
Abstract
Anti-angiogenic drugs, alone or in combination with chemotherapeutics, are increasingly used by medical oncologists. In many cases, however, their mechanism of action and the tailoring of optimal dosage/schedule are still elusive. Circulating endothelial cell (CEC) and progenitor (CEP) number and viability are modulated in a large series of diseases including cancer, and look promising as surrogate biomarkers for the definition of the optimal biological dose of anti-angiogenic drugs and for patients’ stratification. Along with CECs and CEPs, potential EC- and CEP-related surrogate molecular markers such as VE-Cadherin and CD133 are currently under preclinical and clinical investigation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The generation of new and functional vessels is one of the crucial hallmarks of cancer progression [1–2]. New vessels are generated by mature endothelial cells (EC) in a process called angiogenesis, or by bone marrow (BM)-derived EC progenitors in a process called vasculogenesis [3]. Post-natal angiogenesis and vasculogenesis are limited to reproduction and wound healing, and vascular supply seems to be necessary for the growth of neoplastic lesions beyond few millimeters of size. Thus, cancer-related new vessels and EC progenitors have been considered as a convenient target for the therapy of cancer [1–3]. This review will focus on two main issues: The role of circulating ECs (CECs) and circulating EC progenitors (CEPs) in cancer growth and their measurement as biomarkers in cancer therapy.
1.1 Toward a functional and phenotypic definition of CECs and CEPs
Circulating, anucleated cellular carcasses with a EC morphology were first described in mid 1970s [4], but it took almost 30 more years to obtain a procedure able to count this rare cellular population. Using a y-chromosome gene marking approach in recipients of gender-mismatched BM transplants, the Hebbel laboratory [5] was the first able to distinguish between CEPs from the BM (i.e., donor-derived cells), and CECs from vessel walls (i.e., host (recipient)-derived). More than 90% of ECs in the blood were found to be of recipient origin. The remaining endothelial cells, of donor BM-origin, had a relevant functional difference: when cultured in vitro, recipient-genotype ECs expanded only approximately 20-fold, whereas donor-genotype endothelial cells expanded approximately 1,000-fold. Thus, most CECs probably originate from vessel walls and have limited growth capability, whereas a minute CEP subpopulation is responsible, at least in vitro, for the large majority of the EC proliferative potential. These findings, along with previous studies demonstrating that CEPs from peripheral blood could generate mature EC in vitro and in vivo in vascular grafts [6, 7], suggested attempts to differentiate CEPs from CECs by means of monoclonal antibodies.
At the present time, monoclonal antibodies are used for CEC and CEP count in the blood by immunohistochemistry (IHC, ref. [8]) or by flow cytometry (FC, ref. [9]). IHC and FC protocols are undergoing rigorous international standardization trials, and it is becoming clear that the two procedures enumerate two different cellular entities. IHC allows the morphological identification of very rare (about 1–10/mL of blood), large sized, viable and nucleated ECs. FC allows the enumeration of all cellular events that express EC-related antigens. Unfortunately, EC share the very large majority of their antigens with white blood cells (e.g. CD13, CD31, CD34, CD146, VEGFR1 and VEGFR2), so that a multiparametric investigation is mandatory to exclude hematopoietic (i.e. CD45+) cells from the count [9]. Moreover, platelets have a phenotype which is almost identical to EC, so that a further investigation for the presence of DNA (which is almost absent in platelets) is needed. When considering these caveats, FC has identified a population of DNA-containing cellular events with an EC phenotype which is quantitatively more frequent (about 1/µL) than that identified by IHC, shows in most cases apoptosis-related makers and is most likely including apoptotic bodies and fragments resulting from vessel wall turnover. IHC and FC studies have indicated that CECs are increased in many diseases including cancer [10]. As discussed later, CECs measured by FC are modulated by anti-angiogenic treatments both in preclinical models and in cancer patients, and are currently investigated as surrogate biomarkers of angiogenesis and of response to anti-angiogenic treatments [9].
FC studies have also indicated that the very large majority of CECs have a mature phenotype, but a small CEC subpopulation express progenitor-related makers such as CD34, CD133 or low aldehyde dehydrogenase (ALDH) activity [9, 11–13]. These CEPs have been enumerated in the past following two separated approaches. CEP functional properties include the potential to generate mature EC. Thus, functional soft-gel assays for the enumeration of EC colonies generated by single CEPs have been attempted. A recent study, though, has indicated that the true EC-specific output of these colony assays is frequently and largely overestimated, because most of the colonies generated in these assays are hematopoietic and not endothelial [14]. Functional in vivo studies have now defined that the CEP activity is most likely restricted to CD34+ (or, for other authors, to CD133+ cells) with a CD45-negative phenotype [9, 11–14].
1.2 CEPs and cancer
The first evidence of a possible CEP role in cancer progression was reported in 2001 by Lyden et al. [15]. They described that in angiogenic defective Id-mutant mice the transplantation of wild-type BM cells or CEPs was able to restore tumor angiogenesis and the growth of some types of transplanted tumor cell lines. This model had some technical limitations due to the defective endothelial cell sprouting of these knock-out mice, and later some other studies showed negligible or no measurable CEP contribution to tumor vessels [16–17]. More recent studies, though, have indicated that CEP contribution to tumor vasculature significantly varies depending on the organ site and the mouse strain [18–19]. In a clinical study, tumors from six patients who developed cancers after BM transplantation were found to contain an average of 5% of EC from the donor, most likely generated by CEPs [20]. Spring et al. [21] identified a role for CEPs in late stage tumor vascularization, and the Benezra’s group demonstrated that the recruitment of CEPs into tumor vasculature depends on the tumor grade [22]. Yuval Shaked from the Kerbel laboratory [23] found that the administration of a vascular disrupting agent (VDA) to cancer-bearing mice results in a rapid CEP mobilization which contributes to subsequent tumor angiogenesis and re-growth. If the VDA-induced CEP mobilization is prevented by an anti-angiogenic drug, or by administrating the VDA to Id mutant mice (which cannot mobilize CEPs) the anti-tumor effects of VDA therapy are significantly increased. These data suggest to reconsider previous studies showing low levels of CEPs in tumor vasculature, because all past studies have involved untreated, ‘steady state’ tumors. In preclinical studies, maximum tolerable dose (MTD) chemotherapy has been found to cause a short-time suppression of viable CECs and CEPs soon after drug administration, followed by CEC and CEP mobilization [24]. Similar results were reported in breast cancer patients receiving neoadjuvant chemotherapy, where CEP mobilization was significantly increased after the treatment [25].
Taken together, these data suggest that CEPs might be relevant in crucial clinical situations such as relapse after surgery, radiation, chemotherapy, or other therapies, contributing important but transient and reactive host responses. Thus, the addition of an anti-angiogenic agent to or after high-dose chemotherapy might prevent CEP mobilization and their related pro-angiogenic effect. This provides an additional rationale for targeting CEPs as a potentially beneficial anti-cancer and anti-angiogenic treatment strategy, and might—at least in part—explain how anti-angiogenic drugs can augment the efficacy of some standard chemotherapy regimens [26].
In addition to CEPs, there is evidence for the contribution of other BM-derived circulating cell populations in tumor angiogenesis. Cells co-expressing both dendritic cell (DC) and EC markers have been described in mouse and human ovarian carcinomas. Tumor-infiltrating DCs migrated to tumor vessels and contributed to the assembly of the cancer vessels [27]. Adult vasculogenesis may rely on the recruitment of BM-derived circulating cells by secretion of VEGF from the microenvironment. VEGF induction in different tissue led to massive infiltration of cells expressing both CD45 and VEGFR1, but not VEGFR2. These VEGF-recruited cells are predominantly hematopoietic in nature, express the CXCR4 chemokine receptor, and home to tumor perivascular sites [28]. Some CD45+monocytes expressing Tie-2 and CD11b (TEMs) are recruited to tumor sites and promote angiogenesis in a paracrine manner after adhering to newly forming blood vessels [29]. A population of tumor associated stroma cells expressing CD45 and VEGFR2 can colonize the tumor stroma and incorporate (albeit at low frequency) into the lumens of tumor vasculature; these cells can contribute to tumor angiogenesis in a paracrine manner by inducing or enhancing factors stimulating EC recruitment [30]. Moreover, VEGFR1-positive cells able to initiate metastatic niches were recently described [31]. Potentially, these novel cell populations might be investigated as biomarkers along with CECs and CEPs, provided that adequate cellular or molecular markers are identified and enumeration procedures are clinically validated.
1.3 CECs and CEPs as biomarkers in cancer
The broad family of “molecularly targeted” anti-cancer drugs such as monoclonal antibodies and tyrosine-kinase inhibitors has entered mainstream clinical oncology and represents a milestone towards a maximally targeted and minimally invasive therapy of cancer [2, 26]. However, several important handicaps still hamper the development and optimal use of such drugs (including, but not limited to, anti-angiogenic agents). In many cases it is unclear whether a patient’s cancer (or cancer stroma) expresses the drug’s target or if the interaction between the drug and the target has the potential to stop cancer growth. In fact, when using many (if not in all) of these novel therapeutics there is an urgent need for monitoring their biologic activity, selecting and stratifying the patients who are most likely to benefit from treatment, and determining drug optimal biologic dose (OBD). This innovative concept of dose-finding seems particularly appropriate for anti-angiogenic agents because in many cases they stop cancer growth rather than induce a tumor shrinkage; also, there is a lack of dose limiting toxicities (DLT) to define a MTD, and, when a MTD is present, it might be possible that these drugs express their optimal therapeutic activity at doses below the MTD.
Recent randomized clinical trials indicated that in a variety of cancer types the addition of some anti-angiogenic drugs to chemotherapeutics may prolong patients’ survival. However, the mechanism of this “chemosensitizing” effect [26] is presently unclear. When analyzing in detail the results of randomized clinical trials, it looks likely that the clinical benefit of adding anti-angiogenic drugs to chemotherapy is due to an important and prolonged benefit in some patients rather than a more limited benefit in all of the patients.
As said before, empiricism has led so far the way anti-angiogenic drugs have been associated to chemotherapeutics, because no preclinical or clinical data have been available to indicate the OBD or the best sequence schedule of anti-angiogenic and chemotherapy drugs. The tremendous impact on healthcare cost of these very expensive new drugs makes this scenario no more sustainable. Thus, there is an urgent need for biomarkers able to identify patients that are most likely to benefit from these therapies and able to suggest the best sequential anti-angiogenic and chemotherapy drug schedule.
Looking at the list of the available angiogenesis assays, it’s clear that some of them (like the growth factor-induced generation and quantification of new vessels in the cornea, skin or dorsal sac) are not adaptable to patients. The evaluation of microvessel density (MVD) in cancer biopsy samples is invasive and not fully validated for clinical studies [32]. Moreover, tumor tissue heterogeneity implies that MVD of a cancer biopsy might not correlate with MVD of the whole tumor. The circulating levels of angiogenic growth factors such as VEGF, b-FGF or HGF may predict survival in some types of cancer, but there is no proof that these measurement can predict response to anti-angiogenic therapies [33].
Soluble VEGF receptors such as VEGFR1, VEGFR2 and VEGFR3 are studied in a variety of cancer trials with anti-angiogenic therapies in order to understand their potential as surrogate biomarkers. In kidney cancer cell patients treated with the tyrosine kinase inhibitor Sutent, for instance, VEGF-A and PlGF increased after each cycle of drug administration, whereas soluble VEGFR2 decreased. After 2 weeks without treatment, the levels of these biomarkers returned to near basal levels [34]. However, more work seems needed to clarify the clinical relevance of these results, because no clear correlation has been found with response to anti-angiogenic treatments. Functional imaging such as dynamic contrast magnetic resonance imaging (DCE-MRI) might measure subtle changes in tumor blood flow and vascular permeability, but the clinical impact of this type of approach remains unknown and it requires expensive instrumentation available only in few institutions [35].
In murine preclinical models the number of viable CEC correlates well with classical preclinical angiogenesis assays (such as vessel generation in the cornea or in matrigel plugs) that can not be used in the clinic, and CEC kinetic was found to be a useful tool to monitor in vivo the anti-angiogenic effects of different drugs [36]. In fact, in different preclinical cancer models the maximum effect of a given drug over viable CEC count was reached at the same drug dosage that induced the best reduction in cancer volume, with higher drug levels reaching a plateau.
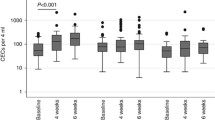
CEC levels are increased in the peripheral blood of some cancer patients at diagnosis, and these cells return to normal values in patients achieving a complete remission [9, 25, 37–40]. In metastatic breast cancer patients treated with low dose metronomic chemotherapy (a therapeutic strategy associated with anti-angiogenic activity) the CEC count after 2 months of continuous therapy was a particularly good predictor of disease-free and overall-survival after a prolonged follow-up of more than 2 years [39]. Similarly to what seen in preclinical models [41], in patients with a clinical benefit the increase in CEC counts was mostly due to an increase in their apoptotic fraction of these cells. Anti-angiogenic agents might reverse cancer vessel abnormalities [42, 43] and part of the remodelling process might cause a shedding of apoptotic CECs from cancer vessels. In preclinical studies, a CEC rise was not observed in cancer-free animals treated with metronomic chemotherapy [39]. Taken together, these findings suggest that the cancer-associated vasculature might most likely be the predominant or even sole source of the rise in apoptotic CECs seen in breast cancer patients treated with metronomic therapy.
1.4 Biomarker-driven tailoring of anti-angiogenic treatments
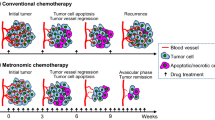
Three major different hypothesis (Fig. 1) are currently used to explain how anti-angiogenic drugs reduce cancer growth and how they synergize with other anti-cancer drugs (and in particular with chemotherapeutics, ref. [26]):
-
(1)
It might be possible that anti-angiogenic drugs induce a normalization of otherwise deficient cancer vessels, thus prompting a better delivery of chemotherapeutics. In fact, one of the possible effects of anti-angiogenic therapy might be on tumor vessel remodeling, i.e., the so called “vessel normalization” hypothesis suggesting that anti-angiogenic agents (alone or in combination with chemotherapeutics) might reverse cancer vessel abnormalities [42, 43]. This remodeling might cause a shedding of apoptotic CECs, perycites and smooth muscle cells from cancer vessels. According to this hypothesis, anti-angiogenic drugs should be administered before (or along with) chemotherapeutic drugs, because they might improve drug delivery to cancer cells.
-
(2)
Anti-angiogenic drugs might reduce vessel-driven tumor repopulation during free break periods of chemotherapy. The consequence of this hypothesis has led to the concept of metronomic chemotherapy (i.e. the close, regular administration of low, non-toxic doses of chemotherapeutic drugs with no breaks, over long periods of time), and this therapeutic strategy is known to have anti-angiogenic activity [44]. According to this hypothesis, anti-angiogenic drugs should be administered after chemotherapeutic drugs, to avoid tumor recurrence during breaks from chemotherapy.
-
(3)
Anti-angiogenic drugs might target proliferating tumor ECs or CEPs. According to this hypothesis, anti-angiogenic drugs should be administered after (or along with) chemotherapeutic drugs, to avoid chemotherapy-induced CEP mobilization.

Hypothesis used to explain how anti-angiogenic drugs may stop cancer growth and synergize with chemotherapeutics: (a) Anti-angiogenic drugs might induce a normalization of otherwise deficient cancer vessels, provoking a better delivery of chemotherapeutics. Part of the remodeling process would likely cause a shedding of apoptotic CECs and potentially of other vessel-related cells. (b) Anti-angiogenic drugs might reduce vessel-driven tumor repopulation during free break periods of chemotherapy. (c) Anti-angiogenic drugs might target proliferating tumor-related ECs and BM-derived CEPs
1.5 The search for molecular markers of angiogenesis and of anti-angiogenic drug activity
As mentioned before, ECs share the very large majority (if not all) of their antigens with other hematopoietic (e.g. CD13, CD31, CD34, CD146, VEGF-receptors) or mesenchymal (e.g. CD90) cells [9]. Thus, the quantification of soluble antigens expressed by EC or the enumeration of their mRNA transcripts will offer promiscuous information, most likely of limited or unspecific clinical predictive value. Many attempts have been made to purify cancer-specific ECs and to screen for genes or proteins expressed only by these cells. The St. Croix laboratory has recently identified 25 transcripts overexpressed in tumor versus normal EC, including 13 that were not found in the EC of regenerating liver [45]. Along a similar line, the Immunicon laboratory [46] identified 61 genes overexpressed in CECs from cancer patients compared to CECs from healthy volunteers, and the Griffioen laboratory [47] identified 17 genes that showed specific overexpression in tumor ECs from colorectal cancer patients compared with angiogenic ECs of normal tissues. Further studies will indicate whether these markers have potential as biomarkers or for therapeutic purposes.
So far, only a very limited number of genes has been considered to be truly EC-restricted or EC-specific. One of these is VE-Cadherin, which, at the present time is considered to be expressed outside of the EC lineage only by stem cells in utero [48] or by some acute leukemia cells [49]. The number of copies of VE-Cadherin RNA in the blood of cancer patients was found to be significantly increased when compared to healthy controls [50]. However, a special care should be considered when using VE-Cadherin to investigate the anti-angiogenic activity of a given drug, because the expression of this gene is markedly reduced (or absent) in apoptotic ECs.
Recent studies have reported an increase in the number of copies of the progenitor-cell associated gene CD133 in patients with colon cancer [51], hepatocellular carcinoma [52] or bone metastases [53]. CD133, however, is expressed by hematopoietic and EC progenitors, so that further studies are needed to better understand what cell population is responsible for these findings. To identify novel biomarkers, Ayers et al. [54] recently investigated cancer bearing mice treated with the anti-angiogenic agent brivanib alaninate, which targets VEGFR-2 and FGFR-1. Tyrosine kinase receptor 1 (Tie-1), collagen type IV alpha1 (Col4a1), complement component 1, q subcomponent receptor 1 (C1qr1), angiotensin receptor-like 1 (Agtrl1), and vascular endothelial-cadherin (Cdh5) were modulated by the treatment, and authors are trying to validate this molecular signature in the clinic.
1.6 Are CECs and CEPs genetically stable?
At variance with cancer cells, tumor-associated vascular cells have been considered for many years to be genetically stable [55]. However, cytogenetically abnormal ECs have been recently described in some preclinical models of cancer [56]. In some non-Hodgkin’s lymphoma patients with specific genetic aberrations, ECs from cancer microvasculature had the same lymphoma-specific chromosomal translocations [57]. Similarly, in myeloma [58] and in some leukemias [59] CECs were found to share the same genetic alterations observed in cancer cells. There are at least 4 different possible explanations for these findings. Cancer-related ECs and neoplastic cells may derive from a common progenitor. Genetically-altered ECs might acquire oncogenes by ‘horizontal’ DNA transfer [60]. Microenvironmental angiogenic factors might dictate a de- (or trans-) differentiation of neoplastic cells toward an EC phenotype. Finally, these observations might be due to fusion between cancer cells and ECs, a phenomenon which has been already reported in the past [61].
2 Conclusions
A variety of promising molecular drugs have entered the anti-cancer arsenal, but there is an urgent need for efficient surrogate biomarkers able to indicate the OBD of these new drugs and to select patients that are likely to benefit from these expensive treatments, alone or in association with other therapies. Molecular markers are in sight but have not yet been fully validated at the bedside. The measurement of cancer-related EC populations looks promising, but standardized protocols are warranted. These measurements might also be of relevant help to predict and manage the unforeseen side effects and toxicity observed after the administration of anti-angiogenic drugs [62].
References
Hanahan, D., & Weinberg, R. A. (2000). The hallmarks of cancer. Cell, 100, 57–70.
Folkman, J. (2007). Angiogenesis: An organizing principle for drug discovery? Naturalist Review Drug Discovery, 6, 273–286.
Conway, E. M., Collen, D., & Carmeliet, P. (2001). Molecular mechanisms of blood vessel growth. Cardiovascular Research, 49, 507–521.
Hladovec, J., & Rossamn, P. (1973). Circulating endothelial cells isolated together with platelets and the experimental modification of their counts in rats. Thrombosis Research, 3, 665–674.
Lin, Y., Weisdorf, D. J., Solovey, A., & Hebbel, R. P. (2000). Origins of circulating endothelial cells and endothelial outgrowth from blood. Journal of Clinical Investigation, 105, 71–77.
Asahara, T., et al. (1997). Isolation of putative progenitor endothelial cells for angiogenesis. Science, 275, 964–967.
Shi, Q., et al. (1998). Evidence for circulating bone marrow-derived endothelial cells. Blood, 92, 362–367.
Dignat-George, F., & Sampol, J. (2000). Circulating endothelial cells in vascular disorders: New insights into an old concept. European Journal of Haematology, 65, 215–220.
Bertolini, F., et al. (2006). The multifaceted circulating endothelial cell in cancer: Towards marker and target identification. Nature Reviews Cancer, 6, 835–845.
Blann, A. D., et al. (2005). Circulating endothelial cells. Biomarker of vascular disease. Thrombosis and Haemostasis, 93, 228–235.
Rafii, S., et al. (2002). Vascular and haematopoietic stem cells: Novel targets for anti-angiogenesis therapy? Nature Reviews Cancer, 2, 826–835.
Melero-Martin, J. M., et al. (2007). In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood, 109, 4761–4768.
Nagano, M., et al. (2007). Identification of functional endothelial progenitor cells suitable for the treatment of ischemic tissue using umbilical cord blood. Blood, 110, 151–160.
Yoder, M. C., et al. (2007). Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood, 109, 1801–1809.
Lyden, D., et al. (2001). Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nature Medicine, 7, 1194–1201.
De Palma, M., et al. (2003). Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nature Medicine, 9, 789–795.
Gothert, J. R., et al. (2004). Genetically tagging endothelial cells in vivo: Bone marrow-derived cells do not contribute to tumor endothelium. Blood, 104, 1769–1777.
Duda, D. G., et al. (2006). Evidence for incorporation of bone marrow-derived endothelial cells into perfused blood vessels in tumors. Blood, 107, 2774–2776.
Monsky, W. L., et al. (2002). Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: Mammary fat pad versus cranial tumors. Clinical Cancer Research, 8, 1008–1013.
Peters, B. A., et al. (2005). Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nature Medicine, 11, 261–262.
Spring, H., et al. (2005). Chemokines direct endothelial progenitors into tumor neovessels. Proceedings of the National Academy of Sciences of the United States of America, 102, 18111–18116.
Ruzinova, M. B., et al. (2003). Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer Cell, 4, 277–289.
Shaked, Y., et al. (2006). Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science, 313, 1785–1787.
Bertolini, F., et al. (2003). Maximum tolerable dose and low-dose metronomic chemotherapy have opposite effects on the mobilization and viability of circulating endothelial progenitor cells. Cancer Research, 63, 4342–4346.
Furstenberger, G., et al. (2006). Circulating endothelial cells and angiogenic serum factors during neoadjuvant chemotherapy of primary breast cancer. British Journal of Cancer, 94, 524–531.
Kerbel, R. S. (2006). Antiangiogenic therapy: A universal chemosensitization strategy for cancer? Science, 312, 1171–1175.
Conejo-Garcia, J. R., et al. (2004). Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of VEGF-A. Nature Medicine, 10, 950–958.
Grunewald, M., et al. (2006). VEGF-induced adult neovascularization: Recruitment, retention, and role of accessory cells. Cells, 124, 175–189.
De Palma, M., et al. (2005). Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cells, 8, 211–226.
Udagawa, T., et al. (2006). Analysis of tumor-associated stromal cells using SCID GFP transgenic mice: Contribution of local and bone marrow-derived host cells. FASEB Journal, 20, 95–102.
Kaplan, R. N., et al. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature, 438, 820–827.
Hlatky, L., et al. (2002). Clinical application of antiangiogenic therapy: Microvessel density, what it does and doesn’t tell us. Journal of the National Cancer Institute, 94, 883–893.
Jain, R. K. (2002). Tumor angiogenesis and accessibility: Role of vascular endothelial growth factor. Seminars in Oncology, 29(6 Suppl 16), 3–9.
Motzer, R. J., et al. (2006). Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology, 24, 16–24.
Morgan, B., et al. (2003). Dynamic contrast-enhanced magnetic resonance imaging as a biomarker for the pharmacological response of PTK787/ZK 222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases, in patients with advanced colorectal cancer and liver metastases: Results from two phase I studies. Journal of Clinical Oncology, 2, 3955–3964.
Shaked, Y., et al. (2005). Genetic heterogeneity of the vasculogenic phenotype parallels angiogenesis: Implications for cellular surrogate marker analysis of antiangiogenesis. Cancer Cells, 7, 101–111.
Mancuso, P., et al. (2001). Resting and activated endothelial cells are increased in the peripheral blood of cancer patients. Blood, 97, 3658–3661.
Zhang, H., et al. (2005). Circulating endothelial progenitor cells in multiple myeloma: Implications and significance. Blood, 105, 3286–3294.
Mancuso, P., et al. (2006). Circulating endothelial cell kinetics and viability predict survival in breast cancer patients receiving metronomic chemotherapy. Blood, 108, 452–459.
Norden-Zfoni, A., et al. (2007). Blood-based biomarkers of SU11248 activity and clinical outcome in patients with metastatic imatinib-resistant gastrointestinal stromal tumor. Clinical Cancer Research, 13, 2643–2650.
Monestiroli, S., et al. (2001). Kinetics and viability of circulating endothelial cells as surrogate angiogenesis marker in an animal model of human lymphoma. Cancer Research, 61, 4341–4344.
Jain, R. K. (2003). Molecular regulation of vessel maturation. Natural Medicines, 9, 685–693.
Batchelor, T. T., et al. (2007). AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cells, 11, 83–95.
Kerbel, R. S., & Kamen, B. A. (2004). The anti-angiogenic basis of metronomic chemotherapy. Nature Reviews Cancer, 4, 423–436.
Seaman, S., et al. (2007). Genes that distinguish physiological and pathological angiogenesis. Cancer Cells, 11, 539–554.
Smirnov, D. A., et al. (2006). Global gene expression profiling of circulating endothelial cells in patients with metastatic carcinomas. Cancer Research, 66, 2918–2922.
van Beijnum, J. R., et al. (2006). Gene expression of tumor angiogenesis dissected: Specific targeting of colon cancer angiogenic vasculature. Blood, 108, 2339–2348.
Kim, I., et al. (2005). CD144 (VE-cadherin). is transiently expressed by fetal liver hematopoietic stem cells. Blood, 106, 903–905.
Wang, L., O’Leary, H., Fortney, J., & Gibson, L. F. (2007). Ph+/VE-cadherin+ identifies a stem-cell like population of acute lymphoblastic leukemia sustained by bone marrow niche cells. Blood, prepublished online 16 July 2007. DOI 10.1182/blood-2007-01-068122.
Rabascio, C., et al. (2004). Assessing tumor angiogenesis: Increased circulating VE-cadherin RNA in patients with cancer indicates viability of circulating endothelial cells. Cancer Research, 64, 4373–4377.
Lin, E. H., et al. (2007). Elevated circulating endothelial progenitor marker CD133 messenger RNA levels predict colon cancer recurrence. Cancer, (Epub ahead of print, Jun 26, 2007).
Yu, D., et al. (2007). Identification and clinical significance of mobilized endothelial progenitor cells in tumor vasculogenesis of hepatocellular carcinoma. Clinical Cancer Research, 13, 3814–3824.
Mehra, N., et al. (2006). Progenitor marker CD133 mRNA is elevated in peripheral blood of cancer patients with bone metastases. Clinical Cancer Research, 12, 4859–4866.
Ayers, M., Fargnoli, J., Lewin, A., Wu, Q., & Platero, J. S. (2007). Discovery and validation of biomarkers that respond to treatment with brivanib alaninate, a small-molecule VEGFR-2/FGFR-1 antagonist. Cancer Research, 67, 6899–6906.
Kerbel, R. S. (1991). Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. BioEssays, 13, 31–36.
Hida, K., et al. (2004). Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Research, 64, 8249–8255.
Streubel, B., et al. (2004). Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. New England Journal of Medicine, 351, 250–259.
Rigolin, G. M., et al. (2006). Neoplastic circulating endothelial cells in multiple myeloma with 13 q14 deletion. Blood, 107, 2531–2535.
Rigolin, G. M., et al. (2007). Neoplastic circulating endothelial-like cells in patients with acute myeloid leukaemia. European Journal of Haematology, 78, 365–373.
Bergsmedh, A., et al. (2001). Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proceedings of the National Academy of Sciences of the United States of America, 98, 6407–6411.
Mortensen, K., Lichtenberg, J., Thomsen, P. D., & Larsson, L. I. (2004). Spontaneous fusion between cancer cells and endothelial cells. Cellular and Molecular Life Sciences, 61, 2125–2131.
van Heeckeren, W. J., Ortiz, J., Cooney, M. M., & Remick, S. C. (2007). Hypertension, proteinuria, and antagonism of VEGF signalling: Clinical toxicity, therapeutic target, or novel biomarker? Journal of Clinical Oncology, 25, 2993–2995.
Acknowledgments
Supported in part by AIRC (Associazione Italiana per la Ricerca sul Cancro), ISS (Istituto Superiore di Sanità) and the Sixth EU Framework Programme (Integrated Project ‘Angiotargeting; contract no. 504743) in the area of ‘Life sciences, genomics and biotechnology for health”. FB is a scholar of the US National Blood Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bertolini, F. Chemotherapy and the tumor microenvironment: the contribution of circulating endothelial cells. Cancer Metastasis Rev 27, 95–101 (2008). https://doi.org/10.1007/s10555-007-9110-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-007-9110-y