
Abstract
Purpose
More than 90% of the breast cancer deaths occur due to the metastasis of the cancer cells to secondary organ sites. Increased Glucose-regulated protein 78 (GRP78) expression is critical for epithelial–mesenchymal transition (EMT) and invasion in breast cancer resulting in poor patient survival outcomes. Therefore, there is an urgent need of potential inhibitors of GRP78 for the abrogation of invasion and metastasis in breast cancer.
Methods
We investigated the effect of IKM5 (2-(1-(1H-indol-3-yl)octyl)-3-hydroxy-6-(hydroxymethyl)-4H-pyran-4-one) (a novel Indolylkojyl methane analogue) on invasion abilities of human breast cancer cells employing invadopodia formation, Matrigel invasion assays, and mouse models for metastasis. The mechanism underlying the anti-invasive effect of IKM5 was examined through molecular docking, immunoblotting, immunocytochemistry, co-immunoprecipitation analysis, siRNA silencing, and sub-cellular fractionation studies.
Results
Treatment with IKM5 at its sub-toxic concentration (200 nM) suppressed invasion and invadopodia formation, and growth factor-induced cell scattering of aggressive human breast cancer MDA-MB-231, MDA-MB-468, and MCF7 cells. IKM5 spontaneously binds to GRP78 (Ki = 1.35 µM) and downregulates its expression along with the EMT markers MMP-2, Twist1, and Vimentin. Furthermore, IKM5 amplified the expression and nuclear translocation of tumor suppressor Par-4 to control NF-kB-mediated pro-EMT activities. Interestingly, IKM5 disrupts the interaction between GRP78 and TIMP-1 by inhibiting GRP78 in a Par-4-dependent manner. Moreover, IKM5 inhibited tumor growth and lung metastasis at a safe dose of 30 mg/kg/body weight.
Conclusion
Our study warrants IKM5, a potential anticancer agent that can abrogate invasion and metastasis, suggesting its clinical development for the treatment of patients with advanced breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most devastating type of cancer among women in developed countries. More than 90% of the breast cancer-related deaths occur due to its local invasion and metastasis to distant organs like, lung, liver, and brain [1,2,3]. Glucose-regulated protein, 78 kDa (GRP78), is an endoplasmic reticulum (ER) stress-regulated protein overexpressed in almost all types of cancer including breast cancer. It escapes cancer cells from undergoing apoptosis and promotes invasion, metastasis; thus, facilitating cancer progression and chemoresistance [4, 5]. Therefore, GRP78 confers a promising therapeutic target for anticancer drug discovery in academia as well as industry. GRP78 has been well documented for promoting invasion and metastasis in most cancers through facilitating the matrix metalloproteases (MMP-2 and MMP-9) and at the same time suppressing the cell-to-cell adhesion molecule E-cadherin; whereas knockdown of GRP78 significantly affects the extracellular matrix degradation, invasion, and metastasis, ensuring attenuated MMP-2 and MMP-9 levels in these cells [6, 7]. Strikingly, GRP78 also favors epithelial–mesenchymal transition (EMT), an early step of cancer metastasis, characterized by prominent mesenchymal features with decreased cell-to-cell adhesion, increased cell scattering and migration [8, 9].
Kojic acid (KA) (5-hydroxy-2-hydroxymethyl-4H-pyran-4-one) is a metabolic by-product produced by several fungal species like; Aspergillus, Acetobacter, Penicillium etc., which is known for its cytotoxic, anti-tumor, antibacterial, anti-inflammatory, and antioxidant activities [10]. KA and its derivatives are known to inhibit cell proliferation by cell cycle arrest (sub-G1) and DNA fragmentation by blocking poly (ADP-ribose) polymerase and caspase cleavage [11]. A study by Yoo et al. reveals that KA derivatives modulate gliomas cell proliferation and Toll-like receptor 4 (TLR4)-mediated functional activation of macrophage-managed tumor microenvironments. These derivatives also attenuate lipopolysaccharide (LPS)-induced nitric oxide production and Interleukin-6 (IL-6) expression [12]. KA also showed its efficacy in skin cancer model wherein the proteomic analysis of the A375, human malignant melanoma cells revealed that KA treatment modulates the expression of various proteins including GRP75, VIME, and 2AA that leads to suppression of melanogenesis and tumorigenesis as well [13]. However, KA lacks potency and target specificity in its anti-tumor activity; hence, better semi-synthetic derivatives of KA could amplify its therapeutic effect.
The present study uncovers the discovery and development of a novel, potential Indolylkojyl methane analogue (IKM5) chemically evolved from less potent natural product KA. The IKM5 abrogates breast cancer proliferation, invasion, and metastasis much more efficiently than KA with excellent in vivo efficacy as well as pharmacokinetics, which are the main themes of this study.
Materials and methods
Chemistry of IKM5
IKM5 (2-(1-(1H-indol-3-yl)octyl)-3-hydroxy-6-(hydroxymethyl)-4H-pyran-4-one), an Indolylkojyl methane analogue was synthesized and characterized in-house in the Natural Product Chemistry Division, CSIR-IIIM, Jammu. Briefly, a mixture of Kojic acid (1 equiv.), octanal (1.2 equiv.), indole (1 equiv.), and Fe–Al pillared clay catalyst calcined at 425 °C (0.5 mol%) in a test tube was heated with stirring at 90 °C for 2 h. After cooling, ethyl acetate was added to the reaction mixture, filtered, and washed (2 × 20 mL) to recover the catalyst. The filtrate was purified by column chromatography (methanol: dichloromethane 0.5:9.5) to afford the pure product. The synthesis procedure, IUPAC name, molecular weight through HRMS, 1H NMR, and 13C NMR characterization and spectra are provided in supporting information file.
Cell culture and reagents
Human breast cancer cell lines used in this study: MDA-MB-231, MDA-MB-468, MCF7, BT474, T47D, and human normal breast epithelial (fR2) cells were obtained from European Collection of Cell Cultures (ECACC). Mouse mammary carcinoma 4T1 cells were the generous gift from Dr. Avinash Bajaj, Regional Centre for Biotechnology, New Delhi, India. MDA-MB-231 cells were grown in L-15 medium and all other cell lines were cultured in RPMI 1640 medium supplemented with 10% FBS (Gibco), 1% penicillin–streptomycin (Sigma) at 37 °C in a humidified incubator (Eppendorf) with 5% CO2. Cells were sub-cultured in each 2–3 days to maintain them in a healthy condition.
Doxorubicin, 6-Thioguanine, paraformaldehyde, phenylmethylsulfonyl fluoride (PMSF), 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), Bradford’s reagent, crystal violet, DMSO, and protease inhibitor cocktail were obtained from Sigma Chemicals. Recombinant human fibroblast growth factor basic, 146aa (bFGF), was purchased from R&D systems (Minneapolis, MN). Fluorescein-5-isothiocyanate (FITC) was procured from Thermo Fischer Scientific, Massachusetts, USA.
Cell viability assay
The cell viability of MDA-MB-231, MDA-MB-468, MCF7, T47D, and fR2 cells was determined in the presence of KA, IKM5, and/or doxorubicin by standard MTT assay method according to the procedure previously described [14, 15].
Automated docking
The crystallographic structure of GRP78 protein bearing PDBID: 3IUC [16] was modified and energy minimization was performed using Swiss PDB viewer [17]. The structure was utilized to study the binding modes of KA and its synthesized variant IKM5. The two compounds were docked into the active site of GRP78 employing AUTODOCK 4.2 tool [18]. The Lamarckian algorithm of the tool was used for the automated docking and calculations of the binding energies. The top five binding modes of the two compounds were analyzed using LIGPLOT+ [19], the interactions were further studied using Pymol [20].
Colony formation and cell scattering assay
Experiments were performed in MCF7, MDA-MB-468, and MDA-MB-231 cells according to the pre-standardized published protocol [21]. For cell scattering assay, bFGF (20 ng/mL) was added to the culture medium to stimulate cell scattering. At least three random colonies from each field were observed under an inverted microscope, counted manually for the number of scattered cells, and photographed at ×20 magnification. The number of scattered cells out of a colony was adjusted to the total cells in that particular colony [22].
Matrigel invasion assay
The effects of IKM5 on invasion capability of MDA-MB-231 cells was evaluated by means of BD Biocoat Tumor Invasion Assay System (BD Bioscience, Bedford, MA, USA) according to the manufacturer’s instruction and our pre-standardized protocol [21, 22].
Invadopodia formation/in situ fluorescent gelatin degradation assay
Matrix degradation ability of invasive MDA-MB-231 cells by invadopodia formation was assessed in the presence of KA or IKM5 as described previously [21, 22]. Imaging of the degraded area/podosomes was carried out with the help of fluorescent microscope (Floid Cell Imaging Station, Life Technologies) at ×20 magnification.
Cell migration assay
MDA-MB-231 cells were examined for their migration capabilities in the presence or absence of vehicle, KA, or IKM5 by means of scratch-motility wound healing assay as per our previously published protocol with minor modifications [14, 21]. Wounded areas were progressively photographed under an inverted microscope with Nikon D3100 camera at ×20 magnification.
Western blotting
Western blotting analyses were carried out with the indicated conditions (mentioned in respective figure legends) according to the pre-standardized and published protocol [21, 23]. The list of antibodies and their working dilutions are provided in Supporting Table S1.
Immunofluorescence analysis
MCF7 and MDA-MB-231 cells were seeded in eight-well chamber slides and treated with indicated agents (please see figure legend). Immunocytochemistry experiments were performed as described previously [21, 22]; list of antibodies and working dilutions are provided in Supporting Table S1. Cells were observed under Floid Cell Imaging Station and images were captured at ×20 magnification.
Transient transfection
The procedure was followed as previously described by our group with minor modifications [15, 22]. Briefly, MDA-MB-468 cells (0.5 × 106 per well) were cultured in 6-well tissue culture plates for 24 h and then transiently transfected with vector or pCMV BiP-Myc-KDEL-wt plasmid construct (Addgene) using Lipofectamine-3000 (Invitrogen, NY, USA) as per the manufacturer’s protocol. Twenty-four hours post-transfection, cells were treated with vehicle or IKM5 for 24 h, and whole-cell lysates were prepared and subjected to western blot analysis.
Co-immunoprecipitation analysis
The experiment was performed on MDA-MB-231 cells with indicated treatments following our standardized protocol described earlier [22, 24].
Preparation of cytoplasmic and nuclear extracts
Cytoplasmic and nuclear fractions were obtained according to the procedure described previously by our group [21]. Briefly, MDA-MB-231 cells following treatment with IKM5 were harvested, washed with ice cold PBS and centrifuged. Cell pellets were then treated with ice cold hypotonic and hypertonic buffers to separate out the cytosolic and nuclear extracts, which were then subjected to western blot analysis to check the expression of various proteins.
siRNA knockdown studies
Small interfering RNA (siRNA) oligonucleotide duplexes targeted against human Par-4 was procured from Sigma-Aldrich. Briefly, MDA-MB-231 cells were seeded in a 90 mm petri dishes and after 24 h transiently transfected with scramble/siRNA Par-4 using Lipofectamine 3000 following the manufacturer’s protocol. After 24 h, the cells were subjected to treatment with vehicle/IKM5; 24 h of post-treatment, cells were harvested and the samples were processed for immunoprecipitation.
Mouse model for tumor growth and metastasis
The experiment was performed according to our established protocol with some modifications [22]. To evaluate the in vivo anti-tumor and anti-metastatic efficacy of KA and IKM5, healthy female Balb/c mice (b.w. 18–23 g) were group-housed under conditions of constant photoperiod (12 h light/12 h dark) with ad libitum (free access to sterilized food and water). Proper care was taken to maintain them in a healthy condition and to avoid any risk of possible pathogenic contaminations. All the in vivo experimental protocols were approved by the Institutional Animal Ethics Committee, CPCSEA, of Indian Institute of Integrative Medicine, Jammu, India. For tumor implantation, the mice were randomized into three groups of 5 mice per group and 1.5 × 106 4T1 cells per 200 µL of serum free RPMI media was injected subcutaneously into the mammary fat pad of each mouse adjacent to the right second mammary gland. A week after the tumor cell implantation or when the tumor volume attained the size of 100–150 mm3, mice were injected intraperitoneally with vehicle (normal saline) or KA (100 mg/kg/b.w.) or IKM5 (30 mg/kg/b.w.) in each alternate day for two weeks. Tumor size was recorded on each alternate day after implantation and body weights were recorded once per week. Mice were sacrificed in a humane way on the 15th day of treatment initiation and tumors were dissected out carefully for its measurement. Lungs of each animal were dissected out and metastatic lung nodules were counted and photographed under a dissecting microscope. Lungs were then minced into fine pieces, suspended in collagenase/DNase solution and incubated in a 37 °C incubator with vigorous shaking for 2 h. Each lung suspension was then passed through the 70 μm cell strainer (BD Biosciences, USA) to get the single cell suspension. Then, the suspension was serially diluted for 3–4 times in selection medium containing 6-thioguanine and incubated at 37 °C and 5% CO2 for the growing of 4T1 cell colonies. After 15 days of incubation, cells growing in the colonies were washed with PBS, fixed with methanol, and stained with 0.2% crystal violet solution for 1 h. The colonies were then observed, counted, and photographed under an inverted microscope at ×10 magnification.
Pharmacokinetic (PK) study
Healthy male Balb/c mice (25–30 grams each) were chosen to access the PK profile of IKM5. There were 11 time points for blood collection and six animals were there in each sampling time point for maintaining accuracy of the experiment. Animals were administered with a single dose of IKM5 (2.5 mg/kg, b.w.) intravenously and control mice were administered with normal saline. A minimum of 100 µL of plasma was collected from each animal at different time points, viz: (0.083, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 16, and 24 h) following treatment with IKM5 and successively 400 µL of acetonitrile was added to precipitate the plasma proteins. The compound was extracted into the solvent, filtered, and analyzed by a 6410B Triple quadrupole LC–MS/MS system (Agilent Technologies, USA). Quantification was performed through multiple reaction monitoring (MRM) separately for all the six samples collected individually at each time point by comparing with the standard calibration curve prepared with the help of Agilent Mass Hunter software (version B.04.00) (see supporting information file). Various PK parameters viz. half life (t1/2) (h), C initial C0 (ng/mL), area under the curve AUC(0–∞) (ng*h/mL), Vd (L/kg), Cl (L/h/kg), and Mean Residence Time (h) were calculated using non-compartmental pharmacokinetic data analysis.
In vitro hemolysis assay
The assay was performed according to the method previously reported and standardized by our group [25]. Briefly, fresh blood was collected from male Balb/c mice and washed three times with isotonic 0.01 M phosphate buffer saline (PBS, pH 7.4) solution by centrifugation at 4000 rpm for 5 min. The red blood cells (RBCs) suspension (0.1 mL) was added with PBS solution (0.9 mL) containing various concentrations (10, 20 and 30 µg/mL) of IKM5. The blank PBS and distilled water were treated as positive and negative control, respectively. The samples were incubated at 37 ± 1 °C for 30 min in shaker incubator, centrifuged at 14,000 rpm for 10 min, and the supernatants were analyzed for absorbance at 540 nm in TECAN microplate reader (Tecan, Infinite M200 Pro, Austria). The percent hemolysis was calculated using the following formula:
where ABS, ABS100, and ABS0 are the absorbance of the samples, negative control (100% hemolysis) and positive control (0% hemolysis), respectively.
Drug potentiation assay
The study was carried out according to the standard procedure with some modification [26]. MDA-MB-231, MCF7, and MDA-MB-468 cells were exposed to IKM5 (200 nM), doxorubicin (200 nM) and in combination of both (1:1 ratio) for 48 h to examine cell viability via MTT method, for 5 days to evaluate colony formation ability and subjected to western blotting for the expression of GRP78 and Par-4. The ability of IKM5 to enhance cell killing was expressed as potentiation index (PI), which is the ratio of the % inhibition of IKM5 alone at the given dose and the % inhibition in combination with doxorubicin. Thus, PI > 1 indicates potentiation and PI < 1 indicates protection.
Statistical analysis
All the data were expressed as mean ± S.D. of at least three independent experiments performed. IC50 values were determined with the help of GraphPad Prism software Version 5.0 (GraphPad Software, Inc., USA) by considering log of inhibitor versus response. For the pharmacokinetic study, a one way ANOVA was used on the values obtained from LC–MS analysis of plasma drug concentrations. Comparisons used Student’s t test and a two sided value of *P < 0.05 was assigned significance.
Results
IKM5, compared to its parent compound KA efficiently abrogates proliferation, invasion, and motility of human breast cancer cells
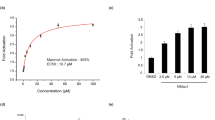
IKM5 (2-(1-(1H-indol-3-yl)octyl)-3-hydroxy-6-(hydroxymethyl)-4H-pyran-4-one) is a potential lead molecule grew out of the strategic semi-synthetic modification of its parent molecule KA (Fig. 1a) (Supporting information file). A patent application has been filed in India, United States, and European countries (International application number: PCT/IN2018/050060) for its superior anti-tumor and anti-metastatic efficacy against breast cancer in comparison to KA. The cytotoxic profile of IKM5 in a panel of breast cancer cell lines revealed that the molecule is modestly active in a lower nanomolar to micro-molar concentration having IC50 values 0.150 ± 0.095, 0.209 ± 0.265, 0.54 ± 0.315 and 3.45 ± 0.255 µM in MCF7, MDA-MB-231, MDA-MB-468, and T47D cells respectively, whereas, the analogue was negligibly toxic towards normal human breast epithelial (fR2) cells (IC50: 74± 2.6) indicating the molecule’s selectivity towards breast cancer cells (Fig. 1b; Supporting Table S2). Next, we examined the effects of IKM5 on proliferation, invasion and migration capabilities of breast cancer cells and the results showed that IKM5 at its sub-toxic concentration (200 nM) abrogated colony formation/proliferation ability of MCF7 and MDA-MB-468 cells substantially in comparison to vehicle (DMSO). However, there was hardly any inhibition of colony formation in presence of parent KA (100 µM) treated cells (Fig. 1c, d). To further envisage the effects of this lead molecule on motility related invadopodia formation ability of aggressive MDA-MB-231 cells, our pre-standardized FITC-gelatin degradation assay unveiled massive degradations on fluorescent gelatin matrix (mimics to the extracellular matrix degradation in physiological condition) in vehicle and KA-treated cells, whereas, IKM5 (200 nM) significantly abrogated the degradation capability of these cells (Fig. 1e, f). We also checked the Matrigel invasion ability through Boyden chamber assay system and the results depicted major inhibition in the invasion capabilities of MDA-MB-231 cells in IKM5-treated wells (Fig. 1g, h). Notably, IKM5 suppressed migration of MDA-MB-231 cells and blocked the bFGF-induced stimulation of cell scattering in MCF7, MDA-MB-231, and MDA-MB-468 cells (Supporting Fig. S1; Fig. 1i, j). These results demonstrate that IKM5, a potential analogue of KA, effectively suppresses cell proliferation, invasion, and migration in breast cancer cells.

IKM5 abrogates proliferation, invasion, and migration abilities in breast cancer cells. a Structure of Indolylkojyl methane analogue (IKM5). b Graph showing the % cell viability in MDA-MB-231, MCF7, MDA-MB-468, T47D, and fR2 cells treated with logarithmic concentrations of IKM5 for 48 h. c Colony formation assay showing the cell proliferation abilities of MCF7 cells exposed to vehicle or KA or IKM5 at indicated concentrations for five days. d Bar graph represents average number of colonies formed in MCF7 and MDA-MB-468 cells in each treatment conditions as quantified from five random fields (n = 3, error bars indicate ± s.d.). **P < 0.01. e MDA-MB-231 cells were treated with indicated concentrations of KA and IKM5 for 48 h and checked for their ability to degrade the gelatin matrix/invadopodia formation over the FITC-gelatin coated coverslips. Blue parts indicate nuclear staining through DAPI mounting media. Images were captured under Floid Cell Imaging Station at ×20 magnification. The threshold area of degradation was determined with the help of Image J software analysis. f Bar graph showing the percent area of degradation as determined through Image J analysis (n = 3, error bars indicate ± s.d.). **P < 0.01. g MDA-MB-231 cells were checked for their ability to invade through the Matrigel barrier in Boyden chamber invasion assay system in the presence or absence of indicated concentrations of KA and IKM5. h Bar graph showing the quantification of number of invading cells (n = 3, error bars indicate ± s.d.). **P < 0.01. i MDA-MB-231, MCF7 and MDA-MB-468 cells were checked for their cell scattering ability in the presence of bFGF (20 nM) alone or bFGF (20 nM) plus IKM5 (200 nM) for 48 h. Cell scattering was observed and photographed under an inverted microscope. Original magnification × 20, scale bar: 50 µm. j Bar graphs showing the quantification of number of scattered cells in each treatment conditions adjusted to the total cells in individual colonies. (n = 3, error bars indicate ± s.d.). *P < 0.05
IKM5 abrogates the GRP78 expression and binds to GRP78 at the molecular level
The expression of GRP78 is considered as a biomarker for the cancer cell’s invasion and metastasis in majority of human cancers including breast cancer [4]. Therefore, we were interested to check the protein expression profile of GRP78 after exposure of breast cancer cells to the IKM5. Interestingly, our western blotting results unveiled that increasing concentrations of IKM5 (50, 100, and 200 nM) for 48 h steadily downregulated the expression of GRP78 in MDA-MB-231, MDA-MB-468, and MCF7 cells. However, we observed biphasic-type of expression of GRP78 in MDA-MB-231 and MDA-MB-468 cells, but the expression was almost diminished at the higher concentrations (Fig. 2a, b). Time-dependent western blot analysis revealed that GRP78 expression was gradually decreased after 6 h exposure of compound IKM5 and diminished at 48 h time point in MDA-MB-231, MDA-MB-468, and MCF7 cell lines (Fig. 2c, d). Further, our immunocytochemistry results implied sufficient cytoplasmic expression and membranous localization of GRP78 in vehicle treated MCF7 cells, whereas, the expression level and cell surface localization was greatly impaired in IKM5-treated MCF7 and MDA-MB-231 cells (Fig. 2e; Supporting Fig. S2). To examine whether IKM5 was interacting directly with GRP78 to exert its inhibitory effects, automated docking analysis was carried out at atomic level. The top five binding modes, their respective binding energies, and inhibition constants were shown in Supporting Fig. S3. The top ranked pose of both the compounds under investigation were further analyzed and Supporting Table S3 shows the data retrieved from them. The ligand-receptor interactions of KA (non-covalent and hydrophobic interactions) were displayed in Fig. 2f. The parent compound (KA) showed a ΔG of − 5.06 kcal/mol and formed eight hydrogen bonds with LYS96, THR37, THR38, GLY227, GLY228, and THR229 of the drug binding pocket. The hydrophobic interacting amino acids of the binding pocket are GLY36, GLY226, and GLU201. The oxygen atoms at the 1st, 7th, 8th, and 10th position of the parent compound were forming the physical non-covalent interactions with the target protein. The in-house synthesized compound IKM5 showed more spontaneous binding with GRP78 protein with ΔG of − 8.03 kcal/mol. Its binding pocket comprises of a total of fourteen amino acids, three out of which (LYS81, LYS296, and ARG297) were forming four hydrogen bonds with the protein. Figure 2g illustrates the three dimensional binding mode of IKM5, the three oxygen atoms at 7th, 8th, and 10th position and the nitrogen atom at 21st position were forming physical non-covalent interactions with the target protein. It’s in silico calculated inhibition constant (Ki) of 1.35 µM is almost 150 times lesser than the KA’s inhibition constant, i.e., 195.08 μM. The in silico insight is giving a clear indication of the in-house synthetic variant of KA being a better inhibitor for targeting GRP78 protein. Together, these results collectively suggest that IKM5 binds to and down-modulates the expression of GRP78 in breast cancer cells.

IKM5 downregulates the protein expression and binds to GRP78 in breast cancer cells. a MDA-MB-231, MDA-MB-468 and MCF7 cells were treated with indicated concentrations of IKM5 for 48 h; whole-cell lysates were prepared and subjected to western blotting for the expression of GRP78. The expression of β-actin was considered as loading control in each western blot experiments performed. b Bar graph showing the relative protein expression of GRP78 as determined by densitometry analysis of bands normalized to the loading control. c Time-dependent expression of GRP78 in MDA-MB-231, MDA-MB-468 and MCF7 cells treated with IKM5 (200 nM) for indicated time points. d Bar graph showing the relative protein expression of GRP78 as determined by densitometry analysis of bands normalized to the loading control. e Immunocytochemistry experiments depicting the expression levels of GRP78 in MCF7 cells after exposure to IKM5 for 48 h. Original magnification ×20. Insets showing magnified view of membrane localization. f, g, In silico AutoDock analysis showing the three dimensional representation of the top poses of binding of KA and its synthesized variant (IKM5) respectively inside the binding pocket of GRP78. The red lines represent the hydrogen bonds. The naked amino acids and the ones with transparent surface symbolize the hydrogen forming and hydrophobic interacting amino acids, respectively
IKM5 negatively regulates matrix metalloproteases (MMPs) and abrogates the expression of EMT-associated markers
To further investigate the molecular mechanism underlying the inhibitory effect of IKM5 on invasion of breast cancer cells, we examined the expression profile of matrix metalloproteases (MMP-2, MMP-9) and associated EMT markers. Our western blotting results clearly showed gradual decrease in expression of MMP-2 and MMP-9, whereas, expression of its physiological negative regulator (TIMP-1) was amplified significantly (> 12-fold) in aggressive MDA-MB-231 cells (Fig. 3a, b). We also examined the effect of IKM5 on HER2+ breast cancer cell line BT474 expressing high levels of GRP78; the results depicted strong inhibition of GRP78 and robust induction of TIMP-1 expression in BT474 cells treated with increasing concentrations of IKM5 for 48 h (Supporting Fig. S4). While investigating the effects of IKM5 on EMT-related markers regulating MMPs, we noticed significant reduction in the expression of Twist1 and Vimentin, along with striking upregulation of epithelial marker E-cadherin (Fig. 3a, b). Since GRP78 plays an important role during ER stress, so we wanted to verify the effects of IKM5 on other ER-stress markers; viz: activating transcription factor 6 (ATF6), inositol requiring enzyme 1 (IRE1) and protein kinase R-like endoplasmic reticulum kinase (PERK). Of note, there was a marked decrease in the expression of these sensor proteins validating that the EMT-inhibiting ability of IKM5 is mutually exclusive to ER-stress activation (Fig. 3c).

IKM5 regulates MMPs and EMT-related markers. a MDA-MB-231 cells were exposed to increasing concentrations of IKM5 for 48 h; whole-cell lysates were prepared and subjected to western blotting analysis for checking the expression of MMP-2, MMP-9, TIMP-1, Twist1, Vimentin, and E-cadherin. The expression of β-actin was checked as a loading control. b Bar graph showing the densitometry analysis of western blots bands after normalization with β-actin expression. c The protein levels of PERK, ATF6, and IRE1 were examined through western blotting using whole-cell lysates of MDA-MB-231 cells exposed to increasing concentrations of IKM5 for 48 h. d Co-immunoprecipitation analysis results depicting that IKM5 disrupts GRP78-TIMP-1 interaction in invasive breast cancer cells. MDA-MB-231 cells were treated with vehicle or IKM5 for 48 h, whole-cell lysates were prepared and subjected to immunoprecipitation with GRP78 and TIMP-1 antibody. The immunoprecipitates and inputs were employed for western blotting analysis to check the expression of GRP78 and TIMP-1
In order to delineate how this inhibition of GRP78 by IKM5 is associated with abrogation of MMPs-mediated invasion and migration in aggressive breast cancer cells, we hypothesized that GRP78 might be involved in some inhibitory interaction with TIMP-1 to facilitate MMPs’ activity. Certainly, our co-immunoprecipitation analysis results uncovered that IKM5 disrupted an important interaction between GRP78 and TIMP-1 consistently in these cells. Indeed, we pulled down sufficient TIMP-1 protein co-immunoprecipitate with GRP78 in vehicle treated MDA-MB-231 cells, whereas, hardly any interaction was observed between GRP78 and TIMP-1 in IKM5 (200 nM) treated cells due to inhibition of GRP78 (Fig. 3d). Collectively, these results imply that IKM5 negatively regulates MMPs as well as EMT-related markers and disrupts the interaction between GRP78 and TIMP-1 in breast cancer cells.
IKM5 induces intracellular Par-4 expression and promotes its nuclear translocation
Prostate apoptosis response 4 (Par-4) is a prominent pro-apoptotic/tumor suppressor protein that resides in the cytoplasm and in nucleus as well [27]. Through its SAC domain (selective for apoptosis in cancer), Par-4 selectively induces apoptosis in cancer cells (not in normal cells) by landing into the nucleus and suppressing NF-kB activity [28]. It is previously elicited that endogenous Par-4 binds to/interacts with GRP78 and this GRP78-Par-4 complex further translocate to the cell surface to activate extrinsic apoptotic pathways in ER-stressed cancer cells [29]. However, there also observed inverse correlation between the expression patterns of GRP78 and Par-4 in cancer cells treated with small molecules/anticancer agents [14]. To examine the effect of inhibition of GRP78 on intracellular Par-4 (one of the major binding partners of GRP78 in ER-stressed cancer cells) level, we carried out western blotting analysis with the whole-cell lysates of IKM5/vehicle treated MDA-MB-231 cells. The results showed that increasing concentrations of IKM5 stimulated the expression of Par-4 in aggressive MDA-MB-231 cells and as a consequence of which NF-kB (p65) expression diminished in a concentration dependent manner (Fig. 4a). IKM5 also blocked the phosphorylation of ERK (upstream regulator of Par-4) at its sub-toxic concentration and suppressed the expression of total ERK1/2 in MDA-MB-231 cells (Fig. 4a). To further investigate the localization of Par-4 following treatment with IKM5 (200 nM), we separated cytosolic and nuclear fractions from the whole-cell lysates of MDA-MB-231 cells and conducted western blotting. Interestingly, while we noticed 2.2-fold increase in the level of cytosolic Par-4, a sharp increase in Par-4 (8.5-fold) was achieved in the nuclear fractions of cells treated with IKM5 (200 nM) compared to the vehicle treated cells (Fig. 4b, c). Additionally, our immunocytochemistry results also confirmed steady increase in the nuclear localization of Par-4 in IKM5 treated MCF7 and MDA-MB-231 cells compared to the vehicle treated cells (Fig. 4d). Since we achieved a marked upregulation in the Par-4 protein levels in IKM5-treated samples; we wanted to investigate whether the IKM5-mediated disruption of GRP78/TIMP-1 interaction was Par-4-dependent. We, therefore, carried out the Par-4 siRNA (siPar-4) knockdown studies and performed immunoprecipitation analysis with TIMP-1 antibody. The results unveiled an intact interaction between GRP78 and TIMP-1 in the absence of Par-4 (siPar-4/siPar-4 + IKM5) condition while the interaction was hindered in the presence of Par-4 (scramble/scramble + IKM5) (Fig. 4e). Further, to investigate whether the overexpression of GRP78 could rescue the effect of IKM5, we transiently transfected aggressive MDA-MB-468 cells with pCMV BiP-Myc-KDEL-wt plasmid construct carrying the gene for expression of GRP78. Our results unveiled that overexpressed GRP78 hampered the inhibitory effect of IKM5 on GRP78 and its ability to induce Par-4 in these cells (Fig. 4f, g). Together, these results demonstrate that IKM5 induces the expression, promotes nuclear localization and regulates upstream/downstream signaling of Par-4 in breast cancer cells. Also, GRP78 overexpression nullifies the effect of IKM5 in invasive breast cancer cells.

IKM5 induces the expression of Par-4 and promotes its nuclear localization. a MDA-MB-231 cells were treated with indicated concentrations of IKM5 for 48 h and the whole-cell lysates were immunoblotted for the expression of Par-4, NF-kB, pERK, and ERK1/2. b MDA-MB-231 cells were treated with vehicle and IKM5 (200 nM) for 48 h; nuclear and cytosolic extracts were prepared according to the procedure described in method section and subjected to western blotting for the expression of Par-4. The expressions of lamin and β-actin were considered as loading controls for nuclear and cytosolic fractions respectively. c Bar graphs indicate densitometry analysis of the bands of Par-4 in nuclear and cytosolic fractions. (n = 3, error bars indicate ± s.d.). d Immunocytochemistry experiments showing the expression levels/nuclear localization of Par-4 after MCF7 and MDA-MB-231 cells were treated with IKM5 for 48 h. Original magnification ×20. e Immunoprecipitation analysis results depicting that IKM5 disrupts GRP78-TIMP-1 interaction in a Par-4-dependent manner. MDA-MB-231 cells were transiently transfected with scramble or siPar-4 and/or treated with IKM5 for 48 h; whole-cell lysates were prepared and processed for immunoprecipitation with TIMP-1 antibody. The immunoprecipitates were employed for western blotting analysis to check the interaction between GRP78 and TIMP-1. f MDA-MB-468 cells were either transiently transfected with vector, GRP78 (pCMV Bip-Myc-KDEL-wt) or treated with IKM5, and/or GRP78 transfection plus IKM5 treatment for 48 h; whole-cell lysates were employed for western blotting to check the expression of GRP78 and Par-4. Beta actin expression was considered as endogenous loading control. g Bar graphs showing the relative protein expression of GRP78 and Par-4 as determined by densitometry analysis of the western blot bands
Toxicity determination, pharmacokinetic assessment, and evaluation of in vivo efficacy of IKM5 on tumor growth and metastasis
Given that some established anticancer drugs like cisplatin and carboplatin cause hemolytic anemia when administered through intravenous route of administration [30], we sought to determine the cytocompatibility, endosmolytic activity of IKM5, and to ascertain whether the molecule is suitable for intravenous administration. We conducted in vitro hemolysis assay by taking various concentrations of IKM5 (10, 20, 30 µg) along with distilled water (negative control) and PBS (positive control). The compound showed slight hemolysis (8.01%) even at its higher concentration (30 µg/mL), indicating IKM5 is non-toxic, cytocompatible, and a suitable candidate for the intravenous formulation (Fig. 5a, Supporting Fig. S5). Next, to check the exposure of the compound in healthy animals, we evaluated the pharmacokinetic profile of IKM5 through intravenous (i.v.) route of administration in Balb/c mice. Following i.v. administration of a single dose of 2.5 mg/kg b.w., the C0 value of 1215.67 ng/mL for the molecule was achieved with an AUC0–∞ value of 406.31 ng*h/mL and t1/2 was 0.43 h. The volume of distribution (Vd) and clearance (Cl) was found to be 7.61 L/kg and 12.31 L/h/kg respectively with mean residence time of 0.45 h (Table 1, Fig. 5b). The pharmacokinetic parameters of IKM5 further divulge good absorption and bio-distribution capability of this lead molecule, which are the essential criteria for drug development.

Pre-formulation, pharmacokinetic study, and in vivo evaluation of IKM5 in inhibiting tumor growth and lung metastasis. a RBC suspension was prepared from the fresh blood collected from male Balb/c mice and incubated with increasing concentrations of IKM5 for 30 min. Hemolysis was checked, compared with PBS (positive control) and distilled water (DW) (negative control). The percent hemolysis was calculated by taking the absorbance of hemoglobin (supernatant after centrifugation) at 540 nm. (n = 2, error bars indicate ± s.d.). b Graph showing the mean plasma concentration versus time curve of IKM5 (2.5 mg/kg/body weight) after intravenous administration in Balb/c mice. c Effect of vehicle (normal saline), KA (100 mg/kg, b.w.), and/or IKM5 (30 mg/kg, b.w.) on tumor growth was studied in 4T1 mouse mammary carcinoma model. d Tumor growth curves showing the quantification of tumor volume measured with the help of a Vernier caliper in regular time intervals. Error bars indicate ± s.d. in tumor volume. *P < 0.05, **P < 0.01. e Lungs of the animals were dissected carefully and observed under an inverted microscope for the formation of metastatic lung nodules and photographed with the help of a NIKON camera (NIKON, D3100). f Bar graph showing the quantification of metastatic lung nodules counted manually from the lungs dissected out from each animal. Error bars indicate ± s.d. **P < 0.01. g Bar graph showing the quantification of 4T1 colonies isolated from the lung suspension of animals from each treatment groups. Error bars indicate ± s.d. **P < 0.01. h Western blotting analysis with the tumor tissue samples from vehicle, KA and IKM5 treated group of animals showing protein expressions of GRP78, Par-4, TIMP-1, and MMP-2. Beta actin expression was considered as loading control. i Bar graph showing relative protein expression of the respected proteins as determined by densitometry analysis of the western blot bands. Error bars indicate ± s.d. *P < 0.05, **P < 0.01
In order to evaluate the in vivo efficacy of IKM5 on the inhibition of tumor growth and metastasis, highly aggressive 4T1 metastatic mouse mammary carcinoma model was employed. IKM5 (30 mg/kg/body weight) treated group of animals showed steady inhibition of tumor growth; there observed 79.2% inhibition in tumor volume at the end point (15th day of treatment initiation) compared to the control/normal saline treated group, whereas, no significant inhibition in tumor volume (23%) in KA (100 mg/kg/body weight) treated group was noticed (Fig. 5c, d). The formation of metastatic lung nodules also steadily abolished (84.5%) in IKM5 treated animal group compared to the KA-treated group, which rendered only 16.7% decrease in metastatic lung nodule formation (Fig. 5e, f). In addition, the colony formation ability of 4T1 cells isolated from the lung suspension of IKM5 treated groups of animals halted drastically compared to the vehicle and KA-treated group (Fig. 5g). Of note, the animals remained healthy throughout the experimental period without any lethality or adverse effects. Further, to validate the signaling changes occurring at the protein level for producing the anti-tumor and anti-metastatic effect of IKM5, we conducted western blotting analysis with the tumor tissue samples isolated from the respective treatment groups of animal. Interestingly, we noticed tremendous downregulation in the protein levels of GRP78 and MMP-2 concomitant with substantial induction of Par-4 and TIMP-1 expression in tumor samples from IKM5-treated group of animals compared to the vehicle and KA-treated group (Fig. 5h, i). Collectively, these results demonstrate that IKM5 is an efficacious lead molecule with negligible toxicity and good pharmacokinetic profile; the molecule inhibits in vivo mammary tumor growth and lung metastasis at a safe and tolerable dose of 30 mg/kg, b.w. much more efficiently than its parent molecule KA.
IKM5 pronounces the effects of doxorubicin against invasive breast cancer cells
Doxorubicin, a topoisomerase inhibitor chemotherapeutic drug, is clinically used to treat early stage, locally advanced, and metastatic breast cancer. The drug is also reported to induce Par-4 and suppresses breast cancer invasion [31, 32]. However, there is ample evidence of expression of GRP78 and doxorubicin resistance in breast cancer; GRP78 protects cancer cells from apoptosis induced by topoisomerase inhibitors and helps them to proceed for metastasis [33,34,35,36]. Therefore, the goal of this study was to curtail invasion in breast cancer and improve doxorubicin sensitivity by inhibiting GRP78. We sought to determine whether IKM5 could enhance the antagonistic effects of doxorubicin against breast cancer cells when treated in combination. In order to examine our hypothesis, MCF7, MDA-MB-231, and MDA-MB-468 cells were treated alone with either 200 nM of doxorubicin or IKM5 (200 nM) or combination of both the molecules in 1:1 ratio. The results showed an increase in sensitivity of doxorubicin: potentiation index (PI) value of 2.29 and 1.98 against MCF7 and MDA-MB-468 cells respectively, when treated in combination with IKM5 in a colony formation assay (Fig. 6a). We further confirmed this combined effect of both the molecules through cell viability assay, in which we found increased potency of doxorubicin; PI value: 1.52 and 1.65 in MDA-MB-231 and MDA-MB-468 cells respectively when treated in combination with IKM5 (Fig. 6b). Further, we checked the expression profile of GRP78 and Par-4 by western blot analysis of whole-cell lysates prepared from the above treatment with each of the molecules alone and in combination. Figure 6c indicates that the expression of GRP78 got severely attenuated and almost vanished, whereas, the expression of Par-4 enhanced robustly, when both IKM5 and doxorubicin were exposed in combination for 24 h compared to the cells treated with each of the molecules alone. Furthermore, densitometry analysis of western blots revealed 5.8-fold increase in the expression of Par-4 and 5.1-fold decrease in the expression of GRP78 in case of combination of both the molecules compared to 2.5-, 3.5-fold increase in Par-4 and 1.8-, 2.6-fold decrease in GRP78 expression in DOXO and IKM5 treatment alone, respectively (Fig. 6d). These results collectively enumerate that IKM5 significantly augmented the effects of doxorubicin in suppressing viability, proliferation, and invasion of breast cancer cells.

IKM5 enhances the effect of doxorubicin against invasive breast cancer cells. a Bar graph showing the number of colonies in MCF7 and MDA-MB-468 cells treated with either doxorubicin (200 nM) or IKM5 (200 nM) individually and/or combination of both (1:1 ratio) for 48 h (n = 3, error bars indicate ± s.d.) *P < 0.05, **P < 0.01. b Bar graph showing the percent cell viability in MDA-MB-231 and MDA-MB-468 cells as determined by MTT method after cells were treated with above treatment conditions for 48 h (n = 3, error bars indicate ± s.d.) *P < 0.05, **P < 0.01. c MDA-MB-231 cells were treated with the same above set of treatment conditions for 48 h; the whole-cell lysates were prepared and checked for the expression of GRP78 and Par-4. d Bar graphs showing the densitometry analysis of the GRP78 and Par-4 western blot bands presented in terms of relative protein expression. (n = 3, error bars indicate ± s.d.). *P < 0.05, **P < 0.01. e Schematic illustration of the proposed mechanism of action of IKM5. IKM5, a potential anti-proliferative and anti-metastatic lead molecule, binds to and inhibits the expression of GRP78 and its associated pro-metastatic genes (MMP-2, MMP-9, Twist1, Vimentin etc.) regulating EMT and invasiveness in breast cancer, whereas, the molecule upregulates the epithelial markers: E-cadherin and TIMP-1. Simultaneously, IKM5 induces the expression of Par-4 (possibly through inhibition of GRP78) and facilitates its nuclear localization, thereby suppressing NF-kB-mediated cell proliferation and invasion. Moreover, IKM5 showed excellent in vivo efficacy in inhibiting tumor growth and metastasis in mouse mammary carcinoma model
Discussion
GRP78, since its critical role in evading apoptosis, facilitating cancer cell proliferation, angiogenesis, tumor growth, and metastasis, has been emerging as a potential druggable target for the discovery of novel anticancer therapeutics [4,5,6,7, 15, 37]. Immunohistochemical as well as genome wide analysis on patient material have unveiled elevated levels of GRP78 in several tumors refractory to therapy like glioma [38], leukemia [39], prostate [40], and breast cancer [41]. Excess of GRP78 seems to be the key feature for cancer cells that unceasingly upregulate the UPR without, however, promoting apoptosis [42]. To override this GRP78 activation, our in vitro, and in vivo experimental approaches uncovered the detailed target based mechanism of action of the lead molecule-IKM5 that effectively suppressed GRP78 signaling in regulating tumor growth, invasion and metastasis in breast cancer.
A handful of natural products have been reported to influence the expression of GRP78 in diverse cancers. For example, -(-) epigallocatechin-3-gallate (EGCG), a green tea polyphenol abrogates the GRP78 induction, thus sensitizes hepatoma cells to chrysin-induced apoptosis [43]. Prunustatin A isolated from Streptomyces violaceoniger 4521-SVS3, found to be a novel inhibitor of GRP78 expression induced by 2-deoxyglucose in human fibro sarcoma (HT1080) cells without showing cytotoxicity in normal nutrient condition [44]. However, none of these inhibitors selectively exhibited their specificity towards GRP78 and therefore lack potency. Since indole group containing phytochemicals such as 3,3′-diindolylmethane (DIM) is known to influence ER-stress sensor GRP78 [45, 46], our recent report demonstrated that 3,3′-bisindolylmethane structural scaffold containing pharmacologically active lead molecule (compound 7d) inhibited cell proliferation, invasion, angiogenesis, and diminished the expression of prosurvival protein GRP78 [14, 15]. Keeping in mind, the importance of bisindolylmethane structural scaffold as inhibitor of GRP78, here we replaced one indole component of bisindolylmethane with KA in an attempt to improve the efficacy, selectivity, and safety profile of our molecule-IKM5. Moreover, our in silico simulation and docking studies demonstrate IKM5 binds to the GRP78 binding pocket more spontaneously with ΔG − 8.03 kcal/mol and inhibition constant (Ki = 1.35 µM), which is almost 150 times lower than the parent KA. This reveals high level of specificity of IKM5 towards GRP78.
GRP78 is basically an ER-resident chaperone protein, also found in mitochondria, cytosol, and nucleus in a cell type and context dependent manner [47]. Upon mild to moderate ER stress, a sub-fraction of GRP78 re-localize to the surface of specific cancer cells and activates oncogenic signaling forming complexes with different cell surface proteins [47,48,49]. In the present study, IKM5 inhibits the endogenous expression of GRP78 in MDA-MB-231, MDA-MB-468, MCF7, and BT474 cells as well as suppresses the activation of ER-stress sensor proteins such as PERK, IRE1, ATF6, as evident from our western blotting analysis. Additionally, IKM5 affected the membrane localization of GRP78 in MCF7 cells as revealed by our immunocytochemistry results. Hence, it can be demonstrated that IKM5 by depleting the cytosolic pool of GRP78 affects its membrane translocation and/or might be creating certain unfavorable environment for the cell surface localization of GRP78 to activate such oncogenic signaling.
The EMT process confers a gaining of invasive phenotype by the deemed to be aggressive cells as characterized by cytoskeletal changes, motility, migration, extracellular matrix degradation and loss of cell–cell adhesion [8]. All these processes also correlate with the changes in gene expression of certain transcription factors like: SNAIL, TWIST, ZEB as well as activation of Vimentin, MMPs, N-cadherin, and loss of E-cadherin [8]. Substantial recent literatures demonstrate that GRP78 is actively involved in favoring EMT and invasion, thus facilitating in cancer progression [5, 7]. Intriguingly, our lead molecule (IKM5) effectively halted the invasion and bFGF-stimulated cell scattering abilities of the breast cancer cells along with down modulation of mesenchymal markers such as MMP-2, MMP-9, Twist1, Vimentin, and simultaneous upregulation of epithelial markers TIMP-1 and E-cadherin (Fig. 6e). However, TIMP-1 has been well documented as an important negative regulator of MMPs. A stoichiometric balance is maintained between the MMPs (MMP-2, MMP-9) and their regulator TIMP-1; alterations in which leads to the downregulation of TIMP-1, upregulation of MMPs, and ultimately confers invasiveness in cancer cells [50, 51]. Although, TIMP-1 contributes controversial role in metastasis/angiogenesis, TIMP-1 binding proteins are known regulators of metastasis cascades [52]. Our co-immunoprecipitation results have clearly illustrated a sufficient GRP78-TIMP-1 binding in untreated cells, but treatment with IKM5 disrupted the GRP78-TIMP-1 complex formation. It could be possible that IKM5 could inhibit the cytosolic pool of GRP78 through direct binding to GRP78 and preventing its complex formation with TIMP-1. Though, there are substantial evidences showing the involvement of TIMP-1 in metastatic switch by inhibiting MMP-2 and MMP-9 [53], limiting factors could be the availability of substrate. The GRP78 might provide some conformational changes in TIMP-1 on binding to it and that complexed TIMP-1 might be insufficient for the cleavage of MMP-2 and MMP-9 to regulate metastasis. Our in vitro data in various aggressive breast cancer cell lines and data from tumor tissue samples rationally correlated the efficacy of IKM5 to enhance the functional redundancy of TIMP-1 by suppressing GRP78 from cytosol.
Prostate apoptosis response-4 (Par-4), a prominent pro-apoptotic leucine zipper protein, lands into the nucleus through its effector domain (SAC domain) and induces apoptosis by suppressing NF-kB activity [29]. The SAC domain of Par-4 also binds to GRP78 independently of its carboxy-terminal leucine zipper domain producing robust ER-stress response involving upregulation and trafficking of GRP78 [29]. Hence, it seems rational that inhibition of GRP78 might release sufficient intracellular Par-4 and facilitate its nuclear entry to block the NF-kB activity. Moreover, the IKM5-meditated tampering of GRP78-TIMP-1 complex was found to be Par-4-dependent. Our recent studies to understand the tumor suppressor functions of Par-4 have uncovered several novel approaches to modulate this vital protein to achieve desired therapeutic effects [21, 23, 47]. Our latest study in this aspect has reported that pharmacological induction of Par-4 can stimulate ALK-2/Smad-4 signaling to abrogate EMT and provoke mesenchymal–epithelial transition (MET) in aggressive pancreatic cancer cells [54]. Therefore, it is well documented that therapeutic modulation of tumor suppressor Par-4 can abolish cancer cell proliferation and invasion. NF-kB is well accepted to promote cell proliferation, EMT and metastasis, which is responsible for breast cancer progression [55]. Elevated levels of NF-kB activity are also associated with upregulation of mesenchymal markers like: Twist1 and Vimentin, and downregulation of epithelial markers like: E-cadherin [56]. Our in vitro experimental results rationally correlated the efficacy of IKM5 to induce endogenous Par-4 (due to suppression of GRP78) and promote its nuclear translocation to block NF-kB activity in breast cancer (Fig. 6e). The in vivo studies further demonstrated that IKM5 is an effective inhibitor of tumor growth and lung metastases at a safe and tolerable dose of 30 mg/kg body weight much more efficiently than KA. Finally, IKM5 possesses a good pharmacokinetic profile and its suitability for intravenous administration suggests the molecule for its therapeutic development.
Conclusions
Concisely, our study provides a novel pharmacological approach to abrogate GRP78-mediated mammary tumor growth and invasion while also enhancing the action of existing cytotoxic agents. Additionally, pharmacokinetic, pre-formulation, and in vivo studies unveil the efficient bio-distribution, safety as well as efficacy of IKM5 in inhibiting tumor growth and lung metastasis in mouse mammary carcinoma model. These findings necessitate IKM5 for its development as a potential therapeutic agent against metastatic breast cancer.
Abbreviations
- EMT:
-
Epithelial-mesenchymal transition
- GRP78:
-
Glucose-regulated protein 78
- MMP-2:
-
Matrix metalloproteinase 2
- TIMP-1:
-
Tissue inhibitor of metalloproteases 1
- Par-4:
-
Prostate apoptosis response 4
- NF-kB:
-
Nuclear factor kappa B
- ER:
-
Endoplasmic reticulum
- KA:
-
Kojic acid
References
Lu J, Steeg PS, Price JE, Krishnamurthy S, Mani SA, Reuben J, Cristofanilli M, Dontu G, Bidaut L, Valero V (2009) Breast cancer metastasis: challenges and opportunities. Cancer Res 69(12):4951–4953
Gupta GP, Massague J (2006) Cancer metastasis: building a framework. Cell 127(4):679–695
Jin X, Mu P (2015) Targeting breast cancer metastasis. Breast Cancer 9:S25460
Lee AS (2007) GRP78 induction in cancer: therapeutic and prognostic implications. Can Res 67(8):3496–3499
Dong D, Stapleton C, Luo B, Xiong S, Ye W, Zhang Y, Jhaveri N, Zhu G, Ye R, Liu Z (2011) A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Can Res 71(8):2848–2857
Zhao G, Kang J, Jiao K, Xu G, Yang L, Tang S, Zhang H, Wang Y, Nie Y, Wu K (2015) High expression of GRP78 promotes invasion and metastases in patients with esophageal squamous cell carcinoma. Dig Dis Sci 60(9):2690–2699
Yuan XP, Dong M, Li X, Zhou JP (2015) GRP78 promotes the invasion of pancreatic cancer cells by FAK and JNK. Mol Cell Biochem 398(1–2):55–62
Lamouille S, Xu J, Derynck R (2012) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15(3):178
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Investig 119(6):1420–1428
Bentley R (2006) From miso, sake and shoyu to cosmetics: a century of science for kojic acid. Nat Prod Rep 23(6):1046–1062
Chen Y-H, Lu P-J, Hulme C, Shaw AY (2013) Synthesis of kojic acid-derived copper-chelating apoptosis inducing agents. Med Chem Res 22(2):995–1003
Yoo DS, Lee J, Choi SS, Rho HS, Cho DH, Shin WC, Cho JY (2010) A modulatory effect of novel kojic acid derivatives on cancer cell proliferation and macrophage activation. Die Pharmazie-An Int J Pharm Sci 65(4):261–266
Nawarak J, Huang-Liu R, Kao S-H, Liao H-H, Sinchaikul S, Chen S-T, Cheng S-L (2008) Proteomics analysis of kojic acid treated A375 human malignant melanoma cells. J Proteome Res 7(9):3737–3746
Sharma DK, Rah B, Lambu MR, Hussain A, Yousuf SK, Tripathi AK, Singh B, Jamwal G, Ahmed Z, Chanauria N (2012) Design and synthesis of novel N, N’-glycoside derivatives of 3, 3’-diindolylmethanes as potential antiproliferative agents. MedChemComm 3(9):1082–1091
Nayak D, Amin H, Rah B, Rasool R, Sharma D, Gupta AP, Kushwaha M, Mukherjee D, Goswami A (2015) A therapeutically relevant, 3, 3’-diindolylmethane derivative NGD16 attenuates angiogenesis by targeting glucose regulated protein, 78 kDa (GRP78). Chem Biol Interact 232:58–67
Wisniewska M, Karlberg T, Lehtio L, Johansson I, Kotenyova T, Moche M, Schuler H (2010) Crystal structures of the ATPase domains of four human Hsp70 isoforms: HSPA1L/Hsp70-hom, HSPA2/Hsp70-2, HSPA6/Hsp70B’, and HSPA5/BiP/GRP78. PLoS ONE 5(1):e8625
Viewer S-PDB (2001) Kaplan W; Littlejohn TG. Brief Bioinform 2(2):195–197
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791
Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51(10):2778–2786
DeLano WL (2002) The PyMOL molecular graphics system. http://pymol.org
Amin H, Nayak D, Chakraborty S, Kumar A, Yousuf K, Sharma PR, Ahmed Z, Sharma N, Magotra A, Mukherjee D (2016) Par-4 dependent modulation of cellular β-catenin by medicinal plant natural product derivative 3-azido Withaferin A. Mol Carcinog 55(5):864–881
Nayak D, Kumar A, Chakraborty S, ur Rasool R, Amin H, Katoch A, Gopinath V, Mahajan V, Zilla MK, Rah B (2017) Inhibition of Twist1-mediated invasion by Chk2 promotes premature senescence in p53-defective cancer cells. Cell Death Differ 24(7):1275
Rah B, Rasool Ru, Nayak D, Yousuf SK, Mukherjee D, Kumar LD, Goswami A (2015) PAWR-mediated suppression of BCL2 promotes switching of 3-azido withaferin A (3-AWA)-induced autophagy to apoptosis in prostate cancer cells. Autophagy 11(2):314–331
Rasool RU, Nayak D, Chakraborty S, Faheem MM, Rah B, Mahajan P, Gopinath V, Katoch A, Iqra Z, Yousuf SK (2017) AKT is indispensable for coordinating Par-4/JNK cross talk in p21 downmodulation during ER stress. Oncogenesis 6(5):e341
Ostacolo L, Marra M, Ungaro F, Zappavigna S, Maglio G, Quaglia F, Abbruzzese A, Caraglia M (2010) In vitro anticancer activity of docetaxel-loaded micelles based on poly (ethylene oxide)-poly (epsilon-caprolactone) block copolymers: do nanocarrier properties have a role? J Control Release 148(2):255–263
Anderson VE, Walton MI, Eve PD, Boxall KJ, Antoni L, Caldwell JJ, Aherne W, Pearl LH, Oliver AW, Collins I (2011) CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res 71(2):463–472
Goswami A, Burikhanov R, de Thonel A, Fujita N, Goswami M, Zhao Y, Eriksson JE, Tsuruo T, Rangnekar VM (2005) Binding and phosphorylation of par-4 by akt is essential for cancer cell survival. Mol Cell 20(1):33–44
El-Guendy N, Zhao Y, Gurumurthy S, Burikhanov R, Rangnekar VM (2003) Identification of a unique core domain of par-4 sufficient for selective apoptosis induction in cancer cells. Mol Cell Biol 23(16):5516–5525
Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM (2009) The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell 138(2):377–388
Maloisel F, Kurtz JE, Andres E, Gorodetsky C, Dufour P, Oberling F (1995) Platin salts-induced hemolytic anemia: cisplatin-and the first case of carboplatin-induced hemolysis. Anticancer Drugs 6(2):324–326
Woodward JKL, Neville-Webbe HL, Coleman RE, Holen I (2005) Combined effects of zoledronic acid and doxorubicin on breast cancer cell invasion in vitro. Anticancer Drugs 16(8):845–854
Chaudhry P, Singh M, Parent S, Asselin E (2012) Prostate apoptosis response 4 (Par-4), a novel substrate of caspase-3 during apoptosis activation. Mol Cell Biol 32(4):826–839
Sharma SP (2006) GRP78 as potential predictor of chemoresistance. Lancet Oncol 7(10):800
Lee E, Nichols P, Spicer D, Groshen S, Mimi CY, Lee AS (2006) GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Can Res 66(16):7849–7853
Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS (2003) Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors role of ATP binding site in suppression of caspase-7 activation. J Biol Chem 278(23):20915–20924
Avril T, Vauleon E, Chevet E (2017) Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 6(8):e373
Kern J, Untergasser G, Zenzmaier C, Sarg B, Gastl G, Gunsilius E, Steurer M (2009) GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood 114(18):3960–3967
Pyrko P, Schönthal AH, Hofman FM, Chen TC, Lee AS (2007) The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Can Res 67(20):9809–9816
Uckun FM, Qazi S, Ozer Z, Garner AL, Pitt J, Ma H, Janda KD (2011) Inducing apoptosis in chemotherapy-resistant B-lineage acute lymphoblastic leukaemia cells by targeting HSPA5, a master regulator of the anti-apoptotic unfolded protein response signalling network. Br J Haematol 153(6):741–752
Daneshmand S, Quek ML, Lin E, Lee C, Cote RJ, Hawes D, Cai J, Groshen S, Lieskovsky G, Skinner DG (2007) Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum Pathol 38(10):1547–1552
Scriven P, Coulson S, Haines R, Balasubramanian S, Cross S, Wyld L (2009) Activation and clinical significance of the unfolded protein response in breast cancer. Br J Cancer 101(10):1692
Roller C, Maddalo D (2013) The molecular chaperone GRP78/BiP in the development of chemoresistance: mechanism and possible treatment. Front Pharmacol 4:10
Sun X, Huo X, Luo T, Li M, Yin Y, Jiang Y (2011) The anticancer flavonoid chrysin induces the unfolded protein response in hepatoma cells. J Cell Mol Med 15(11):2389–2398
Umeda Y, Chijiwa S, Furihata K, Furihata K, Sakuda S, Nagasawa H, Watanabe H, Shin-ya K (2005) Prunustatin A, a novel GRP 78 molecular chaperone down-regulator isolated from Streptomyces violaceoniger. J Antibiot 58(3):206–209
Abdelrahim M, Newman K, Vanderlaag K, Samudio I, Safe S (2006) 3, 3’-diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis 27(4):717–728
Sun S, Han J, Ralph WM Jr, Chandrasekaran A, Liu K, Auborn KJ, Carter TH (2004) Endoplasmic reticulum stress as a correlate of cytotoxicity in human tumor cells exposed to diindolylmethane in vitro. Cell Stress Chaperones 9(1):76–87
Tsai Y-L, Zhang Y, Tseng C-C, Stanciauskas R, Pinaud F, Lee AS (2015) Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface. J Biol Chem 290(13):8049–8064
Kang BR, Yang S-H, Chung B-R, Kim W, Kim Y (2016) Cell surface GRP78 as a biomarker and target for suppressing glioma cells. Sci Rep 6:34922
Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC (2008) GRP78 and Cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor Î2 signaling and enhance cell growth. Mol Cell Biol 28(2):666–677
Zhang S, Li L, Lin J-Y, Lin H (2003) Imbalance between expression of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in invasiveness and metastasis of human gastric carcinoma. World J Gastroenterol 9(5):899–904
Lu H, Cao X, Zhang H, Sun G, Fan G, Chen L, Wang S (2014) Imbalance between MMP-2, 9 and TIMP-1 promote the invasion and metastasis of renal cell carcinoma via SKP2 signaling pathways. Tumor Biol 35(10):9807–9813
Kessenbrock K, Plaks V, Werb Z (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141(1):52–67
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2(10):737–744
Katoch A, Suklabaidya S, Chakraborty S, Nayak D, Rasool RU, Sharma D, Mukherjee D, Faheem MM, Kumar A, Sharma PR (2018) Dual Role of Par-4 in abrogation of EMT and switching on Mesenchymal to Epithelial Transition (MET) in metastatic pancreatic cancer cells. Mol Carcinog 57(9):1102–1115
Gupta SC, Kim JH, Prasad S, Aggarwal BB (2010) Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev 29(3):405–434
Li C-W, Xia W, Huo L, Lim S-O, Wu Y, Hsu JL, Chao C-H, Yamaguchi H, Yang N-K, Ding Q (2012) Epithelial-mesenchymal transition induced by TNF-α requires NF-kB-mediated transcriptional upregulation of Twist1. Can Res 72(5):1290–1300
Acknowledgement
We thank our Director, Dr R. A. Vishwakarma (IIIM, Jammu, India), for his encouragement and support to accomplish this work. The study was funded by institutional internal grant (MLP-6002) from Council of Scientific & Industrial Research (CSIR), Govt. of India with publication number IIIM/2297/2019. The authors acknowledge CSIR and Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India for providing fellowships to the research scholars.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies conducted on human participants by any of the co-authors. Animal studies were approved by the Institutional Animal Ethics Committee (IAEC), CPCSEA, Indian Institute of Integrative Medicine, Jammu, India and performed following all the necessary guidelines of IAEC.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nayak, D., Katoch, A., Sharma, D. et al. Indolylkojyl methane analogue IKM5 potentially inhibits invasion of breast cancer cells via attenuation of GRP78. Breast Cancer Res Treat 177, 307–323 (2019). https://doi.org/10.1007/s10549-019-05301-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-019-05301-0