
Abstract
Purpose
Activation of the phosphoinositide 3-kinase (PI3K) pathway is an important resistance mechanism to anti-HER2 therapies. This study aimed to assess the safety and activity of alpelisib (a PI3Kα isoform-specific inhibitor) with T-DM1 in trastuzumab- and taxane-resistant HER2-positive MBC.
Methods
Patients with HER2-positive MBC that had progressed on trastuzumab-based therapy were treated with alpelisib daily and T-DM1 3.6 mg/kg every 3 weeks. The dose-limiting toxicity (DLT), maximum tolerated dose (MTD), adverse events, overall response rate (ORR), and clinical benefit rate (CBR = CR + PR + SD > 6 months) were assessed with descriptive statistics. Progression-free survival (PFS) was calculated by the Kaplan–Meier method.
Results
Seventeen patients were enrolled with a median of 3 prior therapies for metastatic disease. The DLT was a maculopapular rash and MTD was 250 mg alpelisib daily. The most frequently occurring toxicities included fatigue, rash, gastrointestinal side effects, thrombocytopenia, anemia, elevated liver enzymes, and hyperglycemia. Fourteen patients were evaluable for response with an ORR of 43%. In patients with prior treatment and progression on T-DM1 (n = 10), the ORR was 30%. The CBR was 71% in evaluable patients and 60% in those with prior T-DM1. The median PFS was 8.1 months.
Conclusions
The combination of alpelisib and T-DM1 is tolerable and demonstrates activity in trastuzumab-resistant HER2-positive MBC. Furthermore, activity was observed in T-DM1-resistant disease. These data suggest that PIK3CA inhibition targets an important resistance pathway to anti-HER2 therapy, providing rationale for further study of PI3K inhibition in refractory HER2-positive MBC to validate these results.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
The human epidermal growth factor receptor 2 neu (HER2) is an oncogene overexpressed in 20% of breast cancers. HER2 activation leads to downstream signaling along the PI3K/AKT/mTOR and RAS/RAF/MAPK pathways and promotes cell motility, survival and proliferation, and resistance to apoptosis [1]. HER2 overexpression confers a more aggressive breast cancer phenotype, which prior to the introduction of trastuzumab, was associated with a poor prognosis [2,3,4]. The development of HER2-targeted therapies has resulted in significant improvements in the median progression-free and overall survival (PFS and OS), which is now over 4 years in HER2-positive MBC [5,6,7,8]. The EMILIA and TH3RESA studies demonstrated improvement in PFS and OS with the antibody–drug conjugate T-DM1 when compared to capecitabine and lapatinib or treatment of physician’s choice, establishing T-DM1 as a standard second-line treatment for HER2-positive MBC [9,10,11,12]. Unfortunately, therapy for HER2-positive metastatic disease is not curative and despite an initial response to HER2-targeting, patients eventually develop resistance. In fact, the median PFS for second-line T-DM1 is only 6 months [9, 11].
While there are multiple resistance mechanisms to anti-HER2 therapy, the PI3K/AKT pathway is downstream from the site of action of HER2-targeted drugs, making its activation an important resistance pathway [13]. Activation of the PI3K/AKT pathway is frequently seen in breast cancers that progress on HER2-targeted therapy [14,15,16,17,18,19,20]. Recent clinical trials demonstrated hyperactivity of the PI3K pathway in 47 and 41% of HER2-positive MBC in the trastuzumab-naïve and trastuzumab-resistant setting, respectively. Mutations in PIK3CA were noted in 30% of cases [20]. Furthermore, tumors with PIK3CA mutations are less responsive to chemotherapy and anti-HER2 therapy [21]. This data as well as preclinical studies have raised significant interest in targeting the PI3K/AKT/mTOR pathway in HER2-positive breast cancer [22,23,24,25].
The BOLERO 1 and 3 trials evaluated the mTOR inhibitor everolimus in HER2-positive metastatic breast cancer (MBC), but showed only a small benefit at the cost of significant toxicity [26, 27]. Thus, developing an isoform-specific PI3K inhibitor targeting PI3Kα which is encoded by PIK3CA with the goal of maximizing activity but limited off-target toxicities is of interest. Alpelisib (BYL-719) is an oral selective PI3Kα isoform inhibitor [28, 29]. Clinical trials in patients with advanced solid tumors with PI3KCA alterations and advanced hormone receptor-positive, HER2-negative breast cancer have shown safety and tolerability of alpelisib [30, 31].
We were interested in evaluating alpelisib in trastuzumab-resistant MBC given the compelling data demonstrating the frequency and relevance of PI3K pathway activation in this setting. T-DM1 was chosen as the companion anti-HER2 therapy because of its established role as second-line therapy with more limited activity of trastuzumab in this setting [32, 33]. We conducted a phase I clinical trial to determine the maximum tolerated dose (MTD), safety, and activity of alpelisib with T-DM1 in HER2-positive MBC. We hypothesized that the combination of a specific PI3Kα inhibitor alpelisib with T-DM1 would have acceptable toxicities and demonstrate activity in trastuzumab-refractory HER2-positive breast cancer patients.
Patients and methods
Patient population
Patients had histologically confirmed HER2-positive inoperable locally advanced or MBC. Locally advanced disease must be inoperable and have progressed within 6 months of trastuzumab and/or taxane treatment and MBC must have progressed during or after trastuzumab and taxane therapy. Other key inclusion criteria included ECOG performance status ≤ 2 and adequate bone marrow, renal, and hepatic functions. Patients were not required to have measurable disease. Key exclusion criteria included clinically manifested diabetes (fasting plasma glucose > 140 mg/dL or a history of steroid-induced diabetes), known history of liver disease, or other active medical problems.
Approval was obtained from the ethics committee at Northwestern University and regulatory authorities. All patients gave informed consent. The study followed the Declaration of Helsinki and Good Clinical Practice guidelines.
Study design
This was a phase I, open-label, single-arm, single center, dose escalation study of alpelisib in combination with T-DM1 using a standard 3 + 3 design with a dose expansion cohort at the maximum tolerated dose (MTD). Patients received T-DM1 3.6 mg/kg (based on the FDA-approved dose) on day 1 of each cycle +/− 3 days (1 cycle = 21 days). The first cohort received alpelisib at 300 mg oral daily (25% below the single-agent MTD). Patients were instructed to take alpelisib about 1 h after a light meal at approximately the same time daily. The planned alpelisib escalation tiers were 350 and 400 mg daily and de-escalation tiers were 250, 200, and 150 mg daily.
The primary endpoints were MTD and frequency and severity of toxicities of alpelisib in combination with T-DM1. All patients who received at least 1 dose of the study medication and T-DM1 were considered evaluable for the primary endpoint. The secondary endpoints were preliminary efficacy by assessment of PFS and objective response rate (ORR). All patients who received at least 3 cycles of therapy and had imaging at first response assessment were considered evaluable for the secondary endpoint.
Toxicities were defined using the NCI’s CTCAE version 4.03. Dose-limiting toxicity (DLT) was defined as a clinically significant grade ≥ 3 toxicity being at least possibly related to the study medication, which occurred ≤ 21 days following the first dose of alpelisib or an adverse event grade < 3 that led to dose interruption of alpelisib for greater than 7 days. The MTD was defined as the highest dose at which no more than one-third of patients experienced any DLTs during the first cycle. Patients were treated until disease progression, unacceptable toxicity, investigator decision, or withdrawal of consent.
Safety and efficacy assessments
Patient assessment by clinical and laboratory evaluation was conducted at baseline and on day 1 of each 21-day cycle. ECG and hemoglobin (Hgb) A1c were assessed at baseline and every 3 cycles. PFS was defined as time from the first study treatment to the first occurrence of progression or death. ORR was assessed every 3 cycles with imaging by CT of the chest, abdomen, and pelvis and bone scan or Positron Emission Tomography (PET)/CT. ORR (partial or complete response) was defined as the best response by RECIST v1.1 criteria, and by clinical examination for subjects with only skin disease (followed with serial photographs).
Statistical analysis
The target sample size was 19–28 patients depending on occurrence of DLTs. This 3 + 3 design had a 91% chance of dose escalating when the true toxicity rate for that dose was 10%. Response was analyzed using exact binomial methods and 95% confidence intervals. PFS was analyzed using Kaplan–Meier curves. Exploratory objectives were analyzed by performing a descriptive analysis of the frequency of specific genetic alterations.
Correlative studies
Expression of PTEN and AKT were assessed by immunohistochemistry of archival tumor specimen (from a banked specimen or optional biopsy). PIK3CA gene mutation status was recorded when known from prior next generation sequencing of a primary or metastatic tumor.
Results
Study population
Between May 2014 and February 2015, 17 patients with HER2-positive MBC were enrolled and received at least 1 dose of alpelisib (Table 1). The median age was 53 (range 40–67). The median number of metastatic sites was 3 (1–5) and 82% (n = 14) of patients had visceral disease. The most common sites of metastases were bone (59%), liver (53%), lymph nodes (47%), lung (41%), and brain (29%). Nine patients (53%) had hormone receptor-negative disease. Eight patients (47%) initially presented with de novo metastatic disease. In the metastatic setting patients had received a median of 3 lines (range 0–12) of prior therapy. All patients had received prior trastuzumab and taxane (94% for metastatic disease and 6% in the (neo)adjuvant setting). For treatment of metastatic disease, 94% received pertuzumab, 47% received lapatinib, and 59% received T-DM1. Five (50%) of patients with prior T-DM1 had previously experienced a clinical response to T-DM1. All patients with prior T-DM1 (n = 10) had progression of disease on T-DM1 (after median 8 cycles). T-DM1 was the most recent therapy in 4 patients.
DLT and MTD
Of the 17 patients who received alpelisib, 6 patients received a starting dose of 300 mg daily (cohort 1) and 11 received a starting dose of 250 mg daily (de-escalation cohort − 1). In cohort 1, 1 patient was deemed not evaluable due to death from rapidly progressive disease after only 1 dose of alpelisib. Of the initial 3 evaluable patients receiving 300 mg daily, 1 patient developed grade 3 thrombocytopenia that was attributed to T-DM1 (the patient had thrombocytopenia with prior T-DM1 therapy and this toxicity resolved upon stopping T-DM1). The data monitoring committee recommended enrollment of 3 additional patients at 300 mg daily to ensure safety of the combination. However, the next 2 patients experienced DLTs (both grade 3 rash). Therefore, de-escalation cohort − 1 was opened and enrolled 3 patients at a starting dose of 250 mg daily, in which no DLTs occurred. Accordingly, the MTD and recommended phase II dose for alpelisib in combination with T-DM1 was determined to be 250 mg daily with rash as the DLT at the 300 mg dose. A dose expansion cohort of an additional 8 patients was enrolled for a total of 11 patients who received alpelisib 250 mg daily as the starting dose. Three (27%) of these patients developed a grade 3 rash and 1 (9%) developed grade 3 hyperglycemia during cycle 1 (Table 2).
Adverse events (AE)
The most common treatment-related non-hematologic toxicities were elevation in transaminases (n = 13, 76%), hyperglycemia (n = 9, 53%), fatigue (n = 9, 53%), rash (n = 8, 47%), and nausea (n = 7, 41%) (Table 3). Hematologic toxicities included thrombocytopenia (n = 9, 53%), anemia (n = 7, 41%), and leukopenia (n = 5, 29%). Elevation in transaminases was always grade 1 or 2, was thought to be at least partially related to T-DM1, and did not require any treatment modifications or other management. T-DM1 was thought to be responsible for most hematologic toxicity, particularly in this heavily pre-treated population (Table 4).
Ten patients (59%) experienced a ≥ grade 3 treatment-related toxicity. Grade 3 treatment-related AEs included rash (n = 7), hyperglycemia (n = 3), anorexia (n = 2), hypertension (n = 2), thrombocytopenia (n = 1), weight loss (n = 1), QTC prolongation (n = 1), pancreatitis (n = 1), and anemia/abnormal uterine bleeding (n = 1). One Grade 4 AE occurred (thrombocytopenia) likely due to T-DM1. One grade 5 AE occurred on study, which was thought to be unrelated to the study drug in a patient who received only 1 dose of the study drugs. The patient developed hypoxic respiratory failure with a CT chest showing marked progression of metastatic disease with likely superimposed infection.
Management of serious adverse events
The DLT and most common serious adverse event was a maculopapular rash (n = 7, 41%). No standard premedication was recommended by the study protocol initially. The rash developed cycle 1, days 10 to 15 in 6 of 7 patients with a grade 3 rash. This occurred in both the 300 mg alpelisib and 250 mg alpelisib cohort. The grade 3 rash resolved in a median of 8 days (range 5–15) with alpelisib interruption and dose reduction. Topical steroid cream, oral antihistamines, oral steroids, and dermatology consultation were ordered as needed. Oral steroids (prednisone 30–80 mg daily) were only used in cohort − 1 (n = 4) over a 5- to 21-day course that included a taper for some patients. The rash was biopsied in one patient and demonstrated perivascular lymphocytic dermatitis, suggestive of dermal hypersensitivity reaction such as a drug eruption. After improvement in the grade 3 rash, some patients experienced a grade 1 or 2 rash that was managed with as needed topical steroid cream and oral antihistamines per protocol.
Other grade 3/4 toxicities were managed as follows: hyperglycemia was managed with diet modification, anti-hyperglycemic medications, and endocrinology consultations when necessary; anorexia and weight loss were managed with nutritional supplements and dose reduction; hypertension reaching the 160 s mm Hg systolic, was asymptomatic, and resolved without intervention; QTC prolongation in a patient with a history of prolonged QTC resolved with dose reduction; grade 3 pancreatitis was thought to be possibly alpelisib related and was managed as per hospital protocol; and hematologic toxicities (thrombocytopenia, anemia, and abnormal uterine bleeding) were managed by dose modification or holding T-DM1 and transfusion if needed.
Tolerability
The median number of cycles of treatment in patients who completed at least 1 cycle (n = 14) was 11 (3–19). Three patients in cohort 1 and 5 patients in cohort − 1 required dose reductions in alpelisib for toxicity. Toxicities requiring dose reductions were rash, hyperglycemia, anorexia/weight loss, and prolonged QTC (Table 2). At the final analysis, all patients had discontinued the drug. Of the 17 patients who received at least 1 dose of the drug, treatment was discontinued due to progression of disease in 14 patients and difficulty complying with study procedures and follow-up in 2 patients. One patient was taken off the study due to recurrent hyperglycemia after non-adherence to recommended interventions.
Clinical activity
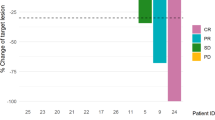
Fourteen patients were evaluable for response (Table 3). Three patients received less than 1 cycle and were not evaluable for response. Two of these patients discontinued treatment due to difficulty complying with study requirements and follow-up visits and one patient died of progressive disease after taking only one dose of the study medication. The ORR was 43%. Exploratory analysis found that clinical benefit rate [CBR; complete response (CR) + partial response (PR) + > 6 mo stable disease (SD)] was experienced by 10 patients (71%).
In the subgroup of 10 patients with prior progression on T-DM1, the ORR was 30% and the CBR was 60%. Only 2 patients, both with prior TDM1 exposure, had progressive disease as their best response, of which one had asymptomatic progression of CNS metastases with stable systemic disease.
The median follow-up was 11.6 months (0.3–19.5). Among all patients evaluable for response, the median PFS (Fig. 1) was 8.1 months (95% CI 3.9–10.8). In the 10 patients with prior T-DM1, the median PFS was 6.3 months (95% CI 1.6–10.5) and in the 4 patients without prior T-DM1 median PFS was 10.8 months (95% CI 8.1–12.5).

Progression-free survival for all patients and by prior T-DM1 exposure
PI3K pathway aberrations
Tumor samples were available from 7 patients, all of whom had a sample from the primary site and 4 had tissue from a metastatic site (Fig. 2). Immunohistochemistry (IHC) showed PTEN loss in 2 patients and AKT increased expression in 3 patients. Six patients had next generation sequencing (NGS) done of a metastatic tumor sample prior to therapy of which 4 had mutations in PIK3CA. In total, 9 of 11 patients that had a tumor sample from which IHC and/or NGS demonstrated aberrations in the PI3K pathway (PTEN loss, AKT overexpression, or PIK3CA mutation).

Aberrations in PI3K pathway and response. a PI3K pathway aberrations by IHC of NGS in patients with evaluable tissue. b Duration of response in patients with evaluable tissue
Of patients with an overall response, two had evaluable tissue for IHC assessment with one showing PTEN loss and the other showing AKT overexpression. Of patients with clinical benefit and evaluable samples (n = 6), 5 had pathway aberrations. In patients with prior T-DM1 exposure and evaluable tissue (n = 8), 6 had pathway aberrations.
Discussion
The results demonstrate the safety and tolerability of the alpelisib and T-DM1 combination with a DLT of rash and a recommended phase II dose of alpelisib 250 mg daily with standard T-DM1 dosing. The study showed activity of the combination, including a median PFS of 8.1 months, ORR of 43%, and a CBR of 71% despite a heavily pre-treated population. Most impressively, even patients with progression on prior T-DM1 demonstrated response (30%) and clinical benefit (60%) with a median PFS of 6.2 months.
In this study although toxicities were common (59% of patients experienced ≥ grade 3 toxicity), they were manageable, with evaluable patients receiving a median of 11 cycles of therapy. Only one patient was taken off study due to recurrent hyperglycemia; however, the patient did not comply with treatment recommendations. The alpelisib-related DLTs of a maculopapular rash occurred in 7 patients at both the 250 and 300 mg dose levels and hyperglycemia occurred in 1 patient. These toxicities were managed with additional medication, dose interruption, and/or dose reduction as described above, and did not preclude further tolerance of or receiving subsequent benefit from the alpelisib and T-DM1 combination. The consistent timing and presentation of the maculopapular rash, may allow for implementation of strategies to mitigate it and improve alpelisib tolerability in future studies.
The study population experienced frequent but mild gastrointestinal toxicities (anorexia, weight loss, nausea, transaminase elevation) and fatigue that was likely due to both alpelisib and T-DM1 and did not result in medication discontinuation. One patient developed grade 3 pancreatitis which has not been previously reported with PI3K inhibitors, but has been reported with T-DM1 [34]. Although hematologic toxicities, particularly thrombocytopenia, are expected with T-DM1 given it contains a cytotoxic agent, the prevalence was higher with this combination than described with T-DM1 monotherapy [9, 30]. This may be related to evaluation in a heavily pre-treated population and is less likely related to alpelisib given that these hematologic toxicities are not common with alpelisib monotherapy [30].
Alpelisib when given with letrozole was tolerated at a higher dose (MTD alpelisib 300 mg daily) with toxicities that included rash (all grades 42%, ≥ grade 3 8%), hyperglycemia (all grades 62%, ≥ grade 3 15%), nausea (all grades 62%, ≥ grade 3 4%), fatigue (all grades 54%, ≥ grade 3 0%), and transaminase elevation (all grades 19%, ≥ grade 3 4%) [30, 31]. Similar toxicities are noted with other PI3K inhibitors, with pan-PI3K also associated with depression and anxiety [35,36,37,38,39,40]. Toxicities of single-agent T-DM1 include thrombocytopenia (all grade 29%, ≥ grade 3 10%), increased AST (all grades 23%, ≥ grade 3 4%), fatigue (all grade 45%, ≥ grade 3 3%) [41].
The combination of a PI3K inhibitor and TDM1, when compared with the studies described above, may have some overlapping low-grade toxicities resulting in higher rates of gastrointestinal toxicity and fatigue than each medication alone. Only one other trial that has reported the use of alpelisib with a cytotoxic agent (paclitaxel), but toxicity prevented further evaluation of this combination [42]. Studies of the pan-PI3K inhibitors buparlisib and pictilisib with chemotherapy showed increased toxicities requiring dose modifications [43,44,45]. It is difficult to assess whether the increased specificity of alpelisib led to a more favorable side effect profile than pan-PI3K inhibitors given its combination with a cytotoxic agent and evaluation in a heavily pre-treated population [35,36,37,38,39,40, 46, 47]. An ongoing phase III study of alpelisib with fulvestrant in HR-positive HER2-negative patients (SOLAR-1) may provide additional insight into the incidence and severity of alpelisib toxicities and how they compare to those of pan-PI3K inhibitors [47, 48].
T-DM1 is the preferred standard therapy after progression on trastuzumab given its superior PFS, OS, and toxicity profile [9]. The safety or efficacy of combining T-DM1 with inhibitors of the PI3K pathway has not been previously reported. This study supports further investigation of combination T-DM1 and alpelisib in extending or enhancing benefit from T-DM1 alone. It is unclear whether the activity demonstrated in this study is primarily from alpelisib or from alpelisib resensitizing the tumor to HER2-targeting with T-DM1 by inhibiting a resistance pathway. Although resistance pathways for T-DM1 are not as well described as those for trastuzumab, their shared binding to HER2 suggests that activation of the downstream PI3K pathway is a possible resistance mechanism to T-DM1 [49].
Inhibitors of the PI3K/AKT/mTOR pathway are currently only investigational in HER2-positive MBC. Adding everolimus (mTOR inhibitor) to trastuzumab and chemotherapy in trastuzumab- and taxane-refractory disease showed modest improvement in PFS in the BOLERO-3 trial, however no benefit was seen in the first-line setting in BOLERO-1 [26, 27]. Early phase studies evaluating PI3K inhibitors (buparlisib and pictilisib) with trastuzumab in HER2-positive disease have suggested activity, safety, and tolerability [24, 25, 35, 36].
The toxicities of PI3K pathway inhibition and the development of other active therapies for HER2-positive disease highlight the importance of identifying biomarkers that might select for patients likely to derive the greatest benefit from therapy. In two large phase III studies of inhibitors of the PI3K pathway in HER2-negative MBC, patients with PI3K pathway aberrations had improved treatment responses [47, 50]. Additionally, subgroup analysis from BOLERO-1 and − 3 suggested only patients with a hyperactive PI3K pathway derived benefit from the addition of everolimus [20]. Our limited correlative studies show that this trastuzumab-resistant population is enhanced for PI3K pathway activation. However, the limited patient samples collected cannot provide definite value in predicting response to alpelisib and T-DM1 therapy. Rigorous evaluation of the PI3K pathway should be an important component of future studies involving PI3K inhibitors.
These are several limitations in this study. A small sample size and single-arm design make it difficult to determine the true impact of clinical or pathologic features on the likelihood of benefit from the combination. Additionally, subgroup analysis of T-DM1 exposed and non-exposed cohorts and calculation of clinical benefit rate was not pre-specified. There was no standardized premedications or a specific protocol for management of the alpelisib-associated rash. If prophylactic measures are initiated in future studies, it potentially could mitigate the DLT. Tissue for correlative studies was only available from a subset of patients. Finally, samples for these correlatives were from archival tissue obtained at variable times during a patient’s course so we do not know if patients had PI3K pathway activation at the time of treatment. Future studies may include biopsy or circulating tumor DNA prior to initiating therapy to more consistently evaluate PI3K pathway abnormalities as a biomarker.
This study provides important evidence for the safety, tolerability, and preliminary efficacy of alpelisib and T-DM1 in trastuzumab-refractory HER2-positive MBC patients. The activity of this combination in trastuzumab- and T-DM1-refractory patients supports further study of the combination with proactive strategies for managing toxicities in phase II studies and highlights the potential of PI3K inhibition in T-DM1-resistant disease.
References
Moasser MM (2007) The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 26(45):6469–6487. https://doi.org/10.1038/sj.onc.1210477
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235(4785):177–182
Tandon AK, Clark GM, Chamness GC, Ullrich A, McGuire WL (1989) HER-2/neu oncogene protein and prognosis in breast cancer. J Clin Oncol 7(8):1120–1128. https://doi.org/10.1200/JCO.1989.7.8.1120
Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN (2009) The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 14(4):320–368. https://doi.org/10.1634/theoncologist.2008-0230
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344(11):783–792. https://doi.org/10.1056/nejm200103153441101
Swain SM, Kim SB, Cortes J, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A, Knott A, Clark E, Ross G, Benyunes MC, Baselga J (2013) Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 14(6):461–471. https://doi.org/10.1016/S1470-2045(13)70130-X
Swain SM, Baselga J, Kim S-B, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero J-M, Schneeweiss A, Heeson S, Clark E, Ross G, Benyunes MC, Cortés J (2015) Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 372(8):724–734. https://doi.org/10.1056/NEJMoa1413513
Mendes D, Alves C, Afonso N, Cardoso F, Passos-Coelho JL, Costa L, Andrade S, Batel-Marques F (2015) The benefit of HER2-targeted therapies on overall survival of patients with metastatic HER2-positive breast cancer – a systematic review. Breast Cancer Res 17(1):140. https://doi.org/10.1186/s13058-015-0648-2
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh D-Y, Diéras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K (2012) Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 367(19):1783–1791. https://doi.org/10.1056/NEJMoa1209124
Diéras V, Miles D, Verma S, Pegram M, Welslau M, Baselga J, Krop IE, Blackwell K, Hoersch S, Xu J, Green M, Gianni L Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol 18 (6):732–742. https://doi.org/10.1016/S1470-2045(17)30312-1
Krop IE, Kim SB, Gonzalez-Martin A, LoRusso PM, Ferrero JM, Smitt M, Yu R, Leung AC, Wildiers H (2014) Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. Lancet Oncol 15(7):689–699. https://doi.org/10.1016/s1470-2045(14)70178-0
Krop IE, Kim S-B, Martin AG, LoRusso PM, Ferrero J-M, Badovinac-Crnjevic T, Hoersch S, Smitt M, Wildiers H Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol 18 (6):743–754. https://doi.org/10.1016/S1470-2045(17)30313-3
Pohlmann PR, Mayer IA, Mernaugh R (2009) Resistance to trastuzumab in breast cancer. Clin Cancer Res 15(24):7479–7491. https://doi.org/10.1158/1078-0432.ccr-09-0636
Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R (2007) A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 12(4):395–402. https://doi.org/10.1016/j.ccr.2007.08.030
Chandarlapaty S, Sakr RA, Giri D, Patil S, Heguy A, Morrow M, Modi S, Norton L, Rosen N, Hudis C, King TA (2012) Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res 18(24):6784–6791. https://doi.org/10.1158/1078-0432.ccr-12-1785
Esteva FJ, Guo H, Zhang S, Santa-Maria C, Stone S, Lanchbury JS, Sahin AA, Hortobagyi GN, Yu D (2010) PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol 177(4):1647–1656. https://doi.org/10.2353/ajpath.2010.090885
D’Amato V, Raimondo L, Formisano L, Giuliano M, De Placido S, Rosa R, Bianco R (2015) Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat Rev 41(10):877–883. https://doi.org/10.1016/j.ctrv.2015.08.001
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D (2004) PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6(2):117–127. https://doi.org/10.1016/j.ccr.2004.06.022
Baselga J, Lewis Phillips GD, Verma S, Ro J, Huober J, Guardino AE, Samant MK, Olsen S, de Haas SL, Pegram MD (2016) Relationship between tumor biomarkers and efficacy in EMILIA, a phase III study of trastuzumab emtansine in HER2-positive metastatic breast cancer. Clin Cancer Res 22(15):3755–3763. https://doi.org/10.1158/1078-0432.ccr-15-2499
André F, Hurvitz S, Fasolo A, Tseng L-M, Jerusalem G, Wilks S, O’Regan R, Isaacs C, Toi M, Burris H, He W, Robinson D, Riester M, Taran T, Chen D, Slamon D (2016) Molecular alterations and everolimus efficacy in human epidermal growth factor receptor 2–overexpressing metastatic breast cancers: combined exploratory biomarker analysis from BOLERO-1 and BOLERO-3. J Clin Oncol 34(18):2115–2124. https://doi.org/10.1200/JCO.2015.63.9161
Loibl S, von Minckwitz G, Schneeweiss A, Paepke S, Lehmann A, Rezai M, Zahm DM, Sinn P, Khandan F, Eidtmann H, Dohnal K, Heinrichs C, Huober J, Pfitzner B, Fasching PA, Andre F, Lindner JL, Sotiriou C, Dykgers A, Guo S, Gade S, Nekljudova V, Loi S, Untch M, Denkert C (2014) PIK3CA mutations are associated with lower rates of pathologic complete response to anti–human epidermal growth factor receptor 2 (HER2) therapy in primary HER2-overexpressing breast cancer. J Clin Oncol 32(29):3212–3220. https://doi.org/10.1200/JCO.2014.55.7876
Nahta R, O’Regan RM (2010) Evolving strategies for overcoming resistance to HER2-directed therapy: targeting the PI3K/Akt/mTOR pathway. Clin Breast Cancer 10 Suppl 3:S72-78. https://doi.org/10.3816/CBC.2010.s.015
Wilks ST (2015) Potential of overcoming resistance to HER2-targeted therapies through the PI3K/Akt/mTOR pathway. Breast 24(5):548–555. https://doi.org/10.1016/j.breast.2015.06.002
O’Brien NA, McDonald K, Tong L, von Euw E, Kalous O, Conklin D, Hurvitz SA, di Tomaso E, Schnell C, Linnartz R, Finn RS, Hirawat S, Slamon DJ (2014) Targeting PI3K/mTOR overcomes resistance to HER2-targeted therapy independent of feedback activation of AKT. Clin Cancer Res 20(13):3507–3520. https://doi.org/10.1158/1078-0432.ccr-13-2769
Yao E, Zhou W, Lee-Hoeflich ST, Truong T, Haverty PM, Eastham-Anderson J, Lewin-Koh N, Gunter B, Belvin M, Murray LJ, Friedman LS, Sliwkowski MX, Hoeflich KP (2009) Suppression of HER2/HER3-mediated growth of breast cancer cells with combinations of GDC-0941 PI3K inhibitor, trastuzumab, and pertuzumab. Clin Cancer Res 15(12):4147–4156. https://doi.org/10.1158/1078-0432.ccr-08-2814
Hurvitz SA, Andre F, Jiang Z, Shao Z, Mano MS, Neciosup SP, Tseng L-M, Zhang Q, Shen K, Liu D, Dreosti LM, Burris HA, Toi M, Buyse ME, Cabaribere D, Lindsay M-A, Rao S, Pacaud LB, Taran T, Slamon D Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): a phase 3, randomised, double-blind, multicentre trial. Lancet Oncol 16 (7):816–829. https://doi.org/10.1016/S1470-2045(15)00051-0
Andre F, O’Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G, Masuda N, Wilks S, Arena F, Isaacs C, Yap YS, Papai Z, Lang I, Armstrong A, Lerzo G, White M, Shen K, Litton J, Chen D, Zhang Y, Ali S, Taran T, Gianni L (2014) Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol 15(6):580–591. https://doi.org/10.1016/s1470-2045(14)70138-x
Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J, Aichholz R, Hamon J, Fabbro D, Caravatti G (2013) Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorganic Med Chem Lett 23(13):3741–3748. https://doi.org/10.1016/j.bmcl.2013.05.007
Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De Pover A, Furet P, Gao H, Ferretti S, Wang Y, Trappe J, Brachmann SM, Maira S-M, Wilson C, Boehm M, Garcia-Echeverria C, Chene P, Wiesmann M, Cozens R, Lehar J, Schlegel R, Caravatti G, Hofmann F, Sellers WR (2014) Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther 13(5):1117–1129. https://doi.org/10.1158/1535-7163.mct-13-0865
Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, Juric D, Solit D, Berger MF, Won HH, Li Y, Cantley LC, Winer E, Arteaga CL (2017) A phase Ib study of alpelisib (BYL719), a PI3Kalpha-specific inhibitor, with letrozole in ER+/HER2- metastatic breast cancer. Clin Cancer Res 23(1):26–34. https://doi.org/10.1158/1078-0432.ccr-16-0134
Juric D, Burris H, Schuler M, Schellens J, Berlin J, Seggewiß-Bernhardt R, Gil-Martin M, Gupta A, Rodon J, Tabernero J, Janku F, Rugo HS, Bootle D, Quadt C, Coughlin C, Demanse D, Blumenstein L, Baselga J (2014) Phase I study of the PI3Kα inhibitor BYL719, as a single agent in patients with advanced solid tumors (AST). Ann Oncol 25 (suppl_4):iv150–iv150. https://doi.org/10.1093/annonc/mdu331.11
Santa-Maria CA, Nye L, Mutonga MB, Jain S, Gradishar WJ (2016) Management of metastatic HER2-positive breast cancer: where are we and where do we go from here? Oncology (Williston Park, NY) 30 (2):148–155
von Minckwitz G, du Bois A, Schmidt M, Maass N, Cufer T, de Jongh FE, Maartense E, Zielinski C, Kaufmann M, Bauer W, Baumann KH, Clemens MR, Duerr R, Uleer C, Andersson M, Stein RC, Nekljudova V, Loibl S (2009) Trastuzumab beyond progression in human epidermal growth factor receptor 2–positive advanced breast cancer: a german breast group 26/breast international group 03–05 study. J Clin Oncol 27(12):1999–2006. https://doi.org/10.1200/JCO.2008.19.6618
Muzaffar M, Jia J, Liles D, Naveed M, Kumari A (2016) Acute pancreatitis associated with ado-trastuzumab emtansine. Am J Ther 23(2):e572-574
Tolaney S, Burris H, Gartner E, Mayer IA, Saura C, Maurer M, Ciruelos E, Garcia AA, Campana F, Wu B, Xu Y, Jiang J, Winer E, Krop I (2015) Phase I/II study of pilaralisib (SAR245408) in combination with trastuzumab or trastuzumab plus paclitaxel in trastuzumab-refractory HER2-positive metastatic breast cancer. Breast Cancer Res Treat 149(1):151–161. https://doi.org/10.1007/s10549-014-3248-4
Saura C, Bendell J, Jerusalem G, Su S, Ru Q, De Buck S, Mills D, Ruquet S, Bosch A, Urruticoechea A, Beck JT, Di Tomaso E, Sternberg DW, Massacesi C, Hirawat S, Dirix L, Baselga J (2014) Phase Ib study of Buparlisib plus trastuzumab in patients with HER2-positive advanced or metastatic breast cancer that has progressed on trastuzumab-based therapy. Clin Cancer Res 20(7):1935–1945. https://doi.org/10.1158/1078-0432.ccr-13-1070
Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, Clarke PA, Raynaud FI, Levy G, Ware JA, Mazina K, Lin R, Wu J, Fredrickson J, Spoerke JM, Lackner MR, Yan Y, Friedman LS, Kaye SB, Derynck MK, Workman P, de Bono JS (2015) First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res 21(1):77–86. https://doi.org/10.1158/1078-0432.ccr-14-0947
Shapiro GI, Rodon J, Bedell C, Kwak EL, Baselga J, Brana I, Pandya SS, Scheffold C, Laird AD, Nguyen LT, Xu Y, Egile C, Edelman G (2014) Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res 20(1):233–245. https://doi.org/10.1158/1078-0432.ccr-13-1777
Patnaik A, Appleman LJ, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Weiss GJ, Sachdev JC, Chadha M, Fulk M, Ejadi S, Mountz JM, Lotze MT, Toledo FGS, Chu E, Jeffers M, Peña C, Xia C, Reif S, Genvresse I, Ramanathan RK (2016) First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol 27(10):1928–1940. https://doi.org/10.1093/annonc/mdw282
Juric D, Krop I, Ramanathan RK, Wilson TR, Ware JA, Sanabria Bohorquez SM, Savage HM, Sampath D, Salphati L, Lin RS, Jin H, Parmar H, Hsu JY, Von Hoff DD, Baselga J (2017) Phase I dose-escalation study of Taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors. Cancer Discov. https://doi.org/10.1158/2159-8290.cd-16-1080
Krop I, Winer EP (2014) Trastuzumab emtansine: a novel antibody–drug conjugate for HER2-positive breast cancer. Clin Cancer Res 20(1):15–20. https://doi.org/10.1158/1078-0432.ccr-13-0541
Rodón J, Curigliano G, Delord JP, Harb W, Azaro A, Donnet V, Han Y, Blumenstein L, Wilke C, Beck JT (2016) A phase Ib dose-finding study of alpelisib (ALP; BYL719) and paclitaxel (PTX) in advanced solid tumors (aST). Ann Oncol 27(suppl_6):375P-375P. https://doi.org/10.1093/annonc/mdw368.18
Vuylsteke P, Huizing M, Petrakova K, Roylance R, Laing R, Chan S, Abell F, Gendreau S, Rooney I, Apt D, Zhou J, Singel S, Fehrenbacher L (2016) Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann Oncol 27(11):2059–2066. https://doi.org/10.1093/annonc/mdw320
Loibl S, de la Pena L, Nekljudova V, Zardavas D, Michiels S, Denkert C, Rezai M, Bermejo B, Untch M, Lee SC, Turri S, Urban P, Kummel S, Steger G, Gombos A, Lux M, Piccart MJ, Von Minckwitz G, Baselga J, Loi S (2017) Neoadjuvant buparlisib plus trastuzumab and paclitaxel for women with HER2 + primary breast cancer: a randomised, double-blind, placebo-controlled phase II trial (NeoPHOEBE). Eur J Cancer 85:133–145. https://doi.org/10.1016/j.ejca.2017.08.020
Martin M, Chan A, Dirix L, O’Shaughnessy J, Hegg R, Manikhas A, Shtivelband M, Krivorotko P, Batista Lopez N, Campone M, Ruiz Borrego M, Khan QJ, Beck JT, Ramos Vazquez M, Urban P, Goteti S, Di Tomaso E, Massacesi C, Delaloge S (2017) A randomized adaptive phase II/III study of buparlisib, a pan-class I PI3K inhibitor, combined with paclitaxel for the treatment of HER2- advanced breast cancer (BELLE-4). Ann Oncol 28(2):313–320. https://doi.org/10.1093/annonc/mdw562
Ma CX, Luo J, Naughton M, Ademuyiwa F, Suresh R, Griffith M, Griffith OL, Skidmore ZL, Spies NC, Ramu A, Trani L, Pluard T, Nagaraj G, Thomas S, Guo Z, Hoog J, Han J, Mardis E, Lockhart C, Ellis MJ (2016) A phase I Trial of BKM120 (Buparlisib) in combination with fulvestrant in postmenopausal women with estrogen receptor-positive metastatic breast cancer. Clin Cancer Res 22(7):1583–1591. https://doi.org/10.1158/1078-0432.ccr-15-1745
Baselga J, Im SA, Iwata H, Cortes J, De Laurentiis M, Jiang Z, Arteaga CL, Jonat W, Clemons M, Ito Y, Awada A, Chia S, Jagiello-Gruszfeld A, Pistilli B, Tseng LM, Hurvitz S, Masuda N, Takahashi M, Vuylsteke P, Hachemi S, Dharan B, Di Tomaso E, Urban P, Massacesi C, Campone M (2017) Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 18(7):904–916. https://doi.org/10.1016/s1470-2045(17)30376-5
Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, Yardley DA, Melichar B, Forero-Torres A, Lee SC, de Boer R, Petrakova K, Vallentin S, Perez EA, Piccart M, Ellis M, Winer E, Gendreau S, Derynck M, Lackner M, Levy G, Qiu J, He J, Schmid P (2016) Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 17(6):811–821. https://doi.org/10.1016/S1470-2045(16)00106-6
Barok M, Tanner M, Köninki K, Isola J (2011) Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res 13(2):R46-R46. https://doi.org/10.1186/bcr2868
Kim S-B, Dent R, Im S-A, Espié M, Blau S, Tan AR, Isakoff SJ, Oliveira M, Saura C, Wongchenko MJ, Kapp AV, Chan WY, Singel SM, Maslyar DJ, Baselga J (2017) Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. The Lancet Oncology 18:1360–1372
Financial support
Novartis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Cristofanilli is a consultant for Novartis, received funding and remuneration from Pfizer. Dr. Santa-Maria received research funding from Medimmune and Pfizer. The remaining authors declare they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Jain, S., Shah, A.N., Santa-Maria, C.A. et al. Phase I study of alpelisib (BYL-719) and trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer (MBC) after trastuzumab and taxane therapy. Breast Cancer Res Treat 171, 371–381 (2018). https://doi.org/10.1007/s10549-018-4792-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-018-4792-0