelial cells are recruited to proliferate, migrate, and organize into tubular structure under the action of specific growth factors, including VEGF, fibroblast growth factor-2 (FGF-2) and angiopoietins. Oxidant species can exert their signaling role (a) at an upstream level, by stimulating the local release of angiogenic factors, or (b) as downstream components of their intracellular cascades in endothelial cells.
Hydrogen peroxide generated by mitochondria of hypoxic tumor cells, or liberated by NOXes in the context of oncogene-initiated redox signaling [168], promotes HIF accumulation through the oxidative inhibition of PDHs, and the HIF-dependent transcription of the VEGF gene. Additionally, ROS may promote HIF activity by stimulating the mTOR/S6-kinase pathway [169, 170], either directly, through a putative redox switch located in the mTOR kinase [171], or indirectly via oxidative inhibition of PTEN [152]. Along different lines of evidence, NO increases HIF transcriptional activity by S-nitrosylation of Cys-800 and increased recruitment of the coactivator p300 [82]. Moreover, NO contributes to HIF stabilization by the normoxic inactivation of PHDs [172], and to HIF synthesis through the MAPK and PI3K pathway [173]. Thus, several oxidation-based mechanisms can contribute to VEGF secretion by cancer cells.
On the endothelial cell side, NO and hydrogen peroxide participate in angiogenesis as signaling intermediates downstream of VEGF [174, 175] and angiopoietin receptors [176]. The role of NOXes in redox signaling by angiogenic factors has been reviewed in depth [177]. Interestingly, NOX activation is also involved in bone marrow-derived endothelial precursor recruitment following hindlimb ischemia [178]. Moreover, ambient oxidants, as generated during hypoxia-reperfusion or by inflammatory cells can directly stimulate endothelial cells to proliferate, migrate and organize into tubular structures [179] and NO promotes the recruitment of perivascular cells (such as pericytes and vascular smooth muscle cells) during angiogenesis, vascular morphogenesis and vessel maturation [180]. Thus, the same pro-oxidant environment may promote release of angiogenic factor by tumor cells and promote or even vicariate their action on endothelial cells in the context of a growing tumor mass.
Invasiveness and metastasis
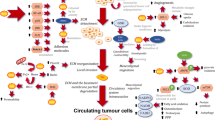
Accumulating lines of evidence points to the role of ROS as promoters of cell invasion and metastatic spread. In a murine model of invasive melanoma, overexpression of c-Met triggers a redox cascade that involves activation of the Rac-1 GTPase and generation of hydrogen peroxide via SOD1-mediated dismutation of superoxide; blockade of this cascade dramatically impairs oncogenic signaling downstream of c-Met and, most notably, inhibits cell capacity to form experimental lung metastases [114]. Along similar lines, in murine breast epithelial cells, exposure to MMP-3/stromelysin-1 (a condition reminiscent of the inflammatory milieu) induces the expression of the Rac-1b splicing variant, which leads to generation of ROS [181]. ROS are in turn responsible for the activation of the Snail factor, a key transcriptional inducer of the EMT program, and additionally induce genomic instability. Importantly, a cell shape change is required for EMT induction by MMP3 and Rac1b [182], in keeping with the notion that Rac-dependent ROS transduce mechanical perturbations into a pro-invasive gene expression program [96].
While the role of the Rac GTPases in the above examples, and the finding that NOXes are concentrated and activated at invadopodia of several types of malignant cells (see above 118, 119) clearly involve NADPH oxidases in invasion-related redox signaling, lines of evidence exist that mitochondria may also contribute as sources of oxidant species in malignant growth. Accordingly, hypoxia triggers EMT in several human tumor cells through the generation of mitochondrial ROS [183]. Along similar lines, Ishikawa et al. have elegantly shown that mutations in mitochondrial DNA favor cancer cell metastasis through the generation of mitochondrial ROS [184]. Of note, in both these examples, invasion and metastasis are associated with elevated intracellular levels of HIF, suggesting a direct link between ROS and the pro-invasive program triggered by this transcription factor.
Signaling roles of oxidants in mesenchymal-like tumor cell movement likely involve, besides gene regulation (e.g., c-Met or lysyl oxidase induction by HIF, E-cadherin down-regulation through snail, expression of MMPs through NF-kB), also a direct modification of cytoskeleton dynamics through actin glutathiolation [106, 107]. In addition, in pancreatic carcinoma cells, secretion and activation of MMP-2 has been reported to be dependent from Rac1 and ROS generation [185]. In general, ROS appear to promote an “explorative” behavior whereby membrane protrusions and fast-turnover focal contacts with ECM prevail over stable focal adhesions and cell contractility. These characteristics are typically observed in invadopodia, where also MMP activity is concentrated. Accordingly, invadopodia formation is impaired in the absence of NOX-derived ROS [119]. An alternative modality of movement of cancer cells, the amoeboid motility, does not require proteolysis but rather relies on actinomyosin contractility to allow cells to squeeze through the pores of the cross-linked matrix network. Chris Marshall and colleagues have shown in melanoma cells that mesenchymal and amoeboid motility styles are interconvertible through the reciprocal regulation of Rac and Rho GTPases [19]. Intriguingly, as illustrated above (Fig. 3), lines of evidence exist for a redox mechanism underlying this molecular cross-talk [103].
The picture of a cell frantically switching back and forth between one movement style to the other while migrating through the matrix provides the clearest demonstration of how finely tuned, transient and localized changes in ROS concentration need to be in order to support tumor cell plasticity. As said before, robust redox buffers (glutathione and thioredoxins) are critical to allow such a “redox plasticity”, thus linking cell invasiveness with glucose metabolism (by which GSH and Trx are regenerated via NADPH) and cell resistance to oxidative stress.
A heretic perspective: ROS as tumor suppressors
The above evidence on the causative role of oxidants in tumor initiation, promotion and progression raise the question of how tumor cells deal with oxidative stress. Oncogene-driven generation of ROS and RNS, coupled with a general reduction of cellular antioxidant enzymes in highly proliferating cells [186], and with transformation-associated genomic instability, lead to a high level of oxidative damage in neoplastic tissues. This pro-oxidant signature of tumor cells likely underlies the relative cancer-selectivity of chemotherapeutics (like doxorubicin) acting at least in part through ROS generation, as well as of chemical compounds (such as 2-methoxyestradiol) specifically selected for their pro-oxidant capacity [187]. Indeed, a large body of evidence indicates that oxidants may also exert important tumor-inhibitory actions (Table 3).
High amount of ROS generated in otherwise normal cells (e.g., primary diploid fibroblasts) by oncogene activation rapidly lead to senescence rather than to transformation. Importantly, replicative senescence due to overexpression of constitutively active H-Ras can be prevented by chemical oxidant scavengers or by mild hypoxia [188]. The p53 and p16/Rb tumor suppressor pathways appear to be crucial for these responses. In fact Ras-triggered superoxide drives mitogenic signaling instead of senescence in immortalized NIH-3T3 cells, which lack p16 [133, 189]. Moreover, co-inactivation of p53 and Rb by a temperature-sensitive SV40 large T antigen allows indefinite proliferation of human fibroblasts at the permissive temperature, while rapid senescence occurs in the same population upon switch to 39°C [190]. Interestingly, in this model, definitive growth arrest requires mitogenic stimulation, which is accompanied by higher levels of intracellular ROS and leads to redox-dependent activation of PKCδ. Along similar lines, a surprising role for Akt in oncogene and oxidative stress-driven senescence has been revealed by studies on Akt-1 and -2 double knock-out mouse embryonic fibroblasts. These cells appear to be resistant to replicative senescence and this correlates with reduced accumulation of intracellular ROS and increased expression of antioxidant enzymes [146].
While senescence effectively prevents cell transformation, successfully transformed cells are still extremely prone to apoptosis triggered by oxidative stress. Mouse embryonic fibroblasts tolerate much higher concentrations of the pro-oxidant drug doxorubicin or of hydrogen peroxide than the same cells transformed by H-Ras and the adenoviral oncogene E1A [191]. This is again dependent on the functional status of the tumor suppressor protein p53, being p53 deficient transformed fibroblasts remarkably resistant to oxidant-induced cell death [192]. Similar to ROS, also NO induces the accumulation of p53 and triggers p53-dependent growth arrest [193]. Conversely, NOS and NO increase malignancy of p53 deficient cells [194], and NOS overexpression is often found in human cancer in association with mutation or deletion of p53 [195], indicating that abrogation of the p53 anti-oncogenic activity converts NO from a tumor suppressor to an endogenous tumor promoter.
Several lines of evidence indicate that cell transformation is accompanied by a general decrease in cellular antioxidant defenses. Akt, a molecule at the crossroad of multiple oncogenic pathways, negatively regulates the expression of key antioxidant enzymes including MnSOD, catalase, peroxiredoxin III and Sestrin 3 [81, 146, 196]. This effect is mediated by the phosphorylation dependent inactivation of FOXO factors, by which the above enzymes are transcriptionally regulated. As a result, Akt protects from many forms of apoptosis, but not from ROS-induced cell death. Interestingly, FoxM1, another member of the forkhead family, is induced by H-Ras in immortalized fibroblasts through a pathway which bypasses Akt and has ROS as intermediate messengers [197]. FoxM1 induces antioxidant enzymes, although less efficiently than FOXO-3a, thus counterbalancing the effect of Akt; interestingly, osteosarcoma cells overexpressing Akt become “addicted” to this factor, to the point that its down-regulation by siRNA technology rapidly elicits a permanent, senescence-like growth arrest.
Likewise, Myc and E2F oncogenes sensitize mouse immortalized fibroblasts and human osteosarcoma cells to serum deprivation by increasing intracellular ROS [198]. This effect appears to be determined by the capacity of these factors to target the 65 kD subunit of NF-kB, another regulator of cell antioxidant response. In particular, Myc prevents NF-kB-dependent up-regulation of MnSOD that in normal cells opposes death by serum starvation. Importantly, NF-kB, as FoxM1, is activated by H-Ras and prevents a Ras-induced, p53 independent pro-apoptotic response, thereby allowing cell transformation [160, 161].
Collectively, these examples demonstrate that cells undergoing oncogenic transformation need a compromise between an increase of intracellular oxidant species that is instrumental to mitogenic and motogenic cascades, and the risk of oxidant-triggered anti-oncogenic responses leading to senescence and/or apoptosis.
A key molecular sensor linking oncogene-induced oxidative stress to the activation of tumor suppressor pathways has been identified in the p38-MAPK [199]. Unlike other members of this kinase family, this molecule displays mainly antiproliferative and pro-apoptopic functions, in part by phosphorylating and activating p53, and by inducing p16 [200]. Accordingly, overexpression of the wild-type p53-induced phosphatase-1 (WIP1) that inactivates p38, is often found in human cancer, while transformation of WIP1 deficient cells is prevented by unbalanced p38 activity and can be restored by inactivation of the p16/Rb pathway [201]. p38 is activated by oxidants and cells lacking p38 are selectively prone to transformation by oncogenes, like H-Ras, N-Ras or Neu, that produce ROS. It is likely that ROS-dependent activation of p38 in this cell context is mediated by the Trx/ASK1 module (see above); in alternative, oxidative inhibition of WIP1 may represent the redox sensor for the cascade. Interestingly, cells lacking p38 were found to accumulate higher ROS levels than p38 proficient cells upon transformation and to grow faster, while the latter underwent apoptosis. Thus, blockade of p38 not only prevents tumor suppressive responses, but also increases oxidative cascades leading to proliferation and genetic instability.
In spite of its anti-oncogenic properties, deletion or inactivation of p38 is rarely found in human malignancy; instead, evidence exists for a positive correlation between the expression of several glutathione S-transferase (GST) family members and cancer progression [202]. GSTm2 inhibits p38 activation by ASK1, and therefore uncouples oxidative stress from p38-dependent tumor suppression, without reducing the overall intracellular level of ROS [199].
A similar survival strategy has been described in E1A/Ras-transformed fibroblasts, where up-regulation of the mitochondrial scavenger MnSOD protects from cell death triggered by doxorubicin or serum deprivation, but without a significant change in the level of oxidative stress associated with these conditions [192]. Instead, indiscriminate removal of ROS by NAC scavenger activates p53 and rapidly kills these transformed cells [203], likely by inhibiting ROS-dependent survival signaling by growth factors.
p53-dependent cell responses are deeply interconnected with the cellular redox homeostasis, since p53 is not only activated by ROS, but in turn exploits ROS to promote cell death [204, 205]. This effect is in part achieved by the up-regulation of mitochondrial enzymes like prolyl oxidase, ferredoxin reductase and p66Shc, that generate ROS in the organelle [67]. Blockade of this redox cascade prevents p53-dependent apoptosis, indicating a possible strategy for cancer cells to avoid p53 dependent cell death. Additionally, p53 represses the expression of MnSOD by inhibiting specificity protein-1-dependent transcription [192, 206, 207], thus favoring the accumulation of mitochondrial superoxide. Levels of MnSOD vary in human malignancies depending on tumor type and stage, but an up-regulation of this enzyme in advanced cancers is being increasingly recognized [208]; moreover, mild overexpression of MnSOD prevents p53-induced cells death in Hela cells engineered to overexpress wild-type p53 [192] and MnSOD is up-regulated by FoxM1 in the context of cell transformation by H-Ras [197]. Thus, besides p38 inactivation by GSTm2, up-regulation of MnSOD may represent an additional strategy for malignant cells to selectively block anti-oncogenic signaling by ROS in mitochondria while preserving, or even enhancing peroxide-dependent mitogenic cascades in the cytosol.
Like p53, also the p16/Rb pathway may use ROS as tumor suppressive effectors. In fact, inhibition of E2F in human diploid fibroblasts, an event mimicking p16 up-regulation, is accompanied by increased levels of oxidant species [146]. Additionally, senescent cells and tissues display signs of oxidative stress, even when senescence appears to be triggered directly by ROS-independent DNA damage, i.e., in a ROS-independent fashion [209].
Taken together, these lines of evidence challenge the unidirectional view of oxidants as inducers of carcinogenesis and tumor progression, provocatively suggesting that elimination of oxidants may under some circumstances help tumor cell survival, and warning against previously underestimated dangers and pitfalls of antioxidant based intervention against cancer.
ROS and cancer stemness
Recent studies aimed at correlating cancer stem cell phenotypes with tumor invasiveness, metastasis, and resistance to therapy further solicit a critical rethinking of the role of oxidants and redox signaling in malignancy (Table 4).
Dihem et al. [210] have shown that both normal mammary stem cells and cancer stem cells isolated from human and murine breast tumors have a lower content of ROS compared to their mature progeny, and that in both cases this difference is critical for maintaining stem cell function. This finding extends to cancer stem cells previous discoveries on the role of intracellular ROS in governing the balance between self-renewal and differentiation of glial precursor cells [211] and of HSCs [209, 212]. In particular, in the hemopoietic system, oxidative stress induced by inactivation of the ataxia teleangectasia mutated kinase promotes precursor proliferation and differentiation into progenitor at expense of self-renewal, and induces premature exhaustion of the stem cell compartment. In an intriguing parallel with tumor suppression by ROS, p38MAPK has been identified as the molecular sensor, and the p16/retinoblastoma (Rb) pathway as the effector for oxidative stress in this model of premature HSC senescence [213]. Additionally, FOXO transcription factors appear to be crucial, through up-regulation of antioxidant enzymes, for HSCs resistance to physiological oxidative stress [214], while, conversely, the nutrient sensitive cascade of mTOR promotes hematopoietic progenitors proliferation and exhaustion through mitochondrial biogenesis and generation of ROS [215].
Like HSCs, breast cancer stem cells also appear to rely on antioxidant defenses to maintain quiescence and self-renewal capacity. Importantly, up-regulation of cell antioxidant capacity (via up-regulation of GSH biosynthetic pathway and of FOXO-1) couples stem cell redox phenotype to radioresistance and presumably also to resistance to radiomimetic, pro-oxidant chemotherapeutics [210]. On the other hand, since maintenance of glutathione redox buffer largely relies on glucose metabolism through the pentose phosphate cycle, these observations may explain, at least in part, the preference of normal and cancer stem cells for a glycolytic metabolism and a hypoxic microenvironment. While these important findings wait to be further confirmed in other cancer models, they may have important implications for new cancer therapies aimed at eradicating tumors by overcoming low ROS levels in CSCs [187], also in view of accumulating evidence on the connection linking tumor stemness with cell metastatic capacity [6].
In line with these emerging concepts, also tumor dormancy, that temporarily halts metastatic growth, but also ensures resistance to therapy and persistence of a minimal residual disease, may represent a cell response to oxidative stress. In fact, activation of p38MAPK, a key stress sensor, plays a pivotal role in the establishment of dormancy [216]; moreover, at a downstream level, p38MAPK activates p53 and down-regulates FoxM1 [217], two molecules capable of modulating in different directions intracellular ROS. Interestingly, dormant cells behave differently with respect to HSCs, in that they do not proliferate and exhaust, but rather arrest and survive in response to excess oxidants or to signaling through the mTOR pathway [218]. Malignant cells that have extravasated at remote hostile sites are likely to be exposed to multiple unfavorable conditions (lack of growth factors and of an appropriate ECM, shortage of nutrients and oxygen) potentially leading to oxidative stress and oxidant-triggered cell death. Dormant cells may therefore represent CSCs positively selected for their capacity to translate oxidative cues into a program of quiescence and to postpone tumor expansion to more favorable times and safer conditions. From this perspective, it would be interesting to determine whether leukemia stem cells responsible for minimal residual disease (MRD) in hematological malignancies share these properties, i.e., they also behave as “dormant” oxidant-resistant cells.
Another important evidence in support of the underappreciated connection linking metastatic growth to cell antioxidant capacity come from experiments performed by Schafer et al. on the role of GSH in the anchorage-independent survival of breast cancer cells [219]. Loss of contact with basement membrane and ECM inhibits general ectopic growth of normal cells and specifically facilitates cavitation of breast acini that are lined by a single layer of basement membrane-attached cells. Malignant breast cells develop resistance to anoikis and therefore survive and proliferate, filling the center of the acini, a histopathological hallmark of breast cancer. Resistant cells are obviously facilitated when leaving their environment and entering the bloodstream to form distant metastases.
One first unexpected finding by these authors was that cell detachment from basement membrane impairs glucose uptake, and that protection of detached cells by Erb-B2 and its downstream effectors PI3K/Akt is accompanied by, and dependent on, increased absorption and metabolism of glucose. Even more surprisingly, glucose appears to be necessary, rather than to directly generate ATP, for maintaining, via the pentose phosphate shunt, the formation of NADPH and the NADPH-dependent regeneration of GSH. Accordingly, antioxidant rescue cells as efficiently as glucose, without affecting glucose uptake. In fact, attenuation of oxidative stress promotes the mitochondrial oxidation of fatty acids and energy independence of the cell. These findings have important implications in that directly link malignant growth with cell metabolism, providing a further rationale for tumor cell hunger for glucose, and for previous observations on glucose-6 phosphate dehydrogenase, the principle enzyme of pentose phosphate pathway, playing a role in cell transformation [220]. Even more importantly, they further support the heretic view that malignant and metastatic growth must involve cell capacity to cope with oxidative stress, and by extension that antioxidants may under some circumstances help, rather than prevent, cancer metastasis [221, 222].
Redox signals versus redox networks
Although provocative, these findings still need to be reconciled with equally solid lines of evidence that: (1) oncogenes use ROS as downstream signaling intermediates, (2) that metastatic cells, and likely CSCs, are highly motile and therefore require oxidants for cytoskeleton dynamics, (3) that NOX-derived ROS in some cancer models actually inhibit cell death and promote anchorage independence and angiogenesis, and (4) that EMT invasive program is generally linked to both achievement of stem cell-like properties and to Rac1b-mediated ROS generation.
Both cell motility/invasion and stemness offer probably the best examples of how these seemly conflicting views could be integrated (Fig. 6). In fact, while NADPH-oxidase-derived ROS play a pivotal role in cell dynamics, acting on PTPs and also directly on cytoskeleton proteins, requirement for NADPH paradoxically connects these dynamics to the NADPH-dependent regeneration of redox buffers glutathione/glutaredoxin and thioredoxin, that are necessary to reset redox switches and allow signal reversibility. Thus, NOX and glutaredoxin/thioredoxin may locally compete for NADPH, inhibiting each other and leading to local, transient micro-oscillations of the redox potential. Additionally, ROS and glutathione may cooperate for the glutathionylation of critical target proteins such as actin, a reaction that involves glutathione oxidation as an intermediate step [223].

A NADPH-dependent network controls oxidation/reduction of molecular switches involved in cell dynamic. a Oxidant generation (by oxidases, in red) and rescue of oxidized targets (by glutathione reductase/GSH and thioredoxin reductase/thioredoxin, in blue) are tightly coupled by the availability of glucose-derived NADPH. NADPH-dependent oxidases include, among the others, NOX and NOS. b Hypothetical coordination of protrusion/retraction dynamics by a redox mechanism. Membrane protrusion at the leading edge of a migrating cell is promoted by a high Rac/Rho ratio and a prevalence of oxidative events (red), with the opposite occurring at the trailing edge (blue). Note that a similar mechanism can be envisaged for localized, explorative membrane protrusion/retraction events that do not lead to oriented migration. See also Fig. 3
In other words, NOXs, glutathione reductases, glutaredoxins and thioredoxin/thioredoxin reductase create a signaling network that may, for instance, sustain rapid protrusion/retraction cycles of invadopodia during cell “explorative” attempts, while preventing apoptosis by loss of ECM contact [219]. Similarly, the same network may coordinate membrane protrusion at the cell front (Rac-dependent) with contraction at the trailing edge (Rho-dependent) during cell migration, or dictate the switch between mesenchymal (“Rac-driven”) and amoeboid (“Rho-driven”) motility styles (Fig. 6).
An important implication of this view is that ROS-dependent cell motility and elevated intracellular reductive capacity are intimately connected, as it is consistent with recent evidence from studies on cancer stem cell biology (see below).
More in general, the idea that malignancy requires a high cellular “redox activity” or “redox plasticity” as opposite to individually considered pro- or antioxidant capacity deserves attention, as it may help reconcile many conflicting evidence on the role of ROS in invasion and metastasis.
A different view. Metastasis as an antioxidant strategy?
Evidence from molecular, cellular, epidemiological and clinical research indicate that cancer is deeply related to inflammation and tissue repair [9, 224]. This relation is complex and multilayered, but could be provocatively summarized in the concept that cancer arises as in an injured tissue, where injury mainly takes the form of DNA damage, and chaotically tries to “repair it” [225]. Like in an over-healing wound, CSCs are recruited and activated, migrate out of their niches and begin to make new tissue. “Neo-plasia” is indeed “New tissue”.
Accumulating knowledge on stem cell biology also indicates that (epithelial) tissue repair can be brought about by bone marrow-derived mesenchymal cells coming from the bloodstream [226]. Likewise, metastasis occurs when cancer stem cells enter the bloodstream and try to reproduce their tissue of origin elsewhere.
Damage to living tissues is often initiated, and almost invariably propagated, by oxidative stress. Injured tissues experience oxidative stress because of inflammation, hypoxia, or the intermittence of hypoxia and re-oxygenation. Molecular cascades governing cell differentiation and proliferation, stromal reactions, tissue remodeling and angiogenesis are, not surprisingly, regulated by oxidants. As illustrated above, this is true also for oncogenic pathways.
Metastasis revisited 1: HIF triggers an integrated antioxidant program
If cancer arises as an aberrant reparative response to (oxidative) tissue damage, one could legitimately see metastasis as cancer (stem?) cell escape from oxidative stress. Molecular knowledge on the role of HIF in metastasis appears to support this view.
A number of eukaryotic transcription factors, including, among the others, NF-kB, AP-1, FOXO, FoxM1 and NRF-2, participate in cellular redox homeostasis by sensing (directly or indirectly) oxidative stress and activating an antioxidant genetic program. Emerging evidence suggests that HIF should be included in this list.
In a typical homeostatic circuitry, well exemplified by the Keap-1/NRF-2 system, a redox sensor, (Keap-1), distinct from the transcription factor, is in the first place oxidized; this event in turn allows NRF-2 activation and trans-activation of critical genes involved in oxidant detoxification (Fig. 7) [227]. In the HIF system, at least two major sensors, Fe++ of PHDs and cysteine on PTEN, can be identified (see above). Additionally, HIF can be activated directly by S-nitrosylation on Cys-800, and indirectly by oxidant-induced de-sumoylation of p300 [228].

HIF activation as a cancer antioxidant response. We propose that HIF activation in cancer cells is part of a typical antioxidant circuitry. Such a circuitry requires that oxidative stress is detected by a ROS sensor, and translated into an antioxidant gene program by one or more sensor-regulated nuclear factors. This is exemplified by the Keap-1/NRF-2 signaling cassette [75, 76]. In the HIF cascade, iron oxidation (Fe+2 → Fe+3) in PDH (leading to HIF stabilization) and cysteine oxidation in PTEN (leading to increased HIF expression) mediate ROS sensing, whereas inhibition of respiration, generation of NADPH and GSH and cell “escape” (the latter by up-regulation of c-Met and lysyl oxidase) provide the effector arm of the response. Redox regulation of mTOR and direct oxidation of HIF at Cys-800 may also participate in this circuitry [82]. ALDH aldehyde dehydrogenase, GSTs glutathione-S transferases, NQO1 NAD:ubiquinone oxidoreductase 1, LOX lysyl oxidase
On the effector arm of the loop, antioxidant effects of HIF activation can be grouped as follows: (a) resolution of hypoxia, that per se represents a source of oxidative stress; (b) inhibition of mitochondrial respiration and respiration-triggered ROS; (c) activation of the glycolytic gene program, which comprises glucose transporters and glycolytic enzymes including glucose 6-phosphate dehydrogenase, the key enzyme of the pentose phosphate cycle. (d) expression of oxidant scavengers like SOD2 (especially activated by HIF-2) and heme oxygenase-1.Interestingly, mice lacking HIF-2 display increased generation of ROS, defective expression of primary antioxidant enzymes and multiorgan failure alleviated by antioxidant treatment [229].
Antioxidant activity of HIF is at least in part related to utilization and metabolism of glucose that maintains intracellular levels of NADPH and GSH through the pentose phosphate shunt and NADPH-dependent glutathione reductase, respectively. Interestingly, malignant cells are specifically dependent on this antioxidant system for survival upon detachment from the substrate [219].
Metastasis revisited 2: HIF, ROS and the escape from danger
HIF-triggered cell motility program largely exploits ROS as signaling intermediates. Transcriptional up-regulation of c-Met and lysyl oxidase is instrumental to invasiveness and triggers a motile response in part mediated by the generation of oxygen species. In the case of c-Met, ROS are likely produced by DPI-sensitive NOXes, i.e., in an NADPH-dependent fashion. Another NADPH-fuelled source of ROS, MICAL, may also have a role in cell scattering triggered by c-Met. In fact, (a) oxidative activity of MICALs mediates axonal repulsion by semaphorins during axon guidance [108]; (b) semaphorin receptors, plexins, are highly homologous to the extracellular portion of c-Met and cooperate with c-Met for invasive growth of epithelial cells [230]. By extension, MICALs may contribute to redox signaling by c-Met during cell migration/invasion, although experimental demonstration of this possibility is still missing. Additionally, c-Met can also be trans-activated by ROS in a ligand-independent fashion [231]. Thus, ROS trigger the expression of lysyl oxidase and c-Met through HIF, and also contribute to the pro-invasive activity of these molecules.
It should not appear too stretched to consider motility/scattering as an additional HIF-dependent strategy whereby malignant cells, simply by moving away, avoid oxidative stress at the primary tumor site. Since ROS increase in hypoxic regions, this same circuitry may as well help cells in oxygen seeking. This response is perfectly integrated with the metabolic profile imposed by HIF (the Warburg’s effect), whereby cells avidly uptake glucose, and reducing equivalents accumulate in the cytosol due to mitochondrial blockade, fuelling NAD(P)H dependent oxidases. In parallel, NADPH reduces glutathione and thioredoxin thus maintaining redox buffers. The resulting phenotype of a cell with active motility and elevated reductive potential is, not surprisingly, what has recently emerged as the picture of a cancer stem cells [6, 210].
The real contribution of redox circuitries to CSC biology is again far to be elucidated. Weinberg’s group reported that breast cancer cells undergoing EMT acquire CSC traits [6]. These data, recently confirmed in prostate carcinoma cells [232], suggest that CSCs are a subpopulation endowed with particular type of plasticity in motility, due to activation of the EMT transcriptional program. Stemness of a tumor population is an instrument to survive in aggressive and strongly oxidative environment due to growth of primitive cancer. In this milieu, quiescent CSCs, upon delivery of motogen stimuli, can regenerate the original cancer in a different site. Therefore, the relationship between cancer stemness and EMT is a first level of pro-survival response, namely a real escape from the primitive tumor site, thereby leading to disseminate motile cancer cells. Although EMT has been correlated with enhanced ROS production, and the quiescent CSC phenotype has been associated with low ROS level, it is likely that environmental increased levels of ROS may “alert” stem cells to enter the cell cycle again. It is therefore likely that tumor hypoxia or stromal cell contact, both reported to affect the milieu in a redox-dependent manner, activate the motogen escaping program of CSC/metastatic cells, via redox-sensitive components of the motility apparatus. In this view CSCs, namely the seed for a new secondary tumor, exploit EMT-related ROS to migrate out of their primary niches attempting to reproduce elsewhere what is perceived as a “damaged” cancer tissue.
2 Conclusion
Knowledge on the molecular mechanism and the biological contexts whereby oxidants (oxygen and nitrogen species) operate as signaling molecules has been expanding at an impressive rate over the last few years [52]. “Redox signaling” has rapidly turned into a major area of “mainstream” cellular and molecular biology, with huge implications for human diseases. Yet, why cells use to regulate basic functions like proliferation/death, motility, and energy metabolism such dangerous compounds as nitrogen and oxygen species, is still far from clear, and it is reasonable to think that safer messenger molecules and molecular devices could serve the same scope.
One attractive possibility is that redox signaling has evolved for the main purpose of allowing cells to sense and avoiding oxidative stress and damage (Fig. 8). At a single cell level, this idea is consistent with the observation that oxygen and nitrogen species play a central role in cell dynamic and in the triggering of antioxidant responses, two different and complementary strategies to reduce exposure to harmful oxidants. Yet cells die if stress is too massive, or if glucose in not available to maintain redox buffers. In a multicellular organism, redox signals gain two additional important functions related to tissue repair: regulation of cell proliferation and angiogenesis. This may as well guarantee replacement of cells killed either directly by environmental oxidants or indirectly by ROS and RNS generated during the inflammatory response to infection or trauma. In multicellular organisms, redox signaling cascades have evolved into a higher level of complexity, being incorporated in the complex network of cell–cell communications systems made of endless ligands and their cognate receptors. Nevertheless, these cascades have maintained their capacity to sense “ambient” oxidative signals, and to engage with them in intricate feed-back and feed-forward circuitries. Accordingly, it is very frequent to observe that these cascades respond to exogenous oxidants almost as powerfully as to the cognate ligand; additionally, often oxidants operate at different levels of the same cascade (e.g., C-Src and H-ras activate NOXes and are in turn redox-regulated), and critical redox-sensitive regulators, like PTPs, respond to multiple parallel pathways, setting the condition for collateral signaling. Finally, oxidants like NO and hydrogen peroxide can act trans-cellularly, thus coordinating complex processes that involve multiple cellular component of a tissue [233, 234].

Antioxidant function of redox signaling and metastasis. Unfavorable environmental conditions that favor metastasis (inflammation, hypoxia, nutrient starvation, etc.) are associated with heavy oxidative stress. We speculate that redox cascades that lead to glycolytic switch, cell proliferation and motility/invasion and are activated in metastasis have originally evolved for the purpose of rescuing normal cells from oxidative damage and allowing tissue repair (left side of the figure). Redox switches may have subsequently been incorporated into ligand (growth factors, ECM, semaphorins)-dependent cascaded that use NOXes, and other oxidases, as components of “normal” cell signaling (right side). Oncogene-driven carcinogenesis hijacks these cascades, leading eventually to metastasis as the malignant counterpart of the original antioxidant response (right side). Metastasis may therefore be part of an integrated strategy whereby malignant cells escape oxidative damage at the primary tumor site
Tumor microenvironment is harshly pro-oxidant: hypoxia and inflammation flood a primary tumor site with ROS and RNS; nutrient shortage depletes glutathione, damaged mitochondria produce oxygen radicals. Cells undertaking the metastatic task can be seen, with many respects, as unicellular organisms escaping from this terrifying scenario. They accomplish this task by activating both the antioxidant program and the motile program, through redox-sensitive transcription factors like HIF, Snail, and NF-kB, and by exploiting the fact that motility and migration are facilitated by ROS. Importantly, extracellular oxidants in turn elicit the generation of endogenous ROS, through the many positive feed-forward loops built in ROS-dependent cascades (Fig. 5).
Escape strategies from unfavorable environments have been extensively studied in bacteria. E. coli and S. typhimurium move away from lack of oxygen and nutrients by activating their flagellar motor through a redox mechanism; Aer, the redox sensor family member that mediates this response is a FAD-binding protein, that undergoes conformational changes based on the oxidation state of its flavin cofactor; in the simplest two state model, FAD is fully reduced under hypoxia and transmits the C-terminal domain of Aer a conformational change that changes the direction of flagellar rotation and triggers a repellent (tumbling) response [235]. Resemblance of this system with cytoskeleton control by NOXes and even more with cell guidance by MICAL, a mammalian FAD-binding proteins mediating repulsive signals, is striking (Fig. 9). Further research in this direction is warranted.

Analogy between aerotaxis in bacteria and redox-regulated motility in eukaryotic (malignant) cells. In bacteria, redox changes triggered by oxygen or nutrient deprivation and excess oxidants are translated into a “tumbling” behavior by the interaction between the Aer sensor, a FAD-binding protein, and the flagellar motor [235]. In eukaryotic cells, MICALs and NOXes may serve a similar function in coupling intracellular oxidations/reductions with migratory behaviors. In eukaryotes a further layer of complexity is added by the fact that FAD-dependent sensors in turn generate ROS, which may regulate the phosphorylation step impinging on the motor structure (cytoskeleton in this case). This attractive model still needs experimental verification
In conclusion, while it is unlikely that bacteria teach us how to fight or prevent metastasis, maybe they can give a hint of why metastasis occurs. No doubts exist that the most unfavorable life in primary tumor is for cancer cells, the higher the risk of metastatic spread [236]. Used to see metastatic cells as fierce invaders, we may have to confront the picture of few cells threatened by oxidant radicals that desperately try to move away. Time will tell whether this paradigm can help our fight against malignancy (Table 5).
References
Steeg, P. S. (2006). Tumor metastasis: mechanistic insights and clinical challenges. Nature Medicine, 12, 895–904.
Nguyen, D. X., & Massague, J. (2007). Genetic determinants of cancer metastasis. Nature Reviews Genetics, 8, 341–352.
Chiang, A. C., & Massague, J. (2008). Molecular basis of metastasis. New England Journal of Medicine, 359, 2814–2823.
Fidler, I. J. (2003). The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature Reviews Cancer, 3, 453–458.
Klein, C. A. (2009). Parallel progression of primary tumours and metastases. Nature Reviews Cancer, 9, 302–312.
Mani, S. A., Guo, W., Liao, M. J., Eaton, E. N., Ayyanan, A., Zhou, A. Y., et al. (2008). The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell, 133, 704–715.
Barnhart, B. C., & Simon, M. C. (2007). Metastasis and stem cell pathways. Cancer and Metastasis Reviews, 26, 261–271.
Bertout, J. A., Patel, S. A., & Simon, M. C. (2008). The impact of O2 availability on human cancer. Nature Reviews Cancer, 8, 967–975.
Mantovani, A. (2009). Cancer: inflaming metastasis. Nature, 457, 36–37.
Comoglio, P. M., & Trusolino, L. (2002). Invasive growth: from development to metastasis. Journal of Clinical Investigation, 109, 857–862.
Kalluri, R., & Weinberg, R. A. (2009). The basics of epithelial–mesenchymal transition. Journal of Clinical Investigation, 119, 1420–1428.
Etienne-Manneville, S., & Hall, A. (2002). Rho GTPases in cell biology. Nature, 420, 629–635.
Hall, A. (2005). Rho GTPases and the control of cell behaviour. Biochemical Society Transactions, 33, 891–895.
Brakebusch, C., Bouvard, D., Stanchi, F., Sakai, T., & Fassler, R. (2002). Integrins in invasive growth. Journal of Clinical Investigation, 109, 999–1006.
Mitra, S. K., & Schlaepfer, D. D. (2006). Integrin-regulated FAK-Src signaling in normal and cancer cells. Current Opinion in Cell Biology, 18, 516–523.
Raftopoulou, M., Etienne-Manneville, S., Self, A., Nicholls, S., & Hall, A. (2004). Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science, 303, 1179–1181.
Yu, D. H., Qu, C. K., Henegariu, O., Lu, X., & Feng, G. S. (1998). Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. Journal of Biological Chemistry, 273, 21125–21131.
Friedl, P. (2004). Prespecification and plasticity: shifting mechanisms of cell migration. Current Opinion in Cell Biology, 16, 14–23.
Sanz-Moreno, V., Gadea, G., Ahn, J., Paterson, H., Marra, P., Pinner, S., et al. (2008). Rac activation and inactivation control plasticity of tumor cell movement. Cell, 135, 510–523.
Habets, G. G., Scholtes, E. H., Zuydgeest, D., van der Kammen, R. A., Stam, J. C., Berns, A., et al. (1994). Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell, 77, 537–549.
Clark, E. A., Golub, T. R., Lander, E. S., & Hynes, R. O. (2000). Genomic analysis of metastasis reveals an essential role for RhoC. Nature, 406, 532–535.
Murakami, M., Meneses, P. I., Knight, J. S., Lan, K., Kaul, R., Verma, S. C., et al. (2008). Nm23-H1 modulates the activity of the guanine exchange factor Dbl-1. International Journal of Cancer, 123, 500–510.
Birchmeier, C., Birchmeier, W., Gherardi, E., & Vande Woude, G. F. (2003). Met, metastasis, motility and more. Nature Reviews Molecular Cell Biology, 4, 915–925.
Douma, S., Van, L. T., Zevenhoven, J., Meuwissen, R., Van, G. E., & Peeper, D. S. (2004). Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature, 430, 1034–1039.
Geiger, T. R., & Peeper, D. S. (2009). Metastasis mechanisms. Biochimica et Biophysica Acta, 1796, 293–308.
Wei, L., Yang, Y., Zhang, X., & Yu, Q. (2004). Altered regulation of Src upon cell detachment protects human lung adenocarcinoma cells from anoikis. Oncogene, 23, 9052–9061.
Aguirre-Ghiso, J. A. (2007). Models, mechanisms and clinical evidence for cancer dormancy. Nature Reviews Cancer, 7, 834–846.
Kim, S., Takahashi, H., Lin, W. W., Descargues, P., Grivennikov, S., Kim, Y., et al. (2009). Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature, 457, 102–106.
Kaplan, R. N., Riba, R. D., Zacharoulis, S., Bramley, A. H., Vincent, L., Costa, C., et al. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature, 438, 820–827.
Bates, R. C., & Mercurio, A. M. (2003). Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Molecular Biology of the Cell, 14, 1790–1800.
Luo, J. L., Tan, W., Ricono, J. M., Korchynskyi, O., Zhang, M., Gonias, S. L., et al. (2007). Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature, 446, 690–694.
Kaelin, W. G., Jr., & Ratcliffe, P. J. (2008). Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molecular Cell, 30, 393–402.
Pouyssegur, J., Dayan, F., & Mazure, N. M. (2006). Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature, 441, 437–443.
Kim, J. W., Tchernyshyov, I., Semenza, G. L., & Dang, C. V. (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab, 3, 177–185.
Fukuda, R., Zhang, H., Kim, J. W., Shimoda, L., Dang, C. V., & Semenza, G. L. (2007). HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell, 129, 111–122.
Dewhirst, M. W., Cao, Y., & Moeller, B. (2008). Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nature Reviews Cancer, 8, 425–437.
Pennacchietti, S., Michieli, P., Galluzzo, M., Mazzone, M., Giordano, S., & Comoglio, P. M. (2003). Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell, 3, 347–361.
Erler, J. T., Bennewith, K. L., Nicolau, M., Dornhofer, N., Kong, C., Le, Q. T., et al. (2006). Lysyl oxidase is essential for hypoxia-induced metastasis. Nature, 440, 1222–1226.
Denko, N. C. (2008). Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nature Reviews Cancer., 8, 705–13.
Vander Heiden, M. G., Cantley, L. C., & Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science, 324, 1029–1033.
Hsu, P. P., & Sabatini, D. M. (2008). Cancer cell metabolism: Warburg and beyond. Cell, 134, 703–707.
Gatenby, R. A., & Gawlinski, E. T. (2001). Mathematical models of tumour invasion mediated by transformation-induced alteration of microenvironmental pH. Novartis Foundation Symposium, 240, 85–96.
Rofstad, E. K., Mathiesen, B., Kindem, K., & Galappathi, K. (2006). Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Research, 66, 6699–6707.
Clarke, M. F., Dick, J. E., Dirks, P. B., Eaves, C. J., Jamieson, C. H., Jones, D. L., et al. (2006). Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Research, 66, 9339–9344.
Cipolleschi, M. G., Rovida, E., Ivanovic, Z., Praloran, V., Olivotto, M., & Dello, S. P. (2000). The expansion of murine bone marrow cells preincubated in hypoxia as an in vitro indicator of their marrow-repopulating ability. Leukemia, 14, 735–739.
Simon, M. C., & Keith, B. (2008). The role of oxygen availability in embryonic development and stem cell function. Nature Reviews Molecular Cell Biology, 9, 285–296.
Ezashi, T., Das, P., & Roberts, R. M. (2005). Low O2 tensions and the prevention of differentiation of hES cells. Proceedings of the National Academy of Sciences of the United States of America, 102, 4783–4788.
Kondoh, H., Lleonart, M. E., Gil, J., Wang, J., Degan, P., Peters, G., et al. (2005). Glycolytic enzymes can modulate cellular life span. Cancer Research, 65, 177–185.
Giuntoli, S., Rovida, E., Gozzini, A., Barbetti, V., Cipolleschi, M. G., Olivotto, M., et al. (2007). Severe hypoxia defines heterogeneity and selects highly immature progenitors within clonal erythroleukemia cells. Stem Cells, 25, 1119–1125.
Das, B., Tsuchida, R., Malkin, D., Koren, G., Baruchel, S., & Yeger, H. (2008). Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells, 26, 1818–1830.
Bjerkvig, R., Johansson, M., Miletic, H., & Niclou, S. P. (2009). Cancer stem cells and angiogenesis. Seminars in Cancer Biology, 19, 279–284.
Janssen-Heininger, Y. M., Mossman, B. T., Heintz, N. H., Forman, H. J., Kalyanaraman, B., Finkel, T., et al. (2008). Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radical Biology and Medicine, 45, 1–17.
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochemical Journal, 417, 1–13.
Paget, M. S., & Buttner, M. J. (2003). Thiol-based regulatory switches. Annual Review of Genetics, 37, 91–121.
Lambeth, J. D. (2004). NOX enzymes and the biology of reactive oxygen. Nature Reviews Cancer, 4, 181–189.
Sundaresan, M., Yu, Z. X., Ferrans, V. J., Irani, K., & Finkel, T. (1995). Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science, 270, 296–299.
Baumer, A. T., Ten, F. H., Sauer, H., Wartenberg, M., Kappert, K., Schnabel, P., et al. (2008). Phosphatidylinositol 3-kinase-dependent membrane recruitment of Rac-1 and p47phox is critical for alpha-platelet-derived growth factor receptor-induced production of reactive oxygen species. Journal of Biological Chemistry, 283, 7864–7876.
Lo, Y. Y., & Cruz, T. F. (1995). Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. Journal of Biological Chemistry, 270, 11727–11730.
Ushio-Fukai, M., Zafari, A. M., Fukui, T., Ishizaka, N., & Griendling, K. K. (1996). p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. Journal of Biological Chemistry, 271, 23317–23321.
Chiarugi, P., Pani, G., Giannoni, E., Taddei, L., Colavitti, R., Raugei, G., et al. (2003). Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. Journal of Biological Chemistry, 161, 933–944.
Inoue, M., Sato, E. F., Nishikawa, M., Park, A. M., Kira, Y., Imada, I., et al. (2003). Mitochondrial generation of reactive oxygen species and its role in aerobic life. Current Medicinal Chemistry, 10, 2495–2505.
Nemoto, S., Takeda, K., Yu, Z. X., Ferrans, V. J., & Finkel, T. (2000). Role for mitochondrial oxidants as regulators of cellular metabolism. Molecular and Cellular Biology, 20, 7311–7318.
Guzy, R. D., Hoyos, B., Robin, E., Chen, H., Liu, L., Mansfield, K. D., et al. (2005). Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab, 1, 401–408.
Cai, J., & Jones, D. P. (1998). Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. Journal of Biological Chemistry, 273, 11401–11404.
Giorgio, M., Migliaccio, E., Orsini, F., Paolucci, D., Moroni, M., Contursi, C., et al. (2005). Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell, 122, 221–233.
Pinton, P., Rimessi, A., Marchi, S., Orsini, F., Migliaccio, E., Giorgio, M., et al. (2007). Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science, 315, 659–663.
Pani, G., Koch, O. R., & Galeotti, T. (2009). The p53-p66shc-Manganese Superoxide Dismutase (MnSOD) network: a mitochondrial intrigue to generate reactive oxygen species. International Journal of Biochemistry and Cell Biology, 41, 1002–1005.
Rhee, S. G., Yang, K. S., Kang, S. W., Woo, H. A., & Chang, T. S. (2005). Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxidants Redox Signaling, 7, 619–626.
Veal, E. A., Day, A. M., & Morgan, B. A. (2007). Hydrogen peroxide sensing and signaling. Molecular Cell, 26, 1–14.
Chiarugi, P., & Cirri, P. (2003). Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends in Biochemical Sciences, 28, 509–514.
Pani, G., Colavitti, R., Bedogni, B., Anzevino, R., Borrello, S., & Galeotti, T. (2000). A redox signaling mechanism for density-dependent inhibition of cell growth. Journal of Biological Chemistry, 275, 38891–38899.
Mannick, J. B., Hausladen, A., Liu, L., Hess, D. T., Zeng, M., Miao, Q. X., et al. (1999). Fas-induced caspase denitrosylation. Science, 284, 651–654.
Gu, Z., Kaul, M., Yan, B., Kridel, S. J., Cui, J., Strongin, A., et al. (2002). S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science, 297, 1186–1190.
Saitoh, M., Nishitoh, H., Fujii, M., Takeda, K., Tobiume, K., Sawada, Y., et al. (1998). Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO Journal, 17, 2596–2606.
Dinkova-Kostova, A. T., Holtzclaw, W. D., Cole, R. N., Itoh, K., Wakabayashi, N., Katoh, Y., et al. (2002). Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proceedings of the National Academy of Sciences of the United States of America, 99, 11908–11913.
Liu, H., Colavitti, R., Rovira, I. I., & Finkel, T. (2005). Redox-dependent transcriptional regulation. Circulation Research, 97, 967–974.
Xanthoudakis, S., & Curran, T. (1992). Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO Journal, 11, 653–665.
Dansen, T. B., Smits, L. M., van Triest, M. H., de Keizer, P. L., van Leenen, D., Koerkamp, M. G., et al. (2009). Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat Chem Biol, 5, 664–672.
van der Horst, A., Tertoolen, L. G., de Vries-Smits, L. M., Frye, R. A., Medema, R. H., & Burgering, B. M. (2004). FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). Journal of Biological Chemistry, 279, 28873–28879.
Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., et al. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell, 96, 857–868.
Nemoto, S., & Finkel, T. (2002). Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science, 295, 2450–2452.
Yasinska, I. M., & Sumbayev, V. V. (2003). S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Letters, 549, 105–109.
Gu, J., Milligan, J., & Huang, L. E. (2001). Molecular mechanism of hypoxia-inducible factor 1alpha -p300 interaction. A leucine-rich interface regulated by a single cysteine. Journal of Biological Chemistry, 276, 3550–3554.
Gerald, D., Berra, E., Frapart, Y. M., Chan, D. A., Giaccia, A. J., Mansuy, D., et al. (2004). JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell, 118, 781–794.
Schroedl, C., McClintock, D. S., Budinger, G. R., & Chandel, N. S. (2002). Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. American Journal of Physiology Lung Cellular and Molecular Physiology, 283, L922–L931.
Lu, H., Dalgard, C. L., Mohyeldin, A., Mcfate, T., Tait, A. S., & Verma, A. (2005). Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. Journal of Biological Chemistry, 280, 41928–41939.
Arbiser, J. L., Petros, J., Klafter, R., Govindajaran, B., McLaughlin, E. R., Brown, L. F., et al. (2002). Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proceedings of the National Academy of Sciences of the United States of America, 99, 715–720.
Wang, F. S., Wang, C. J., Chen, Y. J., Chang, P. R., Huang, Y. T., Sun, Y. C., et al. (2004). Ras induction of superoxide activates ERK-dependent angiogenic transcription factor HIF-1alpha and VEGF-A expression in shock wave-stimulated osteoblasts. Journal of Biological Chemistry, 279, 10331–10337.
Gao, P., Zhang, H., Dinavahi, R., Li, F., Xiang, Y., Raman, V., et al. (2007). HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell, 12, 230–238.
Nott, A., Watson, P. M., Robinson, J. D., Crepaldi, L., & Riccio, A. (2008). S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature, 455, 411–415.
Bedogni, B., Pani, G., Colavitti, R., Riccio, A., Borrello, S., Murphy, M., et al. (2003). Redox regulation of cAMP-responsive element-binding protein and induction of manganous superoxide dismutase in nerve growth factor-dependent cell survival. Journal of Biological Chemistry, 278, 16510–16519.
Riccio, A., Alvania, R. S., Lonze, B. E., Ramanan, N., Kim, T., Huang, Y., et al. (2006). A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Molecular Cell, 21, 283–294.
Ito, K., Hanazawa, T., Tomita, K., Barnes, P. J., & Adcock, I. M. (2004). Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochemical and Biophysical Research Communications, 315, 240–245.
Moldovan, L., Irani, K., Moldovan, N. I., Finkel, T., & Goldschmidt-Clermont, P. J. (1999). The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antioxidants Redox Signaling, 1, 29–43.
Joneson, T., & Bar-Sagi, D. (1998). A Rac1 effector site controlling mitogenesis through superoxide production. Journal of Biological Chemistry, 273, 17991–17994.
Kheradmand, F., Werner, E., Tremble, P., Symons, M., & Werb, Z. (1998). Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science, 280, 898–902.
Werner, E., & Werb, Z. (2002). Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. Journal of Cell Biology, 158, 357–368.
Zhou, C., Ziegler, C., Birder, L. A., Stewart, A. F., & Levitan, E. S. (2006). Angiotensin II and stretch activate NADPH oxidase to destabilize cardiac Kv4.3 channel mRNA. Circulation Research, 98, 1040–1047.
Taddei, M. L., Parri, M., Mello, T., Catalano, A., Levine, A. D., Raugei, G., et al. (2007). Integrin-mediated cell adhesion and spreading engage different sources of reactive oxygen species. Antioxidants Redox Signaling, 9, 469–481.
Nimnual, A. S., Taylor, L. J., & Bar-Sagi, D. (2003). Redox-dependent downregulation of Rho by Rac. Nature Cell Biology, 5, 236–241.
Ng, J., & Luo, L. (2004). Rho GTPases regulate axon growth through convergent and divergent signaling pathways. Neuron, 44, 779–793.
Zondag, G. C., Evers, E. E., ten Klooster, J. P., Janssen, L., van der Kammen, R. A., & Collard, J. G. (2000). Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial–mesenchymal transition. Journal of Cell Biology, 149, 775–782.
Buricchi, F., Giannoni, E., Grimaldi, G., Parri, M., Raugei, G., Ramponi, G., et al. (2007). Redox regulation of ephrin/integrin cross-talk. Cell Adh Migr, 1, 33–42.
Taddei, M. L., Parri, M., Angelucci, A., Onnis, B., Bianchini, F., Giannoni, E., et al. (2009). Kinase-dependent and -independent roles of EphA2 in the regulation of prostate cancer invasion and metastasis. American Journal of Pathology, 174, 1492–1503.
Giannoni, E., Buricchi, F., Raugei, G., Ramponi, G., & Chiarugi, P. (2005). Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Molecular and Cellular Biology, 25, 6391–6403.
Fratelli, M., Demol, H., Puype, M., Casagrande, S., Eberini, I., Salmona, M., et al. (2002). Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proceedings of the National Academy of Sciences of the United States of America, 99, 3505–3510.
Fiaschi, T., Cozzi, G., Raugei, G., Formigli, L., Ramponi, G., & Chiarugi, P. (2006). Redox regulation of beta-actin during integrin-mediated cell adhesion. Journal of Biological Chemistry, 281, 22983–22991.
Terman, J. R., Mao, T., Pasterkamp, R. J., Yu, H. H., & Kolodkin, A. L. (2002). MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell, 109, 887–900.
Ventura, A., & Pelicci, P. G. (2002). Semaphorins: green light for redox signaling? Sci STKE, 2002, e44.
Suzuki, T., Nakamoto, T., Ogawa, S., Seo, S., Matsumura, T., Tachibana, K., et al. (2002). MICAL, a novel CasL interacting molecule, associates with vimentin. Journal of Biological Chemistry, 277, 14933–14941.
Nadella, M., Bianchet, M. A., Gabelli, S. B., Barrila, J., & Amzel, L. M. (2005). Structure and activity of the axon guidance protein MICAL. Proceedings of the National Academy of Sciences of the United States of America, 102, 16830–16835.
Fischer, J., Weide, T., & Barnekow, A. (2005). The MICAL proteins and rab1: a possible link to the cytoskeleton? Biochemical and Biophysical Research Communications, 328, 415–423.
Kanda, I., Nishimura, N., Nakatsuji, H., Yamamura, R., Nakanishi, H., & Sasaki, T. (2008). Involvement of Rab13 and JRAB/MICAL-L2 in epithelial cell scattering. Oncogene, 27, 1687–1695.
Ferraro, D., Corso, S., Fasano, E., Panieri, E., Santangelo, R., Borrello, S., et al. (2006). Pro-metastatic signaling by c-Met through RAC-1 and reactive oxygen species (ROS). Oncogene, 25, 3689–3698.
Jagadeeswaran, R., Jagadeeswaran, S., Bindokas, V. P., & Salgia, R. (2007). Activation of HGF/c-Met pathway contributes to the reactive oxygen species generation and motility of small cell lung cancer cells. American Journal of Physiology Lung Cellular and Molecular Physiology, 292, L1488–L1494.
Li, W., Liu, G., Chou, I. N., & Kagan, H. M. (2000). Hydrogen peroxide-mediated, lysyl oxidase-dependent chemotaxis of vascular smooth muscle cells. Journal of Cellular Biochemistry, 78, 550–557.
Ushio-Fukai, M. (2009). Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxidants Redox Signaling, 11, 1289–1299.
Wu, R. F., Xu, Y. C., Ma, Z., Nwariaku, F. E., Sarosi, G. A., Jr., & Terada, L. S. (2005). Subcellular targeting of oxidants during endothelial cell migration. Journal of Cell Biology, 171, 893–904.
Diaz, B., Shani, G., Pass, I., Anderson, D., Quintavalle, M., & Courtneidge, S. A. (2009). Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci Signal, 2, ra53.
Gianni, D., Diaz, B., Taulet, N., Fowler, B., Courtneidge, S. A., & Bokoch, G. M. (2009). Novel p47(phox)-related organizers regulate localized NADPH oxidase 1 (Nox1) activity. Science Signal, 2, ra54.
Patel, H. H., & Insel, P. A. (2009). Lipid rafts and caveolae and their role in compartmentation of redox signaling. Antioxidants Redox Signaling, 11, 1357–1372.
Gaus, K., Le, L. S., Balasubramanian, N., & Schwartz, M. A. (2006). Integrin-mediated adhesion regulates membrane order. Journal of Cell Biology, 174, 725–734.
Meng, T. C., Fukada, T., & Tonks, N. K. (2002). Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Molecular Cell, 9, 387–399.
Campbell, M., Anderson, P., & Trimble, E. R. (2008). Glucose lowers the threshold for human aortic vascular smooth muscle cell migration: inhibition by protein phosphatase-2A. Diabetologia, 51, 1068–1080.
Leopold, J. A., Walker, J., Scribner, A. W., Voetsch, B., Zhang, Y. Y., Loscalzo, A. J., et al. (2003). Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. Journal of Biological Chemistry, 278, 32100–32106.
Ames, B. N., Gold, L. S., & Willett, W. C. (1995). The causes and prevention of cancer. Proceedings of the National Academy of Sciences of the United States of America, 92, 5258–5265.
Ambrosone, C. B., Freudenheim, J. L., Thompson, P. A., Bowman, E., Vena, J. E., Marshall, J. R., et al. (1999). Manganese superoxide dismutase (MnSOD) genetic polymorphisms, dietary antioxidants, and risk of breast cancer. Cancer Research, 59, 602–606.
Ratnasinghe, D., Tangrea, J. A., Andersen, M. R., Barrett, M. J., Virtamo, J., Taylor, P. R., et al. (2000). Glutathione peroxidase codon 198 polymorphism variant increases lung cancer risk. Cancer Research, 60, 6381–6383.
Zhao, Y., Oberley, T. D., Chaiswing, L., Lin, S. M., Epstein, C. J., Huang, T. T., et al. (2002). Manganese superoxide dismutase deficiency enhances cell turnover via tumor promoter-induced alterations in AP-1 and p53-mediated pathways in a skin cancer model. Oncogene, 21, 3836–3846.
Marnett, L. J. (2000). Oxyradicals and DNA damage. Carcinogenesis, 21, 361–370.
Hussain, S. P., Hofseth, L. J., & Harris, C. C. (2003). Radical causes of cancer. Nature Reviews Cancer, 3, 276–285.
Cerutti, P. A. (1985). Prooxidant states and tumor promotion. Science, 227, 375–381.
Irani, K., Xia, Y., Zweier, J. L., Sollott, S. J., Der, C. J., Fearon, E. R., et al. (1997). Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science, 275, 1649–1652.
Hanahan, D., & Weinberg, R. A. (2000). The hallmarks of cancer. Cell, 100, 57–70.
Szatrowski, T. P., & Nathan, C. F. (1991). Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Research, 51, 794–798.
Lim, S. D., Sun, C., Lambeth, J. D., Marshall, F., Amin, M., Chung, L., et al. (2005). Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate, 62, 200–207.
Mitsushita, J., Lambeth, J. D., & Kamata, T. (2004). The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Research, 64, 3580–3585.
Shinohara, M., Shang, W. H., Kubodera, M., Harada, S., Mitsushita, J., Kato, M., et al. (2007). Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. Journal of Biological Chemistry, 282, 17640–17648.
Lander, H. M., Milbank, A. J., Tauras, J. M., Hajjar, D. P., Hempstead, B. L., Schwartz, G. D., et al. (1996). Redox regulation of cell signalling. Nature, 381, 380–381.
Benhar, M., Dalyot, I., Engelberg, D., & Levitzki, A. (2001). Enhanced ROS production in oncogenically transformed cells potentiates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase activation and sensitization to genotoxic stress. Molecular and Cellular Biology, 21, 6913–6926.
Rhee, S. G., Bae, Y. S., Lee, S. R., & Kwon, J. (2000). Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE, 2000, e1.
Konishi, H., Tanaka, M., Takemura, Y., Matsuzaki, H., Ono, Y., Kikkawa, U., et al. (1997). Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proceedings of the National Academy of Sciences of the United States of America, 94, 11233–11237.
Pani, G., Bedogni, B., Colavitti, R., Anzevino, R., Borrello, S., & Galeotti, T. (2001). Cell compartmentalization in redox signaling. IUBMB Life, 52, 7–16.
Coats, S., Flanagan, W. M., Nourse, J., & Roberts, J. M. (1996). Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science, 272, 877–880.
Medema, R. H., Kops, G. J., Bos, J. L., & Burgering, B. M. (2000). AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature, 404, 782–787.
Nogueira, V., Park, Y., Chen, C. C., Xu, P. Z., Chen, M. L., Tonic, I., et al. (2008). Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell, 14, 458–470.
Itahana, K., Campisi, J., & Dimri, G. P. (2004). Mechanisms of cellular senescence in human and mouse cells. Biogerontology, 5, 1–10.
Parrinello, S., Samper, E., Krtolica, A., Goldstein, J., Melov, S., & Campisi, J. (2003). Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nature Cell Biology, 5, 741–747.
Packer, L., & Fuehr, K. (1977). Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature, 267, 423–425.
Bell, E. L., Klimova, T. A., Eisenbart, J., Schumacker, P. T., & Chandel, N. S. (2007). Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Molecular and Cellular Biology, 27, 5737–5745.
Bermudez, Y., Ahmadi, S., Lowell, N. E., & Kruk, P. A. (2007). Vitamin E suppresses telomerase activity in ovarian cancer cells. Cancer Detection and Prevention, 31, 119–128.
Kwon, J., Lee, S. R., Yang, K. S., Ahn, Y., Kim, Y. J., Stadtman, E. R., et al. (2004). Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proceedings of the National Academy of Sciences of the United States of America, 101, 16419–16424.
Romashkova, J. A., & Makarov, S. S. (1999). NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature, 401, 86–90.
Ozes, O. N., Mayo, L. D., Gustin, J. A., Pfeffer, S. R., Pfeffer, L. M., & Donner, D. B. (1999). NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature, 401, 82–85.
Anderson, M. T., Staal, F. J., Gitler, C., Herzenberg, L. A., & Herzenberg, L. A. (1994). Separation of oxidant-initiated and redox-regulated steps in the NF-kappa B signal transduction pathway. Proceedings of the National Academy of Sciences of the United States of America, 91, 11527–11531.
Sulciner, D. J., Irani, K., Yu, Z. X., Ferrans, V. J., Goldschmidt-Clermont, P., & Finkel, T. (1996). rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Molecular and Cellular Biology, 16, 7115–7121.
Yang, J. Q., Zhao, W., Duan, H., Robbins, M. E., Buettner, G. R., Oberley, L. W., et al. (2001). v-Ha-RaS oncogene upregulates the 92-kDa type IV collagenase (MMP-9) gene by increasing cellular superoxide production and activating NF-kappaB. Free Radical Biology and Medicine, 31, 520–529.
Joneson, T., & Bar-Sagi, D. (1999). Suppression of Ras-induced apoptosis by the Rac GTPase. Molecular and Cellular Biology, 19, 5892–5901.
Lluis, J. M., Buricchi, F., Chiarugi, P., Morales, A., & Fernandez-Checa, J. C. (2007). Dual role of mitochondrial reactive oxygen species in hypoxia signaling: activation of nuclear factor-{kappa}B via c-SRC and oxidant-dependent cell death. Cancer Research, 67, 7368–7377.
Barbie, D. A., Tamayo, P., Boehm, J. S., Kim, S. Y., Moody, S. E., Dunn, I. F., et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature, 462, 108–112.
Mayo, M. W., Wang, C. Y., Cogswell, P. C., Rogers-Graham, K. S., Lowe, S. W., Der, C. J., et al. (1997). Requirement of NF-kappaB activation to suppress p53-independent apoptosis induced by oncogenic Ras. Science, 278, 1812–1815.
Ciani, E., Guidi, S., Della, V. G., Perini, G., Bartesaghi, R., & Contestabile, A. (2002). Nitric oxide protects neuroblastoma cells from apoptosis induced by serum deprivation through cAMP-response element-binding protein (CREB) activation. Journal of Biological Chemistry, 277, 49896–49902.
Hess, D. T., Matsumoto, A., Kim, S. O., Marshall, H. E., & Stamler, J. S. (2005). Protein S-nitrosylation: purview and parameters. Nature Reviews Molecular Cell Biology, 6, 150–166.
Pan, S., & Berk, B. C. (2007). Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circulation Research, 100, 213–219.
Giannoni, E., Fiaschi, T., Ramponi, G., & Chiarugi, P. (2009). Redox regulation of anoikis resistance of metastatic prostate cancer cells: key role for Src and EGFR-mediated pro-survival signals. Oncogene, 28, 2074–2086.
Pani, G., Giannoni, E., Galeotti, T., & Chiarugi, P. (2009). Redox-based escape mechanism from death: the cancer lesson. Antioxidants Redox Signaling, 11, 2791–2806.
Maulik, N. (2002). Redox signaling of angiogenesis. Antioxidants Redox Signaling, 4, 805–815.
Komatsu, D., Kato, M., Nakayama, J., Miyagawa, S., & Kamata, T. (2008). NADPH oxidase 1 plays a critical mediating role in oncogenic Ras-induced vascular endothelial growth factor expression. Oncogene, 27, 4724–4732.
Zundel, W., Schindler, C., Haas-Kogan, D., Koong, A., Kaper, F., Chen, E., et al. (2000). Loss of PTEN facilitates HIF-1-mediated gene expression. Genes and Development, 14, 391–396.
Hudson, C. C., Liu, M., Chiang, G. G., Otterness, D. M., Loomis, D. C., Kaper, F., et al. (2002). Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Molecular and Cellular Biology, 22, 7004–7014.
Sarbassov, D. D., & Sabatini, D. M. (2005). Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. Journal of Biological Chemistry, 280, 39505–39509.
Metzen, E., Zhou, J., Jelkmann, W., Fandrey, J., & Brune, B. (2003). Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Molecular Biology of the Cell, 14, 3470–3481.
Kasuno, K., Takabuchi, S., Fukuda, K., Kizaka-Kondoh, S., Yodoi, J., Adachi, T., et al. (2004). Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. Journal of Biological Chemistry, 279, 2550–2558.
Parenti, A., Morbidelli, L., Cui, X. L., Douglas, J. G., Hood, J. D., Granger, H. J., et al. (1998). Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase1/2 activation in postcapillary endothelium. Journal of Biological Chemistry, 273, 4220–4226.
Colavitti, R., Pani, G., Bedogni, B., Anzevino, R., Borrello, S., Waltenberger, J., et al. (2002). Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. Journal of Biological Chemistry, 277, 3101–3108.
Harfouche, R., Malak, N. A., Brandes, R. P., Karsan, A., Irani, K., & Hussain, S. N. (2005). Roles of reactive oxygen species in angiopoietin-1/tie-2 receptor signaling. FASEB Journal, 19, 1728–1730.
Ushio-Fukai, M. (2006). Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovascular Research, 71, 226–235.
Urao, N., Inomata, H., Razvi, M., Kim, H. W., Wary, K., McKinney, R., et al. (2008). Role of nox2-based NADPH oxidase in bone marrow and progenitor cell function involved in neovascularization induced by hindlimb ischemia. Circulation Research, 103, 212–220.
Lelkes, P. I., Hahn, K. L., Sukovich, D. A., Karmiol, S., & Schmidt, D. H. (1998). On the possible role of reactive oxygen species in angiogenesis. Advances in Experimental Medicine and Biology, 454, 295–310.
Fukumura, D., Kashiwagi, S., & Jain, R. K. (2006). The role of nitric oxide in tumour progression. Nature Reviews Cancer, 6, 521–534.
Radisky, D. C., Levy, D. D., Littlepage, L. E., Liu, H., Nelson, C. M., Fata, J. E., et al. (2005). Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature, 436, 123–127.
Nelson, C. M., Khauv, D., Bissell, M. J., & Radisky, D. C. (2008). Change in cell shape is required for matrix metalloproteinase-induced epithelial-mesenchymal transition of mammary epithelial cells. Journal of Cellular Biochemistry, 105, 25–33.
Cannito, S., Novo, E., Compagnone, A., di Valfre, B. L., Busletta, C., Zamara, E., et al. (2008). Redox mechanisms switch on hypoxia-dependent epithelial–mesenchymal transition in cancer cells. Carcinogenesis, 29, 2267–2278.
Ishikawa, K., Takenaga, K., Akimoto, M., Koshikawa, N., Yamaguchi, A., Imanishi, H., et al. (2008). ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science, 320, 661–664.
Binker, M. G., Binker-Cosen, A. A., Richards, D., Oliver, B., & Cosen-Binker, L. I. (2009). EGF promotes invasion by PANC-1 cells through Rac1/ROS-dependent secretion and activation of MMP-2. Biochemical and Biophysical Research Communications, 379, 445–450.
Oberley, T. D., Schultz, J. L., Li, N., & Oberley, L. W. (1995). Antioxidant enzyme levels as a function of growth state in cell culture. Free Radical Biology and Medicine, 19, 53–65.
Trachootham, D., Alexandre, J., & Huang, P. (2009). Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov, 8, 579–591.
Lee, A. C., Fenster, B. E., Ito, H., Takeda, K., Bae, N. S., Hirai, T., et al. (1999). Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. Journal of Biological Chemistry, 274, 7936–7940.
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D., & Lowe, S. W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell, 88, 593–602.
Takahashi, A., Ohtani, N., Yamakoshi, K., Iida, S., Tahara, H., Nakayama, K., et al. (2006). Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nature Cell Biology, 8, 1291–1297.
Lowe, S. W., Jacks, T., Housman, D. E., & Ruley, H. E. (1994). Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proceedings of the National Academy of Sciences of the United States of America, 91, 2026–2030.
Pani, G., Bedogni, B., Anzevino, R., Colavitti, R., Palazzotti, B., Borrello, S., et al. (2000). Deregulated manganese superoxide dismutase expression and resistance to oxidative injury in p53-deficient cells. Cancer Research, 60, 4654–4660.
Forrester, K., Ambs, S., Lupold, S. E., Kapust, R. B., Spillare, E. A., Weinberg, W. C., et al. (1996). Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proceedings of the National Academy of Sciences of the United States of America, 93, 2442–2447.
Ambs, S., Merriam, W. G., Ogunfusika, M. O., Bennett, W. P., Ishibe, N., Hussain, S. P., et al. (1998). p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells. Nature Medicine, 4, 1371–1376.
Ambs, S., Bennett, W. P., Merriam, W. G., Ogunfusika, M. O., Oser, S. M., Harrington, A. M., et al. (1999). Relationship between p53 mutations and inducible nitric oxide synthase expression in human colorectal cancer. Journal of the National Cancer Institute, 91, 86–88.
Kops, G. J., Dansen, T. B., Polderman, P. E., Saarloos, I., Wirtz, K. W., Coffer, P. J., et al. (2002). Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature, 419, 316–321.
Park, H. J., Carr, J. R., Wang, Z., Nogueira, V., Hay, N., Tyner, A. L., et al. (2009). FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO Journal, 28, 2908–2918.
Tanaka, H., Matsumura, I., Ezoe, S., Satoh, Y., Sakamaki, T., Albanese, C., et al. (2002). E2F1 and c-Myc potentiate apoptosis through inhibition of NF-kappaB activity that facilitates MnSOD-mediated ROS elimination. Molecular Cell, 9, 1017–1029.
Dolado, I., Swat, A., Ajenjo, N., De, V. G., Cuadrado, A., & Nebreda, A. R. (2007). p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell, 11, 191–205.
Kennedy, N. J., Cellurale, C., & Davis, R. J. (2007). A radical role for p38 MAPK in tumor initiation. Cancer Cell, 11, 101–103.
Bulavin, D. V., Phillips, C., Nannenga, B., Timofeev, O., Donehower, L. A., Anderson, C. W., et al. (2004). Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nature Genetics, 36, 343–350.
Helzlsouer, K. J., Selmin, O., Huang, H. Y., Strickland, P. T., Hoffman, S., Alberg, A. J., et al. (1998). Association between glutathione S-transferase M1, P1, and T1 genetic polymorphisms and development of breast cancer. Journal of the National Cancer Institute, 90, 512–518.
Havre, P. A., O'Reilly, S., McCormick, J. J., & Brash, D. E. (2002). Transformed and tumor-derived human cells exhibit preferential sensitivity to the thiol antioxidants. N-acetyl cysteine and penicillamine. Cancer Research, 62, 1443–1449.
Polyak, K., Xia, Y., Zweier, J. L., Kinzler, K. W., & Vogelstein, B. (1997). A model for p53-induced apoptosis. Nature, 389, 300–305.
Johnson, T. M., Yu, Z. X., Ferrans, V. J., Lowenstein, R. A., & Finkel, T. (1996). Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proceedings of the National Academy of Sciences of the United States of America, 93, 11848–11852.
Drane, P., Bravard, A., Bouvard, V., & May, E. (2001). Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis. Oncogene, 20, 430–439.
Dhar, S. K., Xu, Y., Chen, Y., & St Clair, D. K. (2006). Specificity protein 1-dependent p53-mediated suppression of human manganese superoxide dismutase gene expression. Journal of Biological Chemistry, 281, 21698–21709.
Pani, G., Colavitti, R., Bedogni, B., Fusco, S., Ferraro, D., Borrello, S., et al. (2004). Mitochondrial superoxide dismutase: a promising target for new anticancer therapies. Current Medicinal Chemistry, 11, 1299–1308.
Ito, K., Hirao, A., Arai, F., Matsuoka, S., Takubo, K., Hamaguchi, I., et al. (2004). Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature, 431, 997–1002.
Diehn, M., Cho, R. W., Lobo, N. A., Kalisky, T., Dorie, M. J., Kulp, A. N., et al. (2009). Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature, 458, 780–783.
Smith, J., Ladi, E., Mayer-Proschel, M., & Noble, M. (2000). Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proceedings of the National Academy of Sciences of the United States of America, 97, 10032–10037.
Jang, Y. Y., & Sharkis, S. J. (2007). A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood, 110, 3056–3063.
Ito, K., Hirao, A., Arai, F., Takubo, K., Matsuoka, S., Miyamoto, K., et al. (2006). Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nature Medicine, 12, 446–451.
Tothova, Z., Kollipara, R., Huntly, B. J., Lee, B. H., Castrillon, D. H., Cullen, D. E., et al. (2007). FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell, 128, 325–339.
Chen, C., Liu, Y., Liu, R., Ikenoue, T., Guan, K. L., Liu, Y., et al. (2008). TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. Journal of Experimental Medicine, 205, 2397–2408.
Ranganathan, A. C., Adam, A. P., & Aguirre-Ghiso, J. A. (2006). Opposing roles of mitogenic and stress signaling pathways in the induction of cancer dormancy. Cell Cycle, 5, 1799–1807.
Adam, A. P., George, A., Schewe, D., Bragado, P., Iglesias, B. V., Ranganathan, A. C., et al. (2009). Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Research, 69, 5664–5672.
Schewe, D. M., & Aguirre-Ghiso, J. A. (2008). ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proceedings of the National Academy of Sciences of the United States of America, 105, 10519–10524.
Schafer, Z. T., Grassian, A. R., Song, L., Jiang, Z., Gerhart-Hines, Z., Irie, H. Y., et al. (2009). Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature, 461, 109–113.
Kuo, W., Lin, J., & Tang, T. K. (2000). Human glucose-6-phosphate dehydrogenase (G6PD) gene transforms NIH 3T3 cells and induces tumors in nude mice. International Journal of Cancer, 85, 857–864.
The Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. (1994). The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. New England Journal of Medicine, 330, 1029–1035.
Omenn, G. S., Goodman, G. E., Thornquist, M. D., Balmes, J., Cullen, M. R., Glass, A., et al. (1996). Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. New England Journal of Medicine, 334, 1150–1155.
Shackelford, R. E., Heinloth, A. N., Heard, S. C., & Paules, R. S. (2005). Cellular and molecular targets of protein S-glutathiolation. Antioxidants Redox Signaling, 7, 940–950.
Coussens, L. M., & Werb, Z. (2002). Inflammation and cancer. Nature, 420, 860–867.
Schafer, M., & Werner, S. (2008). Cancer as an overhealing wound: an old hypothesis revisited. Nature Reviews Molecular Cell Biology, 9, 628–638.
Phinney, D. G., & Prockop, D. J. (2007). Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair—current views. Stem Cells, 25, 2896–2902.
Ishii, T., Itoh, K., Takahashi, S., Sato, H., Yanagawa, T., Katoh, Y., et al. (2000). Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. Journal of Biological Chemistry, 275, 16023–16029.
Huang, C., Han, Y., Wang, Y., Sun, X., Yan, S., Yeh, E. T., et al. (2009). SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. EMBO Journal, 28, 2748–2762.
Scortegagna, M., Ding, K., Oktay, Y., Gaur, A., Thurmond, F., Yan, L. J., et al. (2003). Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nature Genetics, 35, 331–340.
Conrotto, P., Corso, S., Gamberini, S., Comoglio, P. M., & Giordano, S. (2004). Interplay between scatter factor receptors and B plexins controls invasive growth. Oncogene, 23, 5131–5137.
Fischer, O. M., Giordano, S., Comoglio, P. M., & Ullrich, A. (2004). Reactive oxygen species mediate Met receptor transactivation by G protein-coupled receptors and the epidermal growth factor receptor in human carcinoma cells. Journal of Biological Chemistry, 279, 28970–28978.
Klarmann, G. J., Hurt, E. M., Mathews, L. A., Zhang, X., Duhagon, M. A., Mistree, T., et al. (2009). Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clinical & Experimental Metastasis, 26, 433–446.
O'Dell, T. J., Hawkins, R. D., Kandel, E. R., & Arancio, O. (1991). Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proceedings of the National Academy of Sciences of the United States of America, 88, 11285–11289.
Niethammer, P., Grabher, C., Look, A. T., & Mitchison, T. J. (2009). A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature, 459, 996–999.
Taylor, B. L., Rebbapragada, A., & Johnson, M. S. (2001). The FAD-PAS domain as a sensor for behavioral responses in Escherichia coli. Antioxidants Redox Signaling, 3, 867–879.
Graeber, T. G., Osmanian, C., Jacks, T., Housman, D. E., Koch, C. J., Lowe, S. W., et al. (1996). Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature, 379, 88–91.
Hentze, M. W., & Kuhn, L. C. (1996). Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proceedings of the National Academy of Sciences of the United States of America, 93, 8175–8182.
Acknowledgments
The authors wish to thank Prof. Lido Calorini, as like as members of their laboratories for suggestions and insightful discussions. Authors are supported by the Italian Association for Cancer Research (AIRC) (grant# IG 8634 to G.P. and grant # IG8797 to P.C.), by Istituto Toscano Tumori and Regione Toscana (TRESOR grants and PorCreo grant to P. C.).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Pani, G., Galeotti, T. & Chiarugi, P. Metastasis: cancer cell’s escape from oxidative stress. Cancer Metastasis Rev 29, 351–378 (2010). https://doi.org/10.1007/s10555-010-9225-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-010-9225-4